

Abstract
Adoptive cell transfer (ACT) – based immunotherapies can mediate objective cancer regression in animal models and in up to 70% of patients with metastatic melanoma but it remains unclear whether the tumor vasculature impedes the egress of tumor specific T cells thus hindering this immunotherapy. Disruption of the proangiogenic interaction of vascular endothelial growth factor (VEGF) with its receptor (VEGFR-2) has been reported to “normalize” tumor vasculature, enhancing the efficacy of chemotherapeutic agents by increasing their delivery to the tumor intersitium. We thus sought to determine whether disrupting VEGF/VEGFR-2 signaling could enhance the effectiveness of ACT in a murine cancer model. The administration of an antibody against mouse VEGF synergized with ACT to enhance inhibition of established, vascularised, B16 melanoma (p=0.009) and improve survival (p=0.003). Additive effects of an antibody against VEGFR-2 in conjunction with ACT were seen in this model (p=0.013). Anti VEGF, but not anti VEGFR-2, antibody significantly increased infiltration of transferred cells into the tumor. Thus, normalization of tumor vasculature through disruption of the VEGF/VEGFR-2 axis can increase extravasation of adoptively transferred T cells into the tumor and improve ACT-based immunotherapy. These studies provide a rationale for the exploration of combining antiangiogenic agents with ACT for the treatment of patients with cancer.
Keywords: Adoptive cell therapy, antiangiogenesis, immunotherapy, anti-tumor, melanoma
Introduction
Cell transfer immunotherapy and antiangiogenic therapy are two new biologic approaches to the treatment of cancer. The adoptive transfer of autologous tumor infiltrating lymphocytes (TIL) or lymphocytes genetically engineered to express anti-tumor T cell receptors can mediate the objective regression of cancer in up to 70% of patients with metastatic melanoma (1-4) The integrity of the tumor vasculature and the suppressive nature of the tumor microenvironment can play an important role in modulating the effectiveness of cell based immunotherapies (5, 6) and antiangiogenic approaches can have a profound impact on both of these factors. Thus, in the current study we have explored the interactions and possible synergies between cell transfer and antiangiogenic therapies in a murine cancer model.
Vascular endothelial growth factor (VEGF), a proangiogenic factor secreted by various solid tumors including melanoma has immunomodulatory effects, in part by directly suppressing various immune cells present in the tumor microenvironment (6). VEGF at concentrations similar to those found in cancer patients can contribute to tumor associated immune deficiency and has been reported to negatively regulate antigen presentation by dendritic cells (DC), shift mature DC populations to immature DC precursors, induce apoptotic pathways in CD8+ T cells and induce the activity of regulatory T cells (Tregs) (6-12). VEGF can also alter the tumor endothelium which can further disrupt the infiltration and function of tumor infiltrating T cells (7).
Neutralizing antibodies against VEGF can block the immune suppressive effect of tumor derived supernatant on the function of mature DC's (12). The administration of anti-VEGF antibody (Bevacizumab) to sixteen patients with colorectal cancer significantly decreased the number of immature DCs in peripheral blood (p=0.012). In mixed lymphocyte reaction assays anti-VEGF antibody could enhance the antigen-presenting capacity of DCs (13). Immunotherapy with granulocyte-macrophage colony stimulating factor secreting tumor cells in combination with VEGF inhibition enhanced the number of activated DC and tumor infiltrating effector T cells and reduced the number of Tregs in the tumor microenvironment (14). Other antiangiogenic agents have been shown to inhibit tumor growth and microvessel density and enhance the infiltration of leucocytes and CD8+ cytotoxic T lymphocytes into tumor (15).
Antiangiogenic agents have been used as monotherapy or in combination with cytotoxic chemotherapy in both animal models and in the human with variable results. Mouse anti-VEGF antibody, A4.6.1, was shown to suppress the growth of human rhabdomyosarcoma, glioblastoma, leiomyosarcoma and various other tumors implanted in immunodeficient mice (16, 17). Other antiangiogenic agents targeting VEGFRs and small molecule tyrosine kinase inhibitors have also been used effectively for tumor treatment in several preclinical and clinical models (18, 19). Anti mouse VEGFR-2 antibody (DC101) treatment significantly suppressed the growth of primary murine Lewis lung, 4T1 mammary, and B16 melanoma tumors and completely inhibited the growth of established epidermoid, glioblastoma, pancreatic, and renal human tumor xenografts (18). Monotherapy with anti-VEGF alone has not been successful in human clinical trials but when used in combination with chemotherapy have increased overall survival and/or progression-free survival in colorectal, breast and lung cancer patients (19, 20). VEGF receptor kinase-selective multitargeted agents in combination with various chemotherapeutic agents are in different stages of clinical development (19, 20).
Inhibition of the VEGF/VEGFR-2 axis has been reported to normalize tumor vasculature, decrease interstitial fluid pressure, enhance oncotic pressure gradient, decrease hydrostatic pressure gradient across the tumor vasculature (21) and increase deeper penetration of high molecular weight tetramethyl rhodamine isothiocyanate (TRITC) labeled BSA (22). On the basis of the above observations it has been postulated that judicious application of antiangiogenic agents can “normalize” the abnormal tumor vasculature, resulting in more efficient delivery of drugs and oxygen to targeted cancer cells (23).
The current study was thus proposed to test whether the addition of antiangiogenic agents could significantly enhance the antitumor therapeutic effect of cell transfer therapy. We tested this hypothesis in the pmel-1 TCR transgenic mouse model previously reported from our group (24), which models the use of adoptive cell therapy for the treatment of metastatic melanoma in the clinical setting.
Methods
Mice and Cell Lines
Pmel-1 TCR transgenic (24, 25) and C57BL/6 mice (The Jackson Laboratory) were bred, housed and used according to the guidelines of the National Cancer Institute (NCI) Animal Care and Use Committee. The Vβ-13-pmel-1 TCR recognizes an epitope of the gp100 melanoma/melanocyte differentiation antigen present on the B16 melanoma, a spontaneous murine melanoma in C57BL/6 mice obtained from the National Cancer Institute tumor repository and maintained in culture (24).
In vitro pmel-1 T cell activation and adoptive cell transfer
Seven days prior to adoptive cell transfer, splenocytes from pmel-1 TCR transgenic mice were harvested and depleted of erythrocytes by ACK lysis buffer (GIBCO). Splenocytes were further cultured in complete media (RPMI 1640, 10% heat inactivated fetal bovine serum, penicillin streptomycin, sodium pyruvate, non essential amino acids, L-glutamine and 2-mercaptoethanol) in the presence of 1μM hgp25-33 peptide and 30IU/ml of rhIL-2 (Novartis Corp.) (24, 25). On the day of ACT (day 0, D0) all tumor bearing recipient mice received 500cGy of sublethal radiation as a lymphodepleting regimen prior to cell transfer. 1×106 in-vitro activated pmel-1-Ly5.1 T cell (P) were transferred intravenously (i.v.) along with the injection of 2×107 plaque forming units (pfu) of recombinant vaccinia virus (V) expressing hgp100. All treated mice received an intraperitoneal (i.p.) injection of 600,000IU of rhIL-2 twice daily for a total of six doses. In this paper, groups of mice receiving all three of pmel-1-Ly5.1 cells (P), vaccine (V) and rhIL-2 (I) are referred to as the PVI group.
Antibody administration and in vivo tumor treatment
Anti-mouse VEGF antibody (B20-4.1.1-PHAGE, α-VEGF) was kindly supplied by Genentech (San Francisco, CA) and rat anti-mouse VEGFR-2 (DC101) was kindly supplied by ImClone Systems (New York, NY). For tumor treatment experiments antibodies were injected starting on the day of cell transfer (D0) and continued at an interval of three days. Rat serum Gig (Sigma) at equivalent concentration was used as an isotope control (see figure and figure legends for antibody schedule and number of doses).
C57BL/6 female recipient mice (6-12 weeks of age) were injected subcutaneously with 5 × 105 B16 melanoma cells and tumors were allowed to grow for 12 – 18 days (n = 5 mice per group in all tumor treatment experiments). On the day of cell transfer, experimental groups received either pmel-1-Ly5.1 T cells (P), vaccine (V), IL-2 (I), rat Gig, α-VEGF or DC101 alone or in various combinations (see figure and figure legends for more details). Tumors were measured using calipers, and the products of the perpendicular diameters were recorded. All experiments were performed in a blinded fashion (experimental mouse groups were coded so the investigators measuring the tumor and analyzing the data had no knowledge of the identity of the treatment groups). Error bars represent standard error of mean.
Enumeration and flow kilometric analysis of adoptively transferred tumor infiltrating pmel-1-Ly5.1 cells
For the studies of cell infiltration into the tumor, the experimental groups received a single dose of antibody two days before (-D2) the cell transfer (D0). Mice were euthanized on days 3, 4, 5 and 6 post cell transfers to collect spleen and tumor. Splenocytes were prepared as described above while total tumor cells were harvested by homogenizing the tumor into a single-cell suspension using the rubber end of a 3-cc syringe and a 40-μm filter cup. Tumor infiltrating lymphocytes were enriched by density gradient centrifugation (15,000g/20min), counted by trypan blue exclusion method and analyzed by flow cytometry for the expression of CD3, Ly5.1 and Vβ13. Dead cells were excluded using propidium iodide (PI). We calculated the absolute number of pmel-1-Ly5.1 cells by multiplying the live cell count by the percentage of Ly5.1+PI- cells. Error bars represent the standard error of mean with 4-5 mice in each group.
Measurement of mean tumor vessel area by CD31 immunofluorescence
B16 tumors were collected at various time points from experimental groups (2-5 mice) receiving a single dose of rat IgG, DC101 or α-VEGF alone and frozen in OCT media. Frozen sections were dried overnight after acetone treatment, washed with PBS, blocked with 10% normal goat serum and stained with anti-CD31 antibody in 2.5% normal goat serum. After overnight incubation sections were washed, treated with secondary goat anti-rat-Cy5 antibody in 2.5% normal goat serum and further treated with DAPI containing medium.
Tumor sections were scanned to identify areas of maximal vascular density and 5 different images were obtained per tumor on a LSM 510 microscope. Zeiss LSM Image Software was used to exclude background fluorescence and quantify the image area occupied by the Cy5 fluorescence marker. The vascular area is reported as the mean area of Cy5 fluorescence of the 5 images.
Statistical analyses
We compared tumor growth slopes using the Wilcoxon rank-sum test and the single-measurement comparisons between two groups were tested using unpaired t-tests. Kaplan-Meier analysis was used to assess survival and the logrank (Mantel-Cox) test was used for comparison between two groups.
Results
Administration of anti-VEGF antibody enhances the anti-tumor efficacy of cell transfer therapy and is dependent on prior lymphodepleting total body irradiation (TBI)
In multiple preliminary experiments the administration of anti-VEGF antibody could transiently inhibit growth of subcutaneous B16 melanomas that were about 50 mm2 but had little or no anti-tumor activity on tumors that were greater than or equal to 100 mm2. An example of the efficacy of anti-VEGF antibody alone to treat 50 mm2 and 200 mm2 size tumors in the same experiment is shown in Figure 1A and B. Significant (p=0.009) anti-tumor activity of anti-VEGF antibody was seen on small tumors (∼ ≥ 50 mm2) while lesser impact was seen on large tumors (∼ 200 mm2) (see also Figure 2). Prior published experiments showed that effective ACT of established B16 melanoma (irrespective of tumor size) required the administration of anti-GP100 transgenic pmel-1 T cells plus gp100 vaccine plus IL-2 administration after 5cGy lymphodepleting total body irradiation (24, 25) referred to here as PVI treatment. When small (about 50 mm2) tumors were treated with anti-VEGF antibody a profound antitumor effect was seen and when added to PVI there was little to no additive anti-tumor impact (Figure 1C and D). To better mimic the treatment of human tumors all subsequent experiments testing the impact of combined treatment with this antibody plus ACT utilized mice bearing tumors greater than or equal to 100 mm2.
Figure 1. Anti-tumor effect of ant-VEGF antibody alone and in combination with ACT.

(A) Six doses of 200μg anti-VEGF antibody had a substantial growth inhibitory effect on small (50 mm2) established B16 tumors (p=0.009) and a lesser effect on large (100 mm2) tumors (p=0.028). Closed symbols are groups receiving α-VEGF and open symbols are groups receiving rat IgG. (B) Significant impact was also seen on the survival of mice. (C) Four doses of 200μg α-VEGF exhibited a substantial antitumor effect when given alone or in combination with ACT on small B16 tumors (50 mm2). (D) Significant impact was also seen on the survival of mice.
Figure 2. Combined therapeutic effect of anti-VEGF antibody with ACT on large B16 tumor treatment.

(A) The administration of four, six and eight doses of 100μg of anti-VEGF antibody alone had no impact on the growth of these large established B16 melanomas. A synergistic anti-tumor effect was seen when 6 and 8 doses of anti-VEGF antibody was given in conjunction with ACT (p=0.009). (B) Survival of mice was also improved (p=0.003) (C) Eight doses of 100 μg of α-VEGF alone had no impact on tumor growth but synergized with ACT (p=0.01). The impact of ACT with and without antibody administration was dependent on the administration of 500cGy total body irradiation prior to the cell transfer. (D) Survival of mice correlated with the anti-tumor growth effect.
To determine whether the addition of α-VEGF had any additive or synergistic impact on the therapeutic effect of ACT in treating large established B16 tumors, we treated recipient mice with multiple doses of 100 μg or 200 μg of α-VEGF along with ACT immunotherapy. Two representative experiments combining ACT with the administration of anti-VEGF antibody are shown in Figure 2. Increasing doses (four, six and eight) of 100 μg/dose α-VEGF alone did not have an anti-tumor effect on large established B16 tumors nor did it prolong the survival of these mice when compared to the rat IgG control group (100 μg/dose, eight doses). ACT with PVI alone exhibited significant anti-tumor activity and prolonged survival of mice (solid circles Figure 2A and B). Four doses of α-VEGF had little effect when added to ACT, while six and eight doses of anti-VEGF showed significant (p=0.009) synergistic anti-tumor effects when added to ACT (Figure 2A and B). In repeat experiments, eight doses of α-VEGF consistently enhanced the anti-tumor effect of ACT while lower numbers of doses had a more variable impact.
We also investigated whether a prior lymphodepleting regimen was required for effective ACT when used in combination with α-VEGF (Figure 2C and D). Eight doses of 100μg/dose α-VEGF alone did not have any anti-tumor effect on large B16 tumors when used in the presence or absence of 500cGy radiation compared to corresponding rat IgG controls (Figure 2C and D). ACT immunotherapy of large established B16 tumors using PVI in the absence of prior radiation failed to exhibit any anti-tumor effect when used with or without α-VEGF (Figure 2C). Concordant with our previous reports, host lymphodepletion by total body irradiation (500cGy) significantly enhanced the anti-tumor effect of PVI. The addition of α-VEGF to the PVI group receiving 500cGy TBI further enhanced the anti-tumor effect (p=0.01) and prolonged the survival of mice (p=0.027) (Figure 2C and 2D). Thus, host lymphodepleting conditioning prior to ACT is essential to achieve maximum anti-tumor activity and was further enhanced by the addition of α-VEGF. Hence, in all our further experiments we included 500cGY of radiation as a host lymphodepleting regimen prior to ACT.
Anti-VEGF treatment enhances the infiltration of pmel-1 T cells into B16 tumors
A single dose of antiangiogenic agents (Bevacizumab) in humans and DC101 in mice) has been reported to lead to vascular normalization in tumors (21, 22). Treatment of tumor bearing mice with DC101 resulted in a significantly deeper penetration of tetramethyl rhodamine isothiocyanate (TRITC) labeled BSA molecules into the tumor (22). We thus sought to determine whether the anti-tumor therapeutic effect of the combination therapy of ACT plus anti-VEGF was associated with an enhanced pmel-1 T cell infiltration into the B16 tumor. Ly5.1 pmel-1 T cells were infused into Ly5.2 mice to distinguish transferred from endogenous cells. A single dose of α-VEGF (100 μg, 200 μg or 500 μg) given two days prior to ACT lead to significantly enhanced infiltration of pmel-1-Ly5.1 cells into tumor. In twelve consecutive experimental determinations of cells infiltrating into tumors on days 4 to 6 after cell transfer in mice receiving anti-VEGF antibody two days prior to adoptive cell transfer there were 32.1 ± 6.8% (mean ± SEM) Ly5.1 pmel-1 tumor infiltrating T cells compared to 14.8 ± 5.7% Ly5.1 pmel T cells in mice receiving control rat IgG (p2 = 0.0051, paired t test). In ten consecutive experimental determinations the number of Ly5.1 pmel T cells was also increased in the anti-VEGF group compared to the rat IgG control (12.5 ± 2.8 × 105 vs. 5.8 ± 1.0 × 105, p2 = 0.034). Total tumor cell suspensions were gated on live Ly5.1 cells. In mice treated with a single dose of 200 μg or 500 μg α-VEGF we found significantly higher Ly5.1+ cell (38.4%, p=0.01 and 43.0%, p=0.003 respectively) infiltration into B16 tumor compared to mice receiving 500μg of rat IgG (17%) as early as day 4 post ACT (Figure 3A). In addition we enumerated total live Ly5.1+ cells in tumor and found significantly higher number of cells in mice treated with 200 μg or 500 μg α-VEGF (4.1×105 cells, p=0.03 and 2.2×105 cells, p=0.001 respectively) when compared to mice receiving 500 μg of rat IgG control (6.1×104 cells) (Figure 3B). In a repeat experiment (Figure 3C and D) we investigated T cell infiltration on days 3, 4, 5 and 6 post ACT. Mice receiving a single dose of 200 μg or 500 μg α-VEGF exhibited a higher percent and total number of live Ly5.1+ cells in the tumor when compared to the control group (Figure 3C and D). Thus, the enhanced anti-tumor effects in our combination therapy were associated with and may be explained by the ability of α-VEGF administration to enhance T cell infiltration into the B16 tumor. Of interest no increased cell infiltration was seen when anti-VEGF antibody was given on the day of cell infiltration suggesting that the kinetics of the impact of a single dose of anti-VEGF on tumor vasculature relative to the time of cell infiltration is critical.
Figure 3. Effect of anti-VEGF antibody on pmel-1-Ly5.1 cell infiltration into B16 tumor following ACT.

(A) The percent of infiltrating Ly5.1+PI- cells into B16 melanoma on day 4 post cell transfer was significantly increased in mice administered with 200μg or 500μg anti-VEGF antibody compared to mice receiving 500μg rat IgG. (B) The total number of infiltrating Ly5.1+PI- cells was also increased in mice receiving 200μg and 500μg α-VEGF compared to mice receiving 500μg rat IgG. (C) and (D) A repeat experiment evaluating the infiltration of transferred Ly5.1 pmel-1 cells on days 3, 4, 5 and 6 gave a similar result. Thus, prior administration of α-VEGF to B16 tumor bearing mice receiving ACT significantly enhances tumor infiltration of the adoptively transferred pmel-1 cells.
Effect of anti-VEGFR-2 antibodies along with ACT in treatment of B16 melanoma
Antibodies to VEGFR's also represent attractive antiangiogenic agents. DC101 (rat anti-mouse VEGFR-2) has been successfully used in various preclinical models as a monotherapy or combination therapy for tumor treatment (18, 22). We thus also screened the impact of DC101 administration along with ACT on established B16 tumor. In mice bearing small tumors (25 to 50 mm2) three doses of DC101 alone (800 μg/dose) had some anti-tumor effect and prolonged the survival of mice compared to the rat IgG control (Figure 4A and B). The addition of DC101 to ACT exhibited a significant (p=0.013) additive anti-tumor effect (Figure 4A) while no significant additive impact was observed on prolonging survival (Figure 4B). In mice bearing large tumors (∼100 mm2) higher doses of DC101 (six doses, 800 μg/dose) alone had no anti-tumor effect (Figure 4C). When DC101 was added to ACT we observed a small but reproducible therapeutic impact (Figure 4C, p=0.028) but no effect on survival (Figure 4D). Thus, we next investigated whether treatment of tumor bearing mice with a single dose of DC101 two days prior to ACT augments enhanced cell infiltration in to B16 tumors.
Figure 4. Impact of rat anti-mouse VEGFR-2 antibody (DC101) on tumor treatment and cell infiltration in B16 tumor bearing mice following ACT.

(A and B) Three doses of 800μg DC101 antibody mediated a modest growth inhibitory effect on small established B16 melanoma compared to rat IgG alone. An additive effect of the antibody was seen when combined with ACT (p=0.013). Open symbols are groups receiving antibody alone while closed symbols are groups receiving antibody with ACT (PVI group). (C and D) Six doses of 800μg DC101 had no impact on the growth of large (100 mm2) B16 melanoma but did mediate a modest tumor inhibitory effect when added to ACT. In accord with our previous results vaccination with recombinant vaccinia virus expressing hgp10025-33 peptide was required to see the tumor inhibitory impact of ACT. Mice receiving PI (without vaccination) exhibited no anti-tumor effect with or without anti-VEGFR-2 antibody administration.
In contrast to our results using anti-VEGF administration, data from eight consecutive experimental determinations revealed no impact of the administration of the DC101 antibody on the percentage of live Ly5.1 cells infiltrating into the tumor of mice four to six days after cell administration (p2 = 0.33, paired t test). We could however detect an impact on vessel area within tumors after DC101 administration. The mean tumor vessel area of mice receiving a single dose of DC101 remained relatively stable for at least seven days while vessels increased significantly in mice receiving rat IgG (Figure 5A). Later time points were not tested. In a repeat experiment mice receiving rat IgG showed a significantly increased (p=0.0003, p=0.001 respectively) mean tumor vessel area on days 3 and 4 (CD31 immunofluorescence staining) compared to mice receiving DC101 (Figure 5B). Figure 5C represents a blinded, microscopic observation made on days 0, 3 and 4 in the first experiment. Tumor vasculature on days 3 and 4 was less in mice receiving DC101 than rat IgG. In a preliminary experiment done with α-VEGF and rat IgG a similar pattern was seen (data not shown).
Figure 5. Anti-vascular effect of a single dose of anti-VEGFR-2 antibody (DC101) on B16 tumor vasculature.
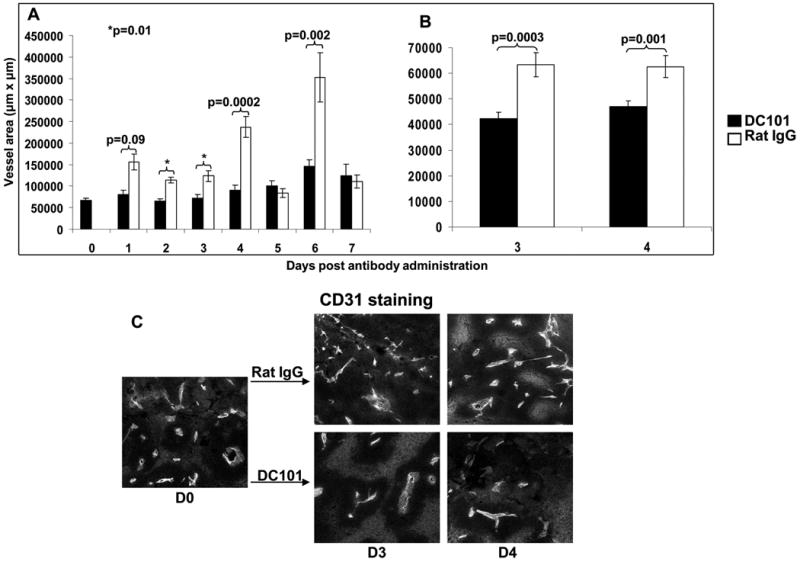
(A and B) Mean tumor vessel area of B16 tumors in mice receiving a single dose of 800μg DC101 was decreased compared to mice receiving rat IgG on various days post antibody administration. Results for each time point are the mean of 5 images for each section/mice and 2 or 5 mice in each group (A and B respectively). Two separate experiments are shown. (C) Representative microscopic sections from mice receiving DC101 antibody or control rat IgG. Vessel area was determined by blinded measurement of CD31 expression. At 3 and 4 days after DC101 administration a significant decrease in tumor vessels was seen compared to mice receiving rat IgG.
Discussion
The adoptive transfer of anti-tumor T cells (ACT) can mediate tumor regression in both murine models and in patients with metastatic melanoma (1-4). A lymphodepleting preparative regimen applied prior to the adoptive transfer profoundly enhances the therapeutic effects of the transferred cells in part by depleting immune regulatory elements such as T regulatory cells and by eliminating competition of the transferred cells for homeostatic cytokines (1-4, 24, 27). Immunosuppressive factors produced by the tumor and the integrity of the tumor vasculature can affect the activity of lymphocytes and limit the extravasation of lymphocytes from tumor vessels into the tumor stroma. VEGF produced by tumor cells can mediate local immune suppression by interfering with the function of antigen presenting cells and can stimulate the growth of tumor vasculature (6-12). In murine models, inhibition of VEGF or its interaction with its predominant receptor VEGFR-2 can overcome the immunosuppressive activity of VEGF (12). Interruption of the VEGF/VEGFR-2 axis can also normalize the tumor vasculature and enhance the extravasation of intravascular components into the tumor stroma. The administration of angiogenesis inhibitors cans upregulate endothelial adhesion molecules in tumor vessels and increases the number of tumor infiltrating leukocytes (15). Several receptors, such as Endothelin B, have been identified as a molecule that can affect lymphocyte infiltration into tumors (28). Several studies have shown that blockade of VEGF or VEGFR-2 can enhance the therapeutic effect of cancer vaccines and increase the numbers of vaccine-induced T cells that infiltrate into tumor (14, 29).
Despite these effects of VEGF blockade antiangiogenic agents have had minimal clinical impact when used alone, an observation probably due to the redundancy of angiogenic pathways. However, antiangiogenic treatments have exhibited modest clinical effectiveness when administered in conjunction with chemotherapy (19-21, 26). This impact has been attributed in large part to the ability of antiangiogenic drugs to increase extravasation of chemotherapy agents into the tumor. The therapeutic effectiveness of ACT therapy in cancer patients as well as the ability of antiangiogenic agents to alter tumor vasculature in ways that could potentially increase the availability of tumor to the transferred T cells thus led us to explore the combination of these approaches in mice with large established B16 melanoma.
We thus evaluated antibodies against VEGF and VEGFR-2, for their ability to enhance ACT. While these antibodies could inhibit the growth of small tumors (about 50 mm2) they had little impact on larger tumors that were ≥ 100 mm2 (Figure 1). In an attempt to more closely mimic the treatment of human tumors we confined our studies to the treatment of larger established vascularized B16 melanomas. When treating these larger tumors the addition of anti-VEGF antibody synergized with ACT therapy. The combination was more effective than either modality utilized alone (Figure 2). Prior studies have shown that a lymphodepleting regimen is an essential component of ACT therapy in this mouse model (24). In our studies little therapeutic impact of ACT with or without anti-VEGF antibody was seen in immunocompetent mice whereas profound combined effects were seen in mice pre-treated with 500cGy whole body irradiation. Multiple doses of anti-VEGF antibody given every three days were essential to observe this synergistic therapeutic effect in conjunction with ACT. A dose of 100μg of anti-VEGF antibody was as effective as higher doses, in accord with prior reports indicating that in mice the 50 to 100 μg dose was optimal for mediating anti-vascular effects (16). The dose and schedule of antiangiogenic agents is a critical factor in the development of effective combination therapies (19, 20).
The mechanism of action of the anti-VEGF mediated anti-tumor activity is a matter of some controversy. Although the inhibition of growth of tumor vessels appears to play a role there is considerable evidence that normalization of the properties of the tumor vasculature can substantially enhance drug penetration into tumor (22, 23). The administration of antiangiogenic agents can sustain a hydrostatic pressure gradient across the tumor vascular by decreasing interstitial fluid pressure and increasing pericyte coverage of tumor endothelium. Antiangiogenic agents can thus enhance infiltration of labeled bovine serum albumin from the vasculature into the tumor stroma (22). To study the mechanism of action of the combination of cell transfer therapy with anti-VEGF therapy we explored whether this combination treatment could have an impact on the extravasation of transferred anti-tumor T cells into the tumor. These experiments clearly showed an increased percentage (p2 = 0.0051) and absolute number (p2 = 0.034) of transferred T cells into tumor that was even apparent at the lowest dose of anti-VEGF used (100 μg per dose) (Figure 3).
The effectiveness of anti-VEGF administration to improve ACT led us to investigate the effects of the administration of an anti-VEGFR-2 antibody, DC101. As was the case for the use of anti-VEGF antibody the administration of anti-VEGFR-2 antibody could mediate an anti-tumor effect on small tumors (50 mm2) but there was little to no impact on large tumors (Figure 4). There was, however, a modest additive effect of anti-VEGFR-2 antibody in conjunction with ACT. In these experiments there was no increase in infiltration of the adoptively transferred cells into tumor (p2 = 0.33) (Figure 4E), possibly accounting for the lesser impact of this antibody compared to anti-VEGF. This may also in part be due to the ability of anti-VEGF antibody to block the binding of VEGF to both VEGFR-1 and VEGFR-2 while anti-VEGFR-2 antibody could only block VEGFR-2 binding. In these experiments the anti-VEGFR-2 antibody did have some impact on angiogenesis since it was able to limit the growth of tumor vasculature following a single dose of antibody (Figure 5).
It should be noted that the ability of anti-VEGF antibody to inhibit tumor growth in conjunction with cell transfer required the administration of multiple doses of antibody (Figures 2A and B) despite the demonstration that increased infiltration of the transferred lymphocytes could be seen after a single dose of antibody. This implies that prolonged impact of anti-VEGF antibody on the tumor vasculature is required to see the anti-tumor effect. Successfully synergy of anti-VEGF antibody with chemotherapy in humans also requires prolonged anti-VEGF administration (26). Anti-VEGF antibody may also be manifesting anti-tumor effects in conjunction with cell transfer by inhibiting the known immunosuppressive impact of VEGF (6-11).
The studies reported here demonstrate that agents capable of blockading the VEGF/VEGFR-2 axis can enhance the anti-tumor activity of adoptively transferred anti-tumor T cells. The therapeutic effectiveness of ACT in patients with metastatic melanoma and the ready availability of clinically approved antiangiogenic agents provide a rationale for the exploration of this combination therapy in the treatment of human cancer.
Acknowledgments
We thank Douglas Palmer, Lindsay Garvin, Robert Reger and David Jones for helping us with the animal studies. Thanks also to Arnold Mixon and Shawn Farid for help with flow cytometry and Don White for statistical analysis.
Footnotes
Authors declare no potential conflict of interest.
References
- 1.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–40. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley ME, Yang JC, Sherry R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–9. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA. Overcoming obstacles to the effective immunotherapy of human cancer. Proc Natl Acad Sci U S A. 2008;105:12643–4. doi: 10.1073/pnas.0806877105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res. 2001;23:263–72. doi: 10.1385/IR:23:2-3:263. [DOI] [PubMed] [Google Scholar]
- 7.Mulligan JK, Day TA, Gillespie MB, Rosenzweig SA, Young MR. Secretion of vascular endothelial growth factor by oral squamous cell carcinoma cells skews endothelial cells to suppress T-cell functions. Hum Immunol. 2009;70:375–82. doi: 10.1016/j.humimm.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 9.Johnson BF, Clay TM, Hobeika AC, Lyerly HK, Morse MA. Vascular endothelial growth factor and immunosuppression in cancer: current knowledge and potential for new therapy. Expert Opin Biol Ther. 2007;7:449–60. doi: 10.1517/14712598.7.4.449. [DOI] [PubMed] [Google Scholar]
- 10.Ohm JE, Gabrilovich DI, Sempowski GD, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–86. doi: 10.1182/blood-2002-07-1956. [DOI] [PubMed] [Google Scholar]
- 11.Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–66. [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Chen HL, Girgis KR, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 13.Osada T, Chong G, Tansik R, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother. 2008;57:1115–24. doi: 10.1007/s00262-007-0441-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li B, Lalani AS, Harding TC, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res. 2006;12:6808–16. doi: 10.1158/1078-0432.CCR-06-1558. [DOI] [PubMed] [Google Scholar]
- 15.Dirkx AE, oude Egbrink MG, Castermans K, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006;20:621–30. doi: 10.1096/fj.05-4493com. [DOI] [PubMed] [Google Scholar]
- 16.Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–4. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 17.Gerber HP, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res. 2005;65:671–80. [PubMed] [Google Scholar]
- 18.Prewett M, Huber J, Li Y, et al. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999;59:5209–18. [PubMed] [Google Scholar]
- 19.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 20.Crawford Y, Ferrara N. VEGF inhibition: insights from preclinical and clinical studies. Cell Tissue Res. 2009;335:261–9. doi: 10.1007/s00441-008-0675-8. [DOI] [PubMed] [Google Scholar]
- 21.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–7. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong RT, Boucher Y, Kozin SV, et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 23.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 24.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 27.Wrzesinski C, Paulos CM, Kaiser A, et al. Increased Intensity Lymphodepletion Enhances Tumor Treatment Efficacy of Adoptively Transferred Tumor-specific T Cells. J Immunother. 2009 Nov 27; doi: 10.1097/CJI.0b013e3181b88ffc. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buckanovich RJ, Facciabene A, Kim S, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14:28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- 29.Manning EA, Ullman JG, Leatherman JM, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951–9. doi: 10.1158/1078-0432.CCR-07-0374. [DOI] [PubMed] [Google Scholar]