

Abstract
The new asymmetric catalytic Strecker reaction of achiral N-phosphonyl imines has been established. Excellent enantioselectivity (95.2–99.7%ee) and yields (89–97%) have been achieved by using primary free natural amino acids as catalysts and Et2AlCN as nucleophile. This work also presents the novel use of non-volatile and inexpensive Et2AlCN in asymmetric catalysis. The N-phosphonyl protecting group enabled simple product purification to be achieved simply by washing the crude products with hexane. It can also be readily cleaved and recycled under mild condition to give a quantitative recovery of N,N′-bis(naphthalen-1-ylmethyl)ethane-1,2-diamine. A new mechanism was proposed for this reaction and was supported by experimental observations.
Keywords: N-Phosphonyl imine, Asymmetric Strecker reaction, Diethylaluminum cyanide, Amino acid catalyst
Introduction
Imine chemistry has been one of the most important and active topics in asymmetric catalysis and synthesis due to its importance in drug discovery and development, which heavily depend on the amine functionality.[1–3] N-protected imines serve as both electrophiles and dienophiles for many asymmetric reactions;[2–3] they are represented by Ar2CH-/Bn-,[4] alkoxycarbonyl,[5] Ar2PO-,[6] Aryl-,[7] and ArSO2 [8] imines in which the protecting groups are often crucial for controlling regio-, enantio- and chemoselectivity for asymmetric reactions.[9] The search for novel imines of general use for both racemic and asymmetric reactions, particularly, for asymmetric catalysis has been challenging.[1–3] In the past several years, we have established chiral N-phosphonyl imine chemistry and successfully utilized it for a variety of asymmetric reactions to produce chiral amino products in good yields and excellent diastereoselectivities.10 During our continuous study of this project, we envisioned that if achiral N-phosphonyl imines are designed and synthesized, they would serve as a new family of C=N electrophiles and dienophiles for general use in advanced synthesis and asymmetric catalysis.
Results and Discussion
N-phosphonyl imines and N-phosphoramides have stronger Lewis basicity as compared with their counterparts.[11] During the reaction process, the resulting N-phosphoramide species can strongly bind to catalytic centers and thus retard catalytic cycles, which consequently makes it extremely difficult to achieve asymmetric catalysis using N-phosphonyl imines as electrophiles. In this report, we would like to describe the new asymmetric catalytic Strecker reaction of N-phosphonyl imines under a concise catalytic system (Scheme 1); In fact, this reaction has recently become an active topic due to its practical use for the synthesis of unnatural α-amino acids and their derivatives and its versatility as a superior model reaction for organocatalysis.[2–4,6] The present N-phosphonyl imine-based Strecker reaction was conducted by using free primary amino acids as catalysts and Et2AlCN[12] as nucleophile. Excellent chemical yields (89–97%) and enantioselectivities (95.2–99.7%ee) have been achieved by performing the reaction at −78 °C in dry toluene in the presence of 4 Å mol sieves (Scheme 1).
Scheme 1.

Our new work described here shows several attractive characteristics as compared with previous known systems: (1) the first asymmetric catalytic reaction of achiral N-phosphonyl imines; (2) the first use of non-volatile and inexpensive Et2AlCN as the cyanide source for catalytic Strecker reaction; (3) N-phosphonyl group can be readily cleaved under mild conditions without racemization; (4) The new N-phosphonyl protection group enabled the pure Strecker products to be obtained simply by washing the crude products with hexane without further purification. (5) The cleavaged N,N-dialkyl diamine auxiliary can be recovered quantitatively for re-use; (6) free primary natural amino acid catalysts are common commercial chemicals; (7) The modifiable flexibility of C2-symmetric structure by changing its ring sizes and N,N-protection groups can result in various C=N electrophiles with changed reactivity for different catalytic reactions.
The reaction is believed to undergo through the mechanism as shown in Scheme 2. The first step is the reaction of amino acid (A) with Et2AlCN to form the catalytic species (B). The evolution of ethane gas and the solubility change of amino acid (which is insoluble in toluene but was dissolved after species (B) was formed) are evidenced of this step occurring. The Lewis acidic center can activat oxygen of N-phosphonyl group of the substrate before species (B) delivers cyanide onto the C=N bond from its Si-face to afford intermediate (C). During the reaction process, Et2AlCN reacts with additive i-PrOH to give species Et(i-PrO)AlCN, its activation of imine nitrogen is also anticipated prior to C=N addition. At the third step, the catalyst (B) is regenerated via the homotransmetallation between species (C) and Et(i-PrO)AlCN which is derived from reaction of Et2AlCN and i-PrOH; simultaneously, the actual catalytic complex (D) was generated, which was followed by deprotonation and quenching to afford the final product.
Scheme 2.

i-PrOH has been proven to be a crucial additive for this reaction, which is similar to other asymmetric Strecker process. [6a.12d] A tiny amount of product was formed without the addition of i-PrOH in reaction mixture. Usually, in aromatic solvents, Et2AlCN exists as tetramers or pentamers[6a] that show lower reactivity. However, its coordination with phosphonyl imine followed by conversion to intermediate (B) and Et(i-PrO)AlCN, which was evidenced by the release of ethane bubbles during the reaction process, led to the present success.
Starting materials, achiral N-phosphonyl imines (1a–k), were readily prepared by following the procedure which is similar to that developed by our group for asymmetric chiral N-phosphonyl imine chemistry.[10] The four-step preparation of N-phosphoramides showed quantitative yields for all steps. Products were obtained as white solids without the need of special purification. The final step of imine formation showed higher yields (76–87%) than the formation of N-phosphonyl imine counterparts. It is somewhat surprising that the design and synthesis of achiral N-phosphonyl imines have not been well documented in literature so far.
Our initial effort on this reaction was made on the use N,N-dibenzyl N-phosphonyl imine as the substrate and free primary amino acid,[13] L-valine, as the catalyst in dry toluene in the presence of 4 Å MS. The reaction was conducted by using Et2AlCN (1.5 equiv, 1.0 M in toluene) together with i-PrOH (1.0 equiv) as an additive at −78 °C for 5 h, followed by quenching with dilute aq HCl to produce product in 75% yield and poor enantioselectivity (22.6 %ee). Next, we attempted to improve the reaction by replacing benzyl with i-propyl in N-phosphonyl imine substrates, which has been proven to be successful for several chiral N-phosphonyl imine-based reactions. However, the resulting N-di-i-propyl phosphonyl imine was found to be insoluble in toluene at −78 °C and even at r.t. We then utilized N,N-naphthalen-1-ylmethyl to replace benzyl, i.e., to use N,N-naphthalen-1-ylmethyl N-phosphonyl imine (1a) as the electrophile. We were pleased to find that not only the solubility problem was solved, but also enantioselectivity and chemical yield were substantially increased to 70 %ee and 98 %, respectively.
Several catalysts were then screened under the above conditions. As revealed in Scheme 3, N-tosyl L-valine resulted in a much lower enantioselectivity of 38 %ee, albeit yield remained high. This was somewhat surprising because nearly all successful asymmetric catalytic reactions utilize amino acids that are based on the use of N-protected ones.[14] N-Tosyl phenylalanine and N-tosyl phenylglycine also showed poor enantioselectivities of 28 %ee and 36 %ee, respectively. We then turned our attention back to free amino acids and found that free L-phenylalanine afforded 63 %ee and 65% yield. Amazingly, the use of free L-phenylglycine (10 mol%) as the catalyst resulted in 99%ee and 95% yield under this new catalytic system.
Scheme 3.

Results of screening catalysts
It should be noted that well-activated 4 Å MS are also very important for this reaction. Much lower yields were observed in the absence of 4 Å MS. Other solvents, such as DCM, THF and CHCl3, proved to be unsuitable for this reaction as they afforded poor yields and %ee’s. During the preparation of catalytic species, it is important to allow chiral amino acids to react with Et2AlCN for at least 15 min at r.t. before the reaction mixture is cooled down to −78 °C. Keeping the reaction at −78 °C was necessary for achieving high enantioselectivity, albeit yield was not affected by temperature. TMSCN was also employed as the nucleophile, but poor yields (<10 %) were obtained.
Having identified the optimal catalyst and catalytic condition, we turned our attention to the substrate scope. As shown in Table 1, the reaction proceeded very well for all eleven substrates that were examined with excellent enantioselectivities (95.2–99.7%ee) and chemical yields (89–97 %). Electron-rich substrates (entries 5 and 6) showed nearly the same reaction rates as other substrates and led to similar results. Strong electron-deficient substrates (entries 2 and 7) that sometimes showed less efficiency in chiral N-phosphonyl imine-based asymmetric reactions [10] resulted in excellent yields (96 and 94 %, respectively) and enantioselectivity (94 and 96.1 %ee, respectively) under the present system. Interestingly, the present N-phosphonyl protection group enabled the pure products to be obtained simply by washing with hexane without further purification. It is noteworthy that resulting halogenated Strecker adducts (entries 3, 4, 8 and 9) can be readily converted to a variety of useful synthetic building blocks via metal catalyzed coupling reactions.[15] 2-Furyl substrate led to complete enantioselectivity (99%ee) and 89% yield, which is important because the furyl functionality can be subjected to other transformations, such as Diels-Alder and oxidative ring opening.[3] Our preliminary results also confirmed that this asymmetric catalytic system showed promising for aliphatic N-dialkoxyphosphoryl imines.[16] More substrates will be under investigation under this system.
Table 1.
Results of the synthesis of N-phosphonyl substituted chiral α-aminonitriles 4a–ka
| Entry | Substrate | R1 | Product | Yield(%)b | eec |
|---|---|---|---|---|---|
| 1 | 1a | H | 4a | 95 | 99.7 |
| 2 | 1b | p-fluoro | 4b | 96 | 94 |
| 3 | 1c | p-bromo | 4c | 97 | 96 |
| 4 | 1d | p-chloro | 4d | 95 | 95.2 |
| 5 | 1e | p-methyl | 4e | 94 | 98.8 |
| 6 | 1f | p-methoxy | 4f | 93 | 97.6 |
| 7 | 1g | o-fluoro | 4g | 94 | 96.1 |
| 8 | 1h | o-bromo | 4h | 92 | 97 |
| 9 | 1i | o-chloro | 4i | 90 | 95.2 |
| 10 | 1j | o-methyl | 4j | 92 | 98.9 |
| 11 | 1k | o-furyl | 4k | 89 | 99 |
All reactions were carried out at −78 °C in 0.06 M solution of imine in toluene.
Combined yields of both isomers.
Enantiomeric ratio has been determined by using chiral HPLC OD-H column (3:7 IPA:Hexane), flow rate = 0.60 ml/min.
The absolute configuration for this asymmetric induction was determined by converting product (4a) to an authentic sample.[17] In this conversion, 4a was subjected to deprotection with 2.0 M aq. HCl at r.t. in methanol followed by in situ t-Boc protection by treating with (t-Boc)2O in the presence of triethylamine (Scheme 5) to yield product 5. Optical rotation of this product was confirmed to be consistent with that of the known sample with S configuration. During this transformation, the cleaved N1,N2-bis(naphthalen-1-ylmethyl)ethane-1,2-diamine was subjected to a one-time extraction with n-butanol from the acidic mixture, prior to t-Boc protection, to give a quantitative recovery yield.
Scheme 5.

Based on the assignment of absolute configuration of the resulting products, a working model has been proposed and shown in Figure 1 to explain the possible source of asymmetric induction. In this model, N-phosphonyl imine approaches the catalytic nucleophilic source of −CN from its less hindered side of hydrogen instead of bulkier phenyl group of the chiral center of the catalyst. Although several conformational arrangements are possible, it is more likely for C=N bond to be arranged on the anti position of Al-CN bond as shown in Figure 1.
Figure 1.

Working model of asymmetric induction in which the nucleophilic species of −CN attacks N-phosphonyl imine from its Si face
Conclusion
The new asymmetric catalytic Strecker reaction of achiral N-phosphonyl imines has been established. Excellent enantioselectivities and yields have been achieved by using primary free natural primary amino acids as catalysts and Et2AlCN as the nucleophile. This work presents the new use of non-volatile Et2AlCN in asymmetric catalysis. N,N′-Protection groups on achiral N-phosphonyl imine were found to play an important role for the present success. This new protection group enabled simple product purification to be achieved simply by washing the crude products with hexane;18 it can also be readily cleaved and recycled under mild condition to give a quantitative recovery of N,N′-bis(naphthalen-1-ylmethyl)ethane-1,2-diamine. A new mechanism has been proposed that is consistent with experimental observations.
Experimental Section
Typical procedure for the synthesis of achiral N-phosphonyl imine (1a–k)
In a dry vial, under inert gas protection, N,N-naphthalen-1-ylmethyl phosphoramide (1.0 equiv.) was taken and dissolved in dry dichloromethane. To the solution, corresponding aldehyde (1.5 equiv.) was added followed by the addition of triethylamine (3.0 equiv.). The reaction was cooled down to 0 °C and titanium (IV) chloride (1.0 M solution in DCM, 0.5 equiv.) was added to the reaction (Scheme 1). The reaction was allowed to stir at room temperature for 36 h and after that the mixture was loaded directly to silica gel. The reaction mixture was purified through column chromatography (Ethyl acetate: Hexane: 1% Et3N). Pure product was obtained by eluting the reaction mixture with ethyl acetate: hexane: triethylamine (60:40:1mL) as white or pale yellow solid in all of the cases reported.
Compound 1a
White solid; yield (0.172 g, 76%); mp 84–86 °C; 1H NMR (500 MHz, CDCl3) δ 9.03 (d, J = 33.5 Hz, 1H), 8.28 (d, J = 9.0 Hz, 2H), 7.86-7.82 (m, 4H), 7.78 (d, J = 8.0 Hz, 2H), 7.53-7.46 (m, 9H), 7.42-7.39 (m, 2H), 4.67-4.64 (m, 2H), 4.55-4.52 (m, 2H), 3.13 (d, J = 9.5 Hz, 4H); 13C NMR (125 MHz, CDCl3) δ 173.5 (d, J = 7.0 Hz), 139.6, 135.8, 135.6 (2C), 133.7, 133.1, 132.7(2C), 132.6, 131.7 (2C), 129.9 (2C), 128.7 (2C), 128.4, 128.3, 126.7 (2C), 126.2, 125.7, 125.4, 125.0 (2C), 123.8, 120.5, 47.43, 47.40 (d, J = 7.3 Hz), 44.9, 42.3. 31P NMR (202 MHz, CDCl3) δ 24.2.
Compound 1b
White foamy solid; Yield (0.214 g, 82%); mp 68–70 °C; 1H NMR (500 MHz, CDCl3) δ 8.87 (d, J = 32.9 Hz, 1H), 8.24 (dd, J = 8.9, 1.8 Hz, 2H), 7.83-7.76 (m, 6H), 7.52-7.46 (m, 6H), 7.41-7.38 (m, 2H), 7.15 (t, J = 8.5 Hz, 2H), 4.66-4.49 (m, 4H), 3.15-3.11 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 172.3 (d, J = 6.45 Hz), 166.8, 164.8, 133.8 (2C), 132.65 (d, J = 6.94 Hz, 2C), 132.18 (d, J = 8.9 Hz, 2C), 131.7 (2C), 128.5 (2C), 128.4 (2C), 126.8 (2C), 126.3 (2C), 125.8 (2C), 125.1 (2C), 123.9 (2C), 116.1, 115.9, 47.5 (d, J = 4.9 Hz, 2C), 45.0 (d, J = 10.4 Hz, 2C). 31P NMR (202 MHz, CDCl3) δ 26.1.
Compound 1c
Pale yellow solid; Yield (0.355 g, 82%); mp 60–62 °C. 1H NMR (500 MHz, CDCl3): 8.83 (d, J = 33.0 Hz, 1H), 8.24 (d, J = 8 Hz, 2H), 7.83 (dd, J = 2.5 Hz, 7.5 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 11 Hz, 2H), 7.51-7.47 (m, 8H), 7.41-7.38 (m, 2H), 4.64 (dd, J = 7.5 Hz, 15.0 Hz, 2H), 4.52 (dd, J = 5.5 Hz, 15.0 Hz, 2H), 3.13 (d, J = 9.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): 172.3 (d, J = 6.9 Hz); 134.6, 134.4, 133.7 (2C), 132.5 (d, J = 6.9 Hz, 2C), 132.0 (2C), 131.7 (2C), 131.5, 131.1 (2C), 128.4 (d, J = 7.5 Hz, 2C), 127.9, 126.8 (2C), 126.3 (2C), 125.7 (2C), 125.1 (2C), 123.8 (2C), 47.4 (d, J = 4.5 Hz, 2C), 45.0 (d, J = 10.9 Hz, 2C); 31P NMR (202 MHz, CDCl3): 26.0.
Compound 1e
White solid; Yield (0.208 g, 87%); mp 64–66 °C; 1H NMR (500 MHz, CDCl3) δ 9.33 (d, J = 33.3 Hz, 1H), 8.27 (d, J = 8.5 Hz, 2H), 8.02 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 2H), 7.78 (d, J = 8.5 Hz, 2H), 7.53-7.39 (m, 9H), 7.31 (t, J = 7.5 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 4.66 (d, J = 7.0 Hz, 2H), 4.55 (d, J = 5.0 Hz, 2H), 3.13 (d, J = 10.0 Hz, 4H), 2.49 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 172.5 (d, J = 12.5 Hz), 140.6 (2C), 133.7 (2C), 132.8 (d, J = 7.4 Hz), 132.6 (2C), 131.7, 131.2 (2C), 129.3 (2C), 128.4 (2C), 128.3 (2C), 126.5 (2C), 126.3 (2C), 126.1, 125.7 (2C), 125.1 (2C), 123.8 (2C), 47.5 (d, J = 4.7 Hz, 2C), 45.0 (d, J = 10.6 Hz, 2C), 19.4. 31P NMR (202 MHz, CDCl3) δ 26.4.
Compound 1f
White solid, yield (0.198 g, 87%); mp 64–66 °C; 1H NMR (500 MHz, CDCl3) δ 8.98 (d, J = 33.3 Hz, 1H), 8.31 (d, J = 8.4 Hz, 2H), 7.88-7.79 (m, 6H), 7.57-7.41 (m, 8H), 7.02 (d, J = 8.7 Hz, 2H), 4.71–4.53 (m, 4H), 3.92 (s, 3H), 3.13 (d, J = 9.6 Hz, 4H). 13C NMR (125 MHz, CDCl3) δ 173.4 (d, J = 6.52 Hz), 164.1 (2C), 134.1 (2C), 133.16 (d, J = 7.4 Hz, 2C), 132.4 (2C), 132.1 (2C), 128.7 (2C), 128.6 (2C), 127.0 (2C), 126.6 (2C), 126.0 (2C), 125.4 (2C), 124.2 (2C), 114.4 (2C), 55.8, 47.8 (d, J = 4.7 Hz, 2C), 45.3 (d, J = 10.6 Hz, 2C). 31P NMR (202 MHz, CDCl3) δ 26.8
Compound 1g
White solid, Yield (0.214 g, 81%); mp 64–66 °C; 1H NMR (500 MHz, CDCl3) δ 9.30 (d, J = 33.0 Hz, 1H), 8.32-8.25 (m, 2H), 8.12-8.07 (m, 1H), 7.88-7.56 (m, 5H), 7.59-7.38 (m, 10H), 4.72-4.49 (m, 4H), 3.13-2.92 (m, 4H). 31P NMR (202 MHz, CDCl3) δ 26.3. HRMS (ESI): m/z calcd for C31H28FN3OP, 508.1954; found, 508.1983.
Compound 1h
Pale yellow solid; Yield (0.362 g, 79%); mp 54–56 °C. 1H NMR (500 MHz, CDCl3) δ 9.32 (d, J = 32.5 Hz, 1H), 8.27 (d, J = 8.5 Hz, 2H), 8.15-8.13 (m, 1H), 7.83 (d, J = 8.25 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H), 7.62-7.60 (m,1H), 7.54-7.44 (m, 6H), 7.42-7.36 (m, 4H), 4.71 (dd, J = 7.0 Hz, 15.0 Hz, 2H), 4.56 (dd, J = 5.0 Hz, 14.5 Hz, 2H), 3.12 (d, J = 10.0 Hz, 4H); 13C NMR (125 MHz, CDCl3) δ 171.4 (d, J = 5.4 Hz), 134.23, 133.9, 133.8, 133.4, 132.6 (d, J = 7.0 Hz, 2C), 131.7 (2C), 129.6, (2C), 128.4 (d, 11.2 Hz, 2C), 127.6 (2C), 127.5 (2C), 126.6 (2C), 126.4 (2C), 125.8 (2C), 125.1 (2C), 123.8 (2C), 47.5 (d, J = 4.5 Hz, 2C), 44.9 (d, J = 10.7 Hz, 2C); 31P NMR (202 MHz, CDCl3) δ 26.0
Compound 1i
White foamy solid, Yield (0.225 g, 85%); mp 66–68 °C; 1H NMR (500 MHz, CDCl3) δ 9.39 (d, J = 32.7 Hz, 1H), 8.43-8.14 (m, 2H), 7.88-7.75 (m, 5H), 7.58-7.29 (m, 11H), 4.74-4.52 (m, 4H), 3.14-2.85 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 167.0 (d, J = 5.7 Hz), 135.1 (d, J = 8.9 Hz), 134.0 (2C), 132.8 (d, J = 6.8 Hz), 131.9 (2C), 128.8, 128.7 (2C), 128.6 (2C), 128.5 (2C), 126.9 (2C), 126.5 (2C), 126.5, 126.0 (2C), 125.4, 125.3 (2C), 124.6, 124.0 (2C), 123.9, 116.4, 47.6 (d, J = 4.8 Hz, 2C), 45.1 (d, J = 10.9 Hz, 2C). 31P NMR (202 MHz, CDCl3) δ 26.2. HRMS (ESI): m/z calcd for C31H28ClN3OP, 524.1659; found, 524.0652.
Compound 1j
Pale yellow solid; Yield (0.284 g, 82%); mp 62–64 °C. 1H NMR (500 MHz, CDCl3): 9.62 (d, J = 33.5 Hz, 1H), 8.29 (d, J = 8.0 Hz, 2H), 7.82-7.76 (m, 6H), 7.53-7.38 (m, 10H), 4.69 (dd, J = 7.0 Hz, 15.0 Hz, 2H), 4.54 (dd, J = 5.0 Hz, 14.5 Hz, 2H), 3.08 (d, J = 10.0 Hz, 4H), 2.04 (s, 3H); 13C NMR (125 MHz, CDCl3): 169.8 (d, J = 5.4 Hz), 134.7 (2C), 133.6 (2C), 132.8 (d, J = 7.3 Hz, 2C), 131.6 (2C), 128.3 (2C), 128.2 (2C), 128.0 (2C), 126.4, (2C), 126.2 (2C), 125.8 (2C), 125.1 (2C), 123.8 (2C), 120.4, 111.1, 47.3 (d, J = 4.5 Hz, 2C), 44.8 (d, J = 10.7, 2C), 14.0; 31P NMR (202 MHz, CDCl3) δ 25.8.
Compound 1k
Yellow color solid; Yield (0.216 g, 90%), mp 68–70 °C; 1H NMR (500 MHz, CDCl3) δ 8.85 (d, J = 34.2 Hz, 1H), 8.29 (d, J = 7.8Hz, 2H), 7.86-δ7.78 (m, 4H), δ7.71 (s, 1H), 7.57-7.41 (m, 8H), 7.07(d, J = 3.6Hz, 1H), 6.62-6.61(m, 1H), 4.71-4.64 (m, 2H), 4.56-4.49 (m, 2H), 3.22-3.09 (m, 4H). 13C NMR (125 MHz, CDCl3) δ161.2 (d, J = 5.9Hz), 152.1, 151.8, 147.4, 133.7 (2C), 132.6 (d, J = 7.3Hz), 131.7 (2C), 128.4 (d, J = 13.7Hz, 2C), 126.7 (2C), 126.3 (2C), 125.7 (2C), 125.1 (2C), 123.8 (2C), 121.2 (2C), 112.7 (2C), 47.2 (d, J = 5.0Hz, 2C), 44.8 (d, J =10.3Hz, 2C). 31P NMR (202 MHz; CDCl3) δ 26.4. HRMS (ESI): m/z calcd for C29H27N3O2P, 480.1841; found, 480.1839.
Typical procedure for the synthesis of N,N-naphthalen-1-ylmethyl N-phosphonyl imine substituted α-aminonitriles
In a dry vial, under inert gas protection, 4 Å MS and the chiral amino acid (3) was added followed by loading dry toluene. A turbid solution was obtained in which diethylaluminium cyanide (2, 1.0 M solution in toluene, 1.50 mmol) was added followed by the addition of i-PrOH (1.0 mmol). The reaction was stirred for 15 min at room temperature, and then cooled to −78 °C. The resulting mixture was stirred at −78 °C for 30 min followed by the addition of achiral N-phosphonyl imine which was dissolved in 3 mL of toluene. The reaction mixture was stirred for 5 h at this temperature before it was quenched by aq. 0.05 M HCl, followed by addition of ethylacetate (10 mL) and water (10 mL). The reaction mixture was filter off through celite. Organic layer was separated (3×10mL of ethyl acetate) and dried over anhydrous sodium sulfate. Sodium sulfate was filtered off, and organic phase was evaporated to obtain crude product, which upon washings with hexane afforded the pure product as white solid without further purification.
Compound 4a
Yield (0.246 g, 92%); mp 112–114 °C; [α]D24 = + 2.52 (c 1.0, CHCl3) 1H-NMR (500 MHz, CDCl3) δ 8.22 (d, J = 9.5 Hz, 2H), 8.15 (d, J = 7.5 Hz, 2H), 7.86 (t, J = 7.0 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 7.51-7.45 (m, 5H), 7.44-7.39 (m, 4H), 7.35-7.34 (m, 2H), 5.42 (t, J = 9.0 Hz, 1H), 4.65-4.55 (m, 4H ), 3.44 (t, J = 9.5 Hz, 1H), 3.05-3.01 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ 135.36, 135.31, 133.8, 133.7, 132.37, 132.31, 131.6 (d, J = 8.0 Hz, 2C), 128.6 ( d, J = 7.3 Hz, 2C), 128.5, 128.4, 128.3, 126.7, 126.6, 126.5, 126.4, 126.3, 125.9, 125.8, 125.24, 125.20, 123.4, 123.3 (2C), 119.98, 119.92, 47.5, 46.8 (d, J = 7.3 Hz), 44.6 (d, J = 7.8 Hz), 44.0, 43.9.31P-NMR (202 MHz, CDCl3) δ 22.9. HRMS (ESI): m/z calcd for C32H30N4OP, 517.2152; found, 517.2156. 99.7 %ee, retention time = 6.77 (major), flow rate = 0.60ml/min, OD-H chiral column (hexane:i-PrOH, v/v = 3:7).
Compound 4b
Off-white solid, Yield (0.172g, 96%); mp: 126–128 °C, [α]D24 = + 7.0 (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.16 (dd, J = 9.2, 27.2 Hz, 2H), 7.82 (dd, J = 8.0, 36.0 Hz, 4H), 7.51-7.44 (m, 6H), 7.40 (q, J = 8.2 Hz, 2H), 7.33 (q, J = 5.1 Hz, 2H), 6.95 (t, J = 8.5 Hz, 2H), 5.36 (t, J = 9.0 Hz, 1H), 4.67-4.52 (m, 4H), 3.82 (q, J = 9.8 Hz, 1H, NH), 3.12-3.02 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 163.9, 161.9, 133.8 (d, J = 8.9 Hz, 2C), 132.3 (d, J = 2.5 Hz), 132.2, 131.5 (d, J = 9.9 Hz), 131.2, 128.7 (d, J = 6.9 Hz, 2C), 128.6 (2C), 128.5, 128.4, 126.7, 126.4 (d, J = 12.4 Hz, 2C), 126.3, 125.9 (d, J = 9.9 Hz, 2C), 125.2 (d, J = 3.9 Hz, 2C), 123.3 (d, J = 12.9 Hz, 2C), 119.7 (d, J = 3.5 Hz), 116.14 (d, J = 21.8 Hz, 2C), 46.84 (d, J = 3.9 Hz), 46.82, 46.1 (d, J = 4.9 Hz), 44.8 (d, J = 12.4 Hz), 44.0 (d, J = 13.4 Hz). 31P NMR (202 MHz, CDCl3) δ 22.1. HRMS (ESI): m/z calcd for C32H28FN4OPNa, 557.1877; found, 557.1884. Ee: 94% (retention time = 6.49 (minor) and 7.17 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4c
White solid; Yield (0.13 5g, 97%); mp 138–140 °C; [α]D25 = +8.0° (c 0.6, CHCl3). 1H NMR (500 MHz, CDCl3): 8.14 (dd, J = 7.5 Hz, 16.5 Hz, 2H), 7.86 (d, J = 7.5 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H), 7.51-7.45 (m, 6H), 7.43-7.36 (m, 4H), 7.20 (d, J = 8.0 Hz, 2H), 5.32 (t, J = 9.0 Hz, 1H), 4.66-4.49 (m, 4H), 3.90 (q, J = 6.5 Hz, 1H), 3.11-3.05 (m, 4H); 13C NMR (125 MHz, CDCl3): 134.4 (d, J = 5.9 Hz), 133.8 (d, J = 7.9 Hz), 132.3 (2C), 132.2, 131.6 (d, J = 8.4 Hz), 128.7 (d, J = 3.5 Hz, 2C), 128.5 (2C), 128.4 (2C), 128.3 (2C), 126.6 (2C), 126.4 (d, J = 7.9 Hz, 2C), 126.2 (2C), 125.9 (d, J = 7.5 Hz, 2C), 125.2 (d, J = 2.9 Hz, 2C), 123.4, 123.3 (d, J = 12.8 Hz), 119.5 (d, J = 3.5 Hz), 46.9, 46.8 (d, J = 4.4 Hz), 46.1 (d, J = 4.9 Hz), 44.8 (d, J = 12.2 Hz), 44.1 (d, J = 13.4 Hz); 31P NMR (202 MHz, CDCl3): 23.0. HRMS (ESI): m/z calcd for C32H29BrN4OP, 595.1257; found, 595.1262. Ee: 96% (retention time = 6.41 (minor) and 6.84 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4d
Pale yellow color solid, Yield (0.128 g, 95%); mp 134–136 °C, [α]D24 = + 8.60 (c 0.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.15 (dd, J = 8.3, 23.4 Hz, 2H), 7.86 (d, J = 7.8 Hz, 2H), 7.78 (d, J = 8.1 Hz, 2H), 7.51-7.45 (m, 6H), 7.40 (q, J = 7.8 Hz, 2H), 7.28-7.22 (m, 4H), 5.35 (t, J = 9.0 Hz, 1H), 4.66-4.51 (m, 4H), 3.77 (bs, 1H, NH), 3.12-3.03 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 135.2, 133.85, 133.81, 133.7, 132.27, 132.25, 132.21, 131.5 (d, J = 9.9 Hz), 129.3 (2C), 128.7 (d, J = 4.9 Hz, 2C), 128.5 (d, J = 17.9 Hz, 2C), 127.9 (2C), 126.7, 126.4 (d, J = 9.9 Hz, 2C), 126.3, 125.9 (d, J = 8.4 Hz, 2C), 125.3 (d, J = 2.9 Hz, 2C), 123.2 (d, J = 14.4 Hz, 2C), 119.5 (d, J = 3.5 Hz), 46.9, 46.8 (d, J = 4.5 Hz), 46.1 (d, J = 5.4 Hz), 44.8 (d, J = 11.9 Hz), 44.1 (d, J = 13.4 Hz). 31P NMR (202 MHz, CDCl3) δ 22.6. HRMS (ESI): m/z calcd for C32H29ClN4OP, 551.1761; found, 551.1768. Ee: 95.2% (retention time = 6.27 (minor) and 6.72 (major flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4e
White solid, Yield (0.110 g, 94%); mp 176–178 °C, [α]D24 = + 7.45 (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 7.0 Hz, 1H), 8.15 (d, J = 7.5 Hz, 1H), 7.87-7.85 (m, 2H), 7.79 (d, J = 5.0 Hz, 1H), 7.31 (d, J = 7.0 Hz, 7H), 7.12 (d, J = 7.0 Hz, 6H), 5.37 (t, J = 8.5 Hz, 1H), 4.66-4.48 (m, 4H), 3.67 (t, J = 10.0 Hz, 1H, NH), 3.07-2.99 (m, 4H), 2.30 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 139.2, 133.7 (d, J = 8.4 Hz), 132.5 (d, J = 6.5 Hz), 132.4 (d, J = 5.5 Hz), 132.3 (d, J = 6.0 Hz), 131.6 (d, J = 8.4 Hz), 129.8 (2C), 128.6 (2C), 128.5 (2C), 128.4 (2C), 128.3, 126.5 (2C), 126.4 (d, J = 6.4 Hz), 126.3, 125.8 (2C), 125.7 (2C), 125.2 (d, J = 4.0 Hz), 123.3 (d, J = 7.5 Hz), 120.0 (d, J = 4.0 Hz), 47.2, 46.6 (d, J = 4.5 Hz), 46.1 (d, J = 5.4 Hz), 44.6 (d, J = 11.9 Hz), 44.0 (d, J = 12.9 Hz), 21.0. 31P NMR (202 MHz, CDCl3) δ 22.5. HRMS (ESI): m/z calcd for C33H31N4O2PNa, 569.2077; found, 569.2070. Ee: 98.8% (retention time = 6.82 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4f
White solid, Yield (0.132 g, 93%); mp 182–184 °C, [α]D24 = + 1.45 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.23-8.19 (m, 1H), 8.16-8.14 (m, 1H), 7.89-7.84 (m, 2H), 7.80-7.78 (m, 2H), 7.52-7.47 (m, 6H), 7.43-7.39 (m, 2H), 7.32-7.28 (m, 2H), 6.83-6.79 (m, 2H), 5.34 (t, J = 8.7 Hz, 1H), 4.66-4.52 (m, 4H), 3.75 (s, 3H), 3.50 (t, J = 9.6 Hz, 1H, NH), 3.09-3.00 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 160.2, 133.8 (d, J = 8.4 Hz), 132.4 (d, J = 6.5 Hz), 131.6 (d, J = 8.4 Hz, 2C), 128.7 (2C), 128.6 (2C), 128.5 (2C), 128.3 (2C), 128.0 (2C), 127.5 (d, J = 6.4 Hz), 126.6, 126.4, 126.3, 125.9 (2C), 125.8 (2C), 125.2 (d, J = 4.0 Hz), 123.4 (d, J = 7.5 Hz), 120.1(d, J = 4.0 Hz), 114.5 (2C), 55.3, 46.9, 46.7 (d, J = 4.5 Hz), 46.1 (d, J = 5.4 Hz), 44.7 (d, J = 11.9 Hz), 44.0 (d, J = 12.9 Hz). 31P NMR (202 MHz, CDCl3) δ 22.5. HRMS (ESI): m/z calcd for C33H31N4O2PNa, 569.2077; found, 569.2070. Ee: 97.6% (retention time = 6.06 (minor) and 6.88 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4g
Off-white solid, Yield (0.101 g, 94%); mp 180–182 °C, [α]D24 = + 3.30 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.21 (d, J = 8.2 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.86-7.83 (m, 2H), 7.79-7.76 (m, 2H), 7.54-7.29 (m, 10H), 7.13-7.05 (m, 2H), 5.64 (t, J = 9.8 Hz, 1H), 4.66-4.39 (m, 4H), 3.95 (t, J = 10.0 Hz, 1H, NH), 2.99 (d, J = 10.7 Hz, 4H). 13C NMR (125 MHz, CDCl3) δ 160.9, 158.9, 133.72 (d, J = 4.9 Hz, 2C), 132.29 (d, J = 6.9 Hz), 132.23 (d, J = 8.4 Hz), 131.6 (d, J = 8.9 Hz), 131.3 (d, J = 8.4 Hz), 128.7 (d, J = 2.5 Hz), 128.6 (d, J = 3.5 Hz, 2C), 128.4 (d, J = 12.4 Hz, 2C), 126.6, 126.4 (2C), 126.3, 126.2 (2C), 125.8 (d, J = 6.9 Hz, 2C), 125.1 (d, J = 4.9 Hz, 2C), 125.0 (d, J = 3.5 Hz), 123.3 (d, J = 10.9 Hz, 2C), 116.2 (d, J = 20.3 Hz), 46.3 (d, J = 4.9 Hz), 46.1 (d, J = 4.9 Hz), 44.1 (dd, J = 20.3, 12.9 Hz, 2C), 42.6. 31P NMR (202 MHz, CDCl3) δ 22.0. HRMS (ESI): m/z calcd for C32H28FN4OPNa, 557.1877; found, 557.1886. Ee: 96.1% (retention time = 6.52 (minor) and 7.04 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4h
White solid; Yield (0.135 g, 94%); mp 128–130 °C; [α]D25 = +7.7° (c 1.1, CHCl3). 1H NMR (500 MHz, CDCl3): 8.19 (d, J = 8.5 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 7.5 Hz, 2 H), 7.77 (d, J = 8.0 Hz, 2H), 7.60-7.57 (m, 2H), 7.54-7.37 (m, 8 H), 7.33-7.30 (m, 1H), 7.21-7.18 (m, 1H), 5.77 (t, J = 9.5 Hz, 1H), 4.64 (dd, J = 6.5 Hz, 14.5 Hz, 1H), 4.57 (t, J = 6 Hz, 2H), 4.36 (dd, J = 6.5 Hz, 15 Hz, 1H), 3.88 (t, J = 10 Hz, 1H), 3.03-2.94 (m, 4H); 13C NMR (125 MHz, CDCl3): 135.0 (d, J = 5.0 Hz), 133.8 (d, J = 8.9 Hz, 2C), 132.4, 132.3 (d, J = 3.4 Hz), 132.2, 131.6 (d, J = 6.4 Hz), 130.9 (s, 2C), 129.3 (s, 2C), 128.6 (d, J = 1.4 Hz, 2C), 128.4 (t, J = 3.0 Hz, 2C), 126.5 (d, J = 4.9 Hz, 2C), 126.3 (s, 2C), 125.8 (d, J = 5.4 Hz, 2C), 125.2 (d, J = 9.9 Hz, 2C), 123.4 (d, J = 5.5 Hz, 2C), 122.6, 118.8 (d, J = 5.4 Hz), 47.8 (d, J = 2 Hz), 46.4 (t, J = 5.9 Hz, 2C), 44.2 (dd, J = 6.0Hz, 12.9 Hz, 2C); 31P NMR (202 MHz, CDCl3): 22.1. HRMS (ESI): m/z calcd for C32H29BrN4OP, 595.1257; found, 595.1253. Ee: 97% (retention time = 6.49 (minor) and 6.95 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4i
White solid, Yield (0.146 g, 90%); mp 220–222 °C, [α]D24 = + 4.50 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 8.2 Hz, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.84 (d, J = 7.8 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H), 7.60 (dd, J = 2.0, 7.2 Hz, 1H), 7.53-7.37 (m, 9H), 7.28-7.22 (m, 2H), 5.76 (t, J = 9.8 Hz, 1H), 4.63 (dd, J = 6.5, 14.8 Hz, 1H), 4.55 (d, J = 6.3 Hz, 2H), 4.36 (dd, J = 6.2, 14.8 Hz, 1H), 4.13 (t, J = 10.0 Hz, 1H, NH), 3.04-2.96 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 133.72 (d, J = 1.5 Hz), 133.44 (d, J = 4.9 Hz), 132.7, 132.3 (d, J = 6.9 Hz), 132.2 (d, J = 7.4 Hz), 131.6 (d, J = 6.9 Hz), 130.6 (d, J = 32.3 Hz, 2C), 129.1 (2C), 128.6 (2C), 128.4 (d, J = 5.4 Hz, 2C), 127.8 (2C), 126.4 (2C), 126.3 (d, J = 20.8 Hz, 2C), 125.8 (d, J = 4.9 Hz, 2C), 125.2 (d, J = 9.4 Hz, 2C), 123.3 (d, J = 7.9 Hz, 2C), 118.8 (d, J = 5.5 Hz), 46.4 (d, J = 4.9 Hz), 46.2 (d, J = 4.9 Hz), 45.6 (d, J = 2.5 Hz), 44.2 (d, J = 2.9 Hz), 44.1 (d, J = 2.9 Hz). 31P NMR (202 MHz, CDCl3) δ 22.3. HRMS (ESI): m/z calcd for C32H29ClN4OP, 551.1761; found, 551.1757. Ee: 95.2% (retention time = 6.28 (minor) and 6.82 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4j
White solid, Yield (0.152 g, 92%); mp 204–206 °C, [α]D24 = + 3.50 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.31 (d, J = 7.2 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.88-7.78 (d, J = 7.8 Hz, 7H), 7.50-7.26 (m, 10H), 5.56 (t, J = 9.8 Hz, 1H), 4.46-4.14 (m, 4H), 3.52 (t, J = 10.0 Hz, 1H, NH), 3.04-2.96 (m, 4H), 2.41 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 135.6, 133.7 (d, J = 4.9 Hz), 132.3, 131.7 (d, J = 6.9 Hz), 131.4 (2C), 129.4 (2C), 128.6 (2C), 128.4 (d, J = 5.4 Hz, 2C), 128.2, 126.9 (d, J = 7.5 Hz, 2C), 126.6 (2C), 126.4 (d, J = 20.8 Hz, 2C), 126.3 (d, J = 4.9 Hz, 2C), 125.8, 125.1 (d, J = 9.0 Hz, 2C), 123.6, 123.4, 123.3, 46.9 (d, J = 4.9 Hz), 46.6 (d, J = 4.9 Hz), 46.3 (d, J = 2.5 Hz), 44.3 (t, J = 2.9 Hz), 43.9 (d, J = 3.0 Hz), 19.0. 31P NMR (202 MHz, CDCl3) δ 22.3. Ee: 98.9% (retention time = 6.13 (minor) and 6.68 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Compound 4k
White solid; yield (0.156 g, 89%), mp 148–150 °C [α]D25 + 1.74 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.25(d, J = 8.5 Hz, 1H), 8.19-8.17 (m, 1H), 7.88-7.84 (m, 2H), 7.79 (t, J = 7.0 Hz, 2H), 7.55-7.47 (m, 6H), 7.43-7.39 (m, 2H), 7.37-7.36 (m, 1H), 6.46-6.45 (m, 1H), 6.35-6.34(m, 1H), 5.60(t, J = 9.5 Hz, 1H), 4.65-4.53 (m, 4H), 3.62 (t, J = 10.0 Hz, 1H), 3.03-2.93 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 147.5 (d, J = 6.4 Hz), 143.8, 133.8 (d, J = 7.8 Hz), 132.3, 131.7 (d, J = 7.3 Hz), 128.7, 128.4, 126.9, 126.5 (d, J = 13.4 Hz), 125.9 (d, J = 7.8 Hz), 125.2 (d, J = 5.9 Hz), 123.4 (d, J = 9.3 Hz), 117.9 (d, J = 4.4 Hz), 46.6 (d, J = 5.0 Hz), 46.1 (d, J = 5.3 Hz), 44.4 (d, J = 12.8 Hz), 43.9 (d, J = 13.3 Hz), 41.9 (d, J = 1.0 Hz). 31P NMR (202 MHz; CDCl3) δ 21.6. HRMS (ESI): m/z calcd for C30H27N4O2PNa, 529.1764; found, 529.1761. Ee: 99% (retention time = 6.55 (minor) and 6.83 (major), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system).
Absolute Configuration Determination
The absolute configuration for this asymmetric induction was determined by converting a product (4a) to an authentic sample.[2] In this conversion, 4a was subjected to deprotection with 2.0 M aq. HCl at r.t. in methanol followed by in situ t-Boc protection by treating with (t-Boc)2O in the presence of Et3N to give product 5. The optical rotation of this product was confirmed to be consistent with that of the known sample with S configuration. During this transformation, the cleaved N1, N2-bis(naphthalen-1-ylmethyl)ethane-1,2-diamine (6) was extracted with n-butanol from the acidic mixture prior to t-Boc protection to give a quantitative yield. [α]24D = −1.71 (c = 1.2, CHCl3) {Literature value [α]24D −1.82 (c = 1.1, CHCl3)}.
Supplementary Material
Scheme 4.

Acknowledgments
We would like to thank the Robert A. Welch Foundation (D-1361) for providing us with financial assistance. Partial support from NIH (R03DA026960) and NSFC (20928001) is also acknowledged. We thank our co-workers, Teng Ai, Venkatesh Kattamuri, Hao Sun, Austin Ballew and Raizada Milles, for their assistance.
Footnotes
Supporting Information Available. 1H and 13C NMR spectra of all pure products 1a–1k and 4a–4k are available free of charge via the Internet at http://pubs.acs.org.
Notes and References
- 1.(a) Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]; (b) Aschwanden P, Carreira EM. In: Acetylene Chemistry: Chemistry, Biology and Material Science. Diederich F, Stang PJ, Tykwinski RR, editors. Wiley; Weinheim: 2005. pp. 101–138. [Google Scholar]
- 2.(a) Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394–1401. doi: 10.1021/ar7001123. [DOI] [PubMed] [Google Scholar]; (b) Cannon SJ. Angew Chem Int Ed. 2008;47:1176–1178. doi: 10.1002/anie.200703879. [DOI] [PubMed] [Google Scholar]; (c) Sweeney J. Eur J Org Chem. 2009;29:4911–4919. [Google Scholar]; (d) Ferreira F, Botuha C, Chemla F, Perez-Luna A. Chem Soc Rev. 2009;38:1162–1186. doi: 10.1039/b809772k. [DOI] [PubMed] [Google Scholar]; (e) Friestad GK, Mathies AK. Tetrahedron. 2007;63:2541–2569. doi: 10.1016/j.tet.2007.06.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Enders D, Shilvock JP. Chem Soc Rev. 2000;29:359–373. [Google Scholar]; (b) Williams RM, Hendrix AJ. Chem Rev. 1992;92:889–917. [Google Scholar]; (c) Bloch R. Chem Rev. 1998;98:1407–1438. doi: 10.1021/cr940474e. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069–1094. doi: 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN. Nature. 2009;461:968–970. doi: 10.1038/nature08484. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sigman MS, Vachal P, Jacobsen EN. Angew Chem Int Ed Engl. 2000;39:1279–1281. doi: 10.1002/(sici)1521-3773(20000403)39:7<1279::aid-anie1279>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Benowitz AB, Sprengeler PA, Barbosa J, Guzman MC, Hirschmann R, Schweiger EJ, Bolin DR, Nagy Z, Campbell RM, Cox DC, Olson GL. J Am Chem Soc. 1999;121:9286–9298. [Google Scholar]; (d) Pan SC, List B. Org Lett. 2007;9:1149–1151. doi: 10.1021/ol0702674. [DOI] [PubMed] [Google Scholar]
- 5.(a) Cowen BJ, Saunders LB, Miller SJ. J Am Chem Soc. 2009;131:6105–6107. doi: 10.1021/ja901279m. [DOI] [PubMed] [Google Scholar]; (b) Yang JW, Stadler M, List B. Angew Chem Int Ed. 2007;46:609–611. doi: 10.1002/anie.200603188. [DOI] [PubMed] [Google Scholar]; (c) Zuend SJ, Jacobsen EN. J Am Chem Soc. 2009;131:15358–15374. doi: 10.1021/ja9058958. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Uraguchi D, Koshimoto K, Ooi T. J Am Chem Soc. 2008;130:10878–10879. doi: 10.1021/ja8041004. [DOI] [PubMed] [Google Scholar]; (d) Wieland LC, Vieira EM, Snapper ML, Hoveyda AH. J Am Chem Soc. 2009;131:570–576. doi: 10.1021/ja8062315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Yin L, Kanai M, Shibasaki M. J Am Chem Soc. 2009;131:9610–9611. doi: 10.1021/ja9036675. [DOI] [PubMed] [Google Scholar]; (b) Abell JP, Yamamoto H. J Am Chem Soc. 2009;131:15118–15119. doi: 10.1021/ja907268g. [DOI] [PubMed] [Google Scholar]
- 7.(a) Chowdari NS, Ahmad M, Albertshofer K, Tanaka F, Barbas CF., III Org Lett. 2006;8:2839–2842. doi: 10.1021/ol060980d. [DOI] [PubMed] [Google Scholar]; (b) Kawamura K, Fukuzawa H, Hayashi M. Org Lett. 2008;10:3509–3512. doi: 10.1021/ol801259u. [DOI] [PubMed] [Google Scholar]; (c) Gille S, Cabello N, Kizirian JC, Alexakis A. Tetrahedron: Asymmetry. 2006;17:1045–1047. [Google Scholar]
- 8.(a) Abermil N, Masson G, Zhu J. J Am Chem Soc. 2008;130:12596–12597. doi: 10.1021/ja805122j. [DOI] [PubMed] [Google Scholar]; (b) Aggarwal VK, Alonso E, Ferrara M, Spey SE. J Org Chem. 2002;67:2335–2344. doi: 10.1021/jo016312h. [DOI] [PubMed] [Google Scholar]; (c) Okamoto K, Hayashi T, Rawal VH. Chem Commun. 2009:4815–4817. doi: 10.1039/b904624k. [DOI] [PubMed] [Google Scholar]; (d) Shi M, Chen LH, Li CQ. J Am Chem Soc. 2005;127:3790–3800. doi: 10.1021/ja0447255. [DOI] [PubMed] [Google Scholar]
- 9.(a) Storer RI, Carrera DE, Ni Y, MacMillan DWC. J Am Chem Soc. 2006;128:84–86. doi: 10.1021/ja057222n. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Silverman SM, Stambuli JP. J Am Chem Soc. 2007;129:12398–12399. doi: 10.1021/ja0753389. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wieland LC, Vieira EM, Snapper ML, Hoveyda AHJ. Am Chem Soc. 2009;131:570–576. doi: 10.1021/ja8062315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Corey EJ, Grogan MJ. Org Lett. 1999;1:157–160. doi: 10.1021/ol990623l. [DOI] [PubMed] [Google Scholar]; (e) Simon L, Goodman JM. J Am Chem Soc. 2009;131:4070–4077. doi: 10.1021/ja808715j. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kaur P, Shakya G, Sun H, Pan Y, Li G. Org Biomol Chem. 2010;8:1091. doi: 10.1039/b923914f. [DOI] [PubMed] [Google Scholar]; (b) Kaur P, Nguyen T, Li G. Eur J Org Chem. 2009:912–916. [Google Scholar]; (c) Ai T, Li G. Bioorg Med Chem Lett. 2009;19:3967–3969. doi: 10.1016/j.bmcl.2009.03.001. [DOI] [PubMed] [Google Scholar]; (d) Ai T, Han J, Chen ZX, Li G. Chem Biol Drug Des. 2009;73:203–208. doi: 10.1111/j.1747-0285.2008.00771.x. [DOI] [PubMed] [Google Scholar]; (e) Kattuboina A, Li G. Tetrahedron Lett. 2008;49:1573–1577. [Google Scholar]
- 11.Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 12.(a) Nagata W, Yoshioka M, Hirai S. J Am Chem Soc. 1972;94:4635–4643. [Google Scholar]; (b) Benedetti F, Berti F, Norbedo S. Tetrahedron Lett. 1999;40:1041–1044. [Google Scholar]; (c) Nakamura S, Sato N, Sugimoto M, Toru T. Tetrahedron: Asymmetry. 2004;15:1513–1516. [Google Scholar]; (d) Davis FA, Zhang H, Lee SH. Org Lett. 2001;3:759–762. doi: 10.1021/ol015520t. [DOI] [PubMed] [Google Scholar]
- 13.Paradowska J, Stodulski M, Mlynarski J. Angew Chem Int Ed. 2009;48:4288–4297. doi: 10.1002/anie.200802038. [DOI] [PubMed] [Google Scholar]; (b) Córdova A, Zou W, Ibrahem I, Reyes E, Engqvist M, Liao WW. Chem Commun. 2005:3586–3588. doi: 10.1039/b507968n. [DOI] [PubMed] [Google Scholar]
- 14.Corey EJ, Loh TP, Roper TD, Azimioara MD, Noe MC. J Am Chem Soc. 1992;114:8290–8292. [Google Scholar]; (b) Li G, Wei HX, Phelps BS, Purkiss DW, Kim SH. Org Lett. 2001;3:823–826. doi: 10.1021/ol000377+. [DOI] [PubMed] [Google Scholar]
- 15.Fu GC. Acc Chem Res. 2008;41:1555–1564. doi: 10.1021/ar800148f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guram AS, Buchwald SL. J Am Chem Soc. 1994;116:7901–7902. [Google Scholar]; (c) Paul F, Patt J, Hartwig JF. J Am Chem Soc. 1994;116:5969–5970. [Google Scholar]
-
16.An aliphatic N-phosphonyl substituted imine was prepared in situ and subject to asymmetric strecker reaction using the optimized conditions.
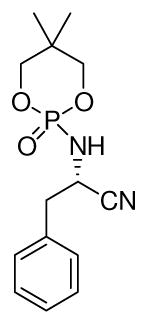 Yellow oil, yield (0.090g, 92%); [α]D24 = + 12.0 (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.16–4.08 (m, 4H), 3.96–3.84 (m, 3H), 3.35 (bs, 1H), 1.32 (t, J = 5.5 Hz, 1H), 1.25 (s, 3H), 1.07 (s, 3H), 0.93 (s, 3H), 0.85 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 128.7, 76.3 (2C), 68.0, 45.5, 38.5, 21.5, 21.49, 21.43, 20.8, 19.9. 31P NMR (202 MHz, CDCl3) δ 22.1. Ee: 92% (retention time = 5.43 (major) and 5.86 (minor), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system). HPLC conditions need to be further optimized (peaks’ overlapping still exists).
Yellow oil, yield (0.090g, 92%); [α]D24 = + 12.0 (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.16–4.08 (m, 4H), 3.96–3.84 (m, 3H), 3.35 (bs, 1H), 1.32 (t, J = 5.5 Hz, 1H), 1.25 (s, 3H), 1.07 (s, 3H), 0.93 (s, 3H), 0.85 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 128.7, 76.3 (2C), 68.0, 45.5, 38.5, 21.5, 21.49, 21.43, 20.8, 19.9. 31P NMR (202 MHz, CDCl3) δ 22.1. Ee: 92% (retention time = 5.43 (major) and 5.86 (minor), flow rate = 0.60 ml/min, OD-H chiral column (7:3 hexane:IPA solvent system). HPLC conditions need to be further optimized (peaks’ overlapping still exists).
- 17.Kuo C-W, Zhu J-L, Wu J-D, Chu C-M, Yao CF, Shia K-S. Chem Comm. 2007;3:301–303. doi: 10.1039/b614061k. [DOI] [PubMed] [Google Scholar]
- 18.We tentatively define this observation as the “GAP” synthesis (group-assisted-precipitation synthesis) since both achiral and chiral N-phosphonyl groups, and chiral N-phosphinyl group have resulted in similar simple purification and recovery of auxiliary quantitatively for re-use for several reactions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.