

Abstract
Proton-translocating ATPases are ubiquitous protein complexes that couple ATP catalysis with proton translocation via a rotary catalytic mechanism. The peripheral stalks are essential components that counteract torque generated from proton translocation during ATP synthesis or from ATP hydrolysis during proton pumping. Despite their essential role, the peripheral stalks are the least conserved component of the complexes, differing substantially between subtypes in composition and stoichiometry. We have determined the crystal structure of the peripheral stalk of the A-type ATPase/synthase from Thermus thermophilus consisting of subunits E and G. The structure contains a heterodimeric right-handed coiled coil, a protein fold never observed before. We have fitted this structure into the 23-Å resolution electron microscopy density of the intact A-ATPase complex, revealing the precise location of the peripheral stalk and new implications for the function and assembly of proton-translocating ATPases.
Proton-translocating ATPases (H+-ATPases) are rotary enzymes that couple proton (or Na+) translocation across membranes with ATP synthesis or hydrolysis1–4. There are three evolutionarily related subtypes of these protein complexes that are categorized as F-, V- and A-type ATPases on the basis of their function and taxonomic origin. F-type ATPases, better known as F1Fo ATP synthases, use energy from proton translocation across an electrochemical gradient to synthesize ATP3. In contrast, vacuolar or V-type ATPases work in reverse by actively pumping protons through membranes using energy derived from ATP hydrolysis1. Although eukaryotes contain both F- and V-ATPases, each highly specialized in its physiological function, archaea and eubacteria typically contain only one complex. Most eubacteria contain F-type ATPases, but some eubacteria and all known archaea contain complexes that are evolutionary closer to V-ATPases and are referred to as A-ATPases/synthases. Although these complexes are simpler in subunit composition than their eukaryotic counterparts, they are functionally more versatile, as they are able to operate in either direction5,6.
The overall architecture of all H+-ATPases is conserved and includes a water-soluble ATPase-active F1,V1, or A1 domain and a membrane-bound proton-translocating Fo, Vo, or Ao domain1,3,7. The T. thermophilus A1 domain is composed of a head group that contains a trimer of nucleotide-binding AB dimers and a central stalk composed of subunits C, D and F8–10. The proton-translocating Ao domain contains a ring of L proteolipids and a single copy of the amphipathic subunit I that is located adjacent to the ring, thus forming the proton path11 (Fig. 1a). ATP synthesis and hydrolysis are coupled to proton translocation via the rotation of the central stalk and L-ring relative to the nucleotide-binding domain and subunit I. However, an additional stator component is required to counteract the rotation of the nucleotide-binding domain during ATP synthesis and subunit I during hydrolysis. This essential component, known as the peripheral stalk or stator stalk, is composed of subunits E and G in A-ATPases and links the nucleotide-binding domain to subunit I (refs. 12–14; Fig. 1).
Figure 1.

Architecture and nomenclature of the intact T. thermophilus A-ATPase/ synthase. (a) Side view of the A-ATPase. The stator subunits (A, B, E, G and I) are labeled in white and the rotor subunits (D, F, C and L) are labeled in black. (b) View from above. The black arrow indicates the rotational direction of the central stalk during ATP synthesis and the white arrows indicate the torque in the A3B3 nucleotide-binding domain that subunit E counteracts.
Despite its central role in all H+-ATPases, the peripheral stalk is the least conserved component of these complexes, and EM and MS have revealed distinct configurations in different H+-ATPase subtypes. In F-type ATP synthase, there is only one peripheral stalk15–19, whereas A- and V-ATPases display two and three peripheral stalks, respectively, each made of an EG heterodimer11–13,20–24.
The high-resolution structure of the mitochondrial F-type ATP synthase single peripheral stalk complex consisting of subunits b, F6 and d25 and the NMR structure of the oligomycin sensitivity-conferring protein26 have been determined. However, these subunits show little to no sequence homology to either A- or V-type peripheral stalk subunits, for which only the structure of the C-terminal domain of Pyrococcus horikoshii subunit E27 is known.
We have solved the crystal structure of the complete peripheral stalk complex formed by subunits E and G from T. thermophilus at 3.1-Å resolution and have fitted our structure into the 23-Å resolution EM density of intact T. thermophilus A-ATPase along with other known high-resolution structures. This has provided the most complete composite model of any A- or V-ATPase so far to our knowledge and has revealed several functionally important features of these complexes.
First, as in the central stalk, coiling in the peripheral stalk suggests that it provides a second avenue for these types of H+-ATPases to store transient elastic energy. Second, heterodimerization of the peripheral stalk allows each subunit to have distinct roles, in which subunit E mediates the interaction with the nucleotide-binding domain and subunit G strengthens the peripheral stalk. Third, unlike the central stalk, the peripheral stalk is a right-handed coiled coil, which places subunit G in a mechanically robust position to act as an obstruction and prevent bowing of subunit E during ATP synthesis, hence increasing the lateral rigidity of the peripheral stalk. Finally, the two peripheral stalks in A-ATPases appear to be in different conformations in the intact A-ATPase density, supporting proposed models of conformational flexibility in the stator subunits. This is further corroborated by conformational differences between the C-terminal domains of T. thermophilus and P. horikoshii27 E subunits that highlight the potential of an interdomain loop region to act as a hinge.
RESULTS
Overall structure of the E–G heterodimer
Subunits E and G assemble into an elongated heterodimeric complex (Fig. 2 and Supplementary Fig. 1). Both subunits have a peculiar, highly repetitive sequence that is enriched in the amino acids alanine (21%), leucine (15%), glutamate (18%) and arginine (10%). The structure of the heterodimer reveals how these create a strong surface complementarity between the E–G subunits. The leucine and alanine residues form the majority of a large hydrophobic subunit interface (2560 Å2; Fig. 3c). The glutamate and arginine residues form part of a charged, solvent-exposed surface as well as at least ten intersubunit salt bridges that act to further stabilize the heterodimer.
Figure 2.
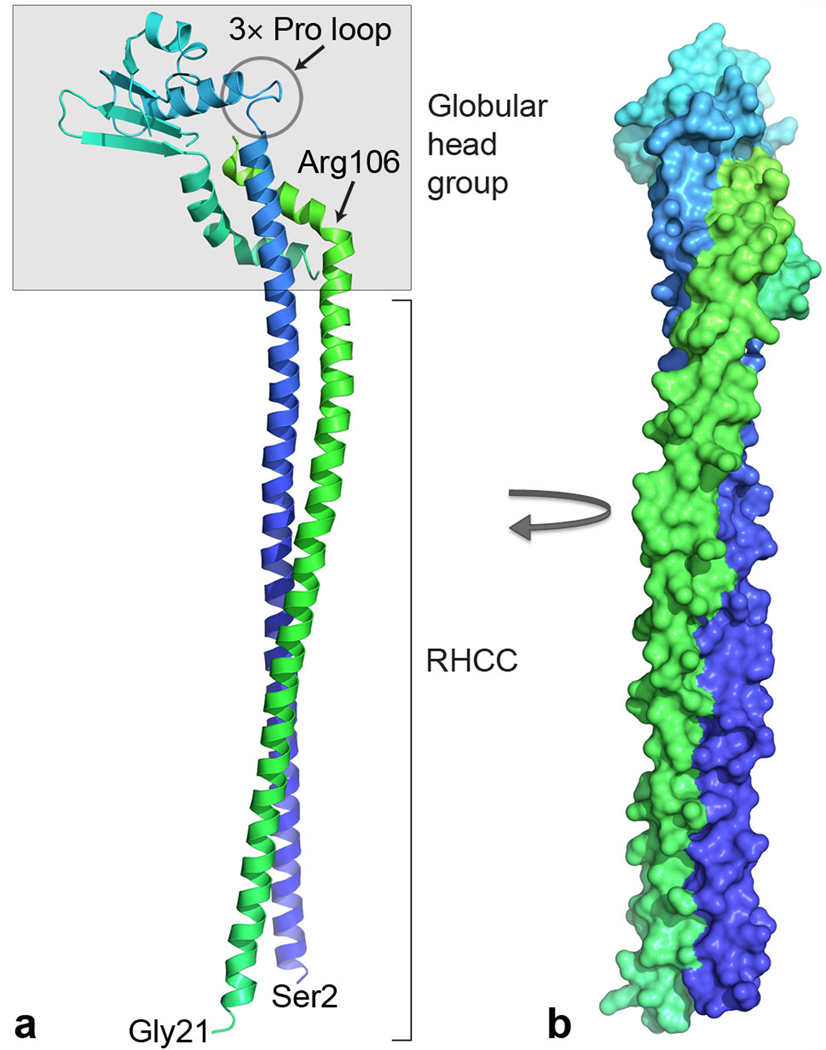
Structure of the EG peripheral stalk complex. (a) Ribbon representation with subunit E (blue to cyan) from N to C terminus and subunit G (green). The C termini of both subunits are shaded in gray and form a globular head group, whereas the N-terminal helices form an RHCC. These domains are tethered via flexible loops, one containing three proline residues (3× Pro loop) in subunit E and the other consisting of a kink at Arg106 in subunit G. Bottom, N-terminal residues of subunits E (Ser2) and G (Gly21). (b) Surface representation of the complex rotated clockwise by 90° (same colors as in a).
Figure 3.

Hydrophobic repeats forming the RHCC interface between subunits E and G. (a,b) The sequence of subunit E (a) and subunit G (b), formatted to highlight the hendecad and quindecad hydrophobic repeats that mediate the interaction between the two subunits. Residues at the intersubunit interface are highlighted in yellow; residues forming intersubunit hydrogen bonds or salt bridges are in red. Purple highlighting of Arg106 marks the position of the kink in chain G. (c) Surface representation of the EG dimer with subunit E in blue, subunit G in green and residues at the intersubunit interface in yellow. Below, regions of the RHCC with a hendecad and quindecad repeat in subunit G.
The heterodimer contains two distinct domains, a 140-Å long right-handed coiled coil (RHCC) and a globular head group (Fig. 2). The coiled coil is formed by the long N-terminal helices from both subunits that are staggered with residue Ser2 from chain E being roughly aligned with residue Ser25 from chain G, which is well defined in the electron density from residue Gly21 (Supplementary Fig. 2). Although this suggests that subunit G may extend beyond the RHCC, N-terminal sequencing and MS of the intact T. thermophilus A-ATPase indicates that subunit G is N-terminally truncated by 17 residues11,13, which suggests that the structure is an almost complete representation of the peripheral stalk. The C-terminal half of subunit E (Leu94–Gly188) forms the majority of the head group and consists of a globular domain, which contains a four-stranded b-sheet and two a-helices followed by a C-terminal a-helical tail. Subunit G is entirely a-helical with a long N-terminal helix and a short C-terminal helix that are separated by a sharp kink at Arg106, which changes the helical axis by approximately 50°. This short helix is packed tightly against the long helix and C-terminal tail of subunit E, with extensive hydrophobic contacts, and constitutes the remainder of the head group. In subunit E, the beginning of the globular domain is marked by a loop containing three proline residues (Pro95, Pro98 and Pro101; Fig. 2). This loop and the kink in subunit G seem to be two flexible joints that tether the head group to the coiled coil, perhaps indicative of a hinge that could facilitate movement of the head group.
Indeed, the crystal structure of the C-terminal half of subunit E from P. horikoshii, containing residues Glu81–Glu198, (24% sequence identity to T. thermophilus; Protein Data Bank accession code 2dma)27 has a similar fold with the central b-sheet and helix 2 aligning well (Fig. 4). However, the relative angle between the N-terminal helical axis and the globular domain is much sharper in P. horikoshii (~60°) than in T. thermophilus (~90°). Although this angle is clearly dependent on the hydrophobic interactions with the small helix in subunit G, the conformational difference corroborates the potential of the three-proline loop to act as a hinge. A further difference between the two structures is in their C-terminal a-helical tails. Both helices contain a kink, which is much sharper in the T. thermophilus structure, presumably induced by the contact with the short helix of subunit G (Fig. 4).
Figure 4.

Superposition of the P. horikoshii subunit E C-terminal domain and T. thermophilus subunit E. The C-terminal domain of subunit E from P. horikoshii (PDB accession code 2dma)27 is presented in yellow to red from the N to C terminus; T. thermophilus subunit E is presented in dark blue to light blue from the N to C terminus; and T. thermophilus subunit G is in green. Top, location of the three-proline loop of T. thermophilus subunit E, which forms a 90° bend in between helices 1 and 2, whereas the angle in between P. horikoshii helices 1 and 2 is closer to 60°. Left, Arg106, which forms a 50° kink in subunit G.
Structure of the coiled-coil domain
The coiled coil of EG exhibits some structural properties that have not previously been observed. Coiled coils are typically left-handed (LHCC) and their fold is dictated by the heptad repeat (a-b-c-d-e-f-g)n, with hydrophobic residues at positions a and d that lie at the interface between the two helices28. Less commonly found are right-handed coiled coils (RHCCs) that display either an 11-residue (hendecad) repeat or a 15-residue (quindecad) repeat with hydrophobic residues at (a-d-e-h) or (a-d-e-h-l), respectively. So far there are only two other crystal structures of naturally occurring RHCCs, one made from a hendecad29 and the other made from a quindecad repeat30. Both of those structures form homotetramers, and the EG heterodimer is the first heteromeric RHCC structure. A consequence of the heteromer is that unlike previously observed RHCC structures, the coiling of each helix is not shared equally between the two chains. Although the N-terminal helix of subunit E (Ser2–Ala93; 138 Å) is almost straight, subunit G is wrapped around subunit E in a right-handed fashion (Fig. 2). Furthermore, the RHCC in the E–G heterodimer contains both a hendecad repeat and a quindecad repeat and this is reflected in its structure (Fig. 3). On the basis of the periodicity of hydrophobic residues, the pitch of an ideal RHCC with a hendecad repeat is very large, at around +55 nm, leading to structures that seem essentially straight29–31. In contrast, the pitch of an ideal RHCC with a 15-residue repeat is similar in magnitude to that of an LHCC, but in the reverse direction, at around +18.5 nm30. The RHCC in the EG structure is formed from a hendecad repeat at the N terminus of both subunits, resulting in a pitch of approximately +45 nm, as calculated by the coiled-coil analysis software TWISTER32. However, the hendecad repeat in subunit G changes to a quindecad repeat at residue Ala66, and the pitch of the RHCC consequently tightens to around +15 nm toward the C terminus (Fig. 3). This tighter coiling toward the C terminus is further accentuated by the kink in subunit G, which brings the C terminus to the opposite side of subunit E relative to the N terminus (Fig. 2). The long helix in subunit E is considerably straighter than subunit G and contains two breaks in its hendecad repeat that are known as stutters and that allow subunit G to coil around it (Fig. 3).
Interestingly, all H+-ATPases/synthases use an LHCC in the central stalk, which is thought to store transient elastic energy during the rotary catalytic cycle10,33–35. The E–G structure is the first experimental evidence to our knowledge of right-handed coiling in the peripheral stalk establishing the intriguing phenomenon of opposite coiling in the central and peripheral stalks. This phenomenon has been predicted previously from bacterial F-type ATP synthases on the basis of conserved hendecad repeats in the homodimeric peripheral stalk subunit b36. The presence of opposite coiling in different H+-ATPase subtypes suggests that this opposite coiling may be a conserved feature among most subtypes of H+-ATPases. To further investigate this, we searched for hydrophobic periodicities in the first 100 residues of the E and G subunits of a selection of A and V-type ATPases, using Fourier analysis as previously described37. As expected, both subunits E and G from A-ATPases exhibit a single clear peak at a periodicity of 3.66 and 3.75, which is indicative of a hendecad RHCC with three hydrophobic repeats (11/3) and a quindecad RHCC with four hydrophobic repeats (15/4), respectively (Supplementary Fig. 3). Remarkably, eukaryotic V-type ATPase subunits E and G exhibit the same periodicity as their archaeal counterparts (Supplementary Fig. 3) and, notably, there is no prominent peak at a periodicity of 3.5 as would be expected for a LHCC with a heptad repeat containing two hydrophobic side chains (7/2). Thus, the peripheral stalks of eukaryotic V-ATPases indeed seem to be RHCCs, which suggests that the opposite coiling of the peripheral and central stalks is also important in proton pumping.
Docking of E–G complex into the EM density of intact A-ATPase
EM and MS studies of intact A-ATPases have demonstrated that there are two peripheral stalks per A-ATPase complex, each composed of one E–G heterodimer11–14,20–23,38. Known high-resolution structures of A-ATPase subunits or their homologs have been fitted into the 23-Å EM reconstruction of intact T. thermophilus A-ATPase9– 11,39 to generate partial composite models of the complete complex. Recently, the X-ray structure of the A1 domain of A-ATPase from the same organism was elucidated10. Fitting of this complex into the EM density in place of individually fitted homologs created a more applicable composite model. Further, by matching prominent asymmetric features between the X-ray structure and the EM density, we were able to confidently determine the correct handedness of the EM density that is consistent with the experimentally determined handedness of eukaryotic V-ATPase20. The remaining density identified the shape and position of the structurally unknown stator components, consisting of subunits E and G, as well as the amphipathic subunit I, which contains a transmembrane domain that forms the ion channel and a soluble domain (Isol) that crosslinks the two peripheral stalks (Fig. 1a).
Despite having no equivalent in A-ATPases, the eukaryotic V-ATPase subunit Vma5p is functionally equivalent to Isol, as it cross-links peripheral stalks two and three of the three peripheral stalks in V-ATPases20. Peripheral stalks one and two in V-ATPases are crosslinked by the eukaryotic homolog of subunit I. Both small-angle X-ray scattering and EM11,20,40 indicate that Vma5p and Isol have similar dumbbell-shaped structures. Although they show no detectable sequence homology, docking of the crystal structure of Vma5p41 into the EM density illustrates a remarkable consistency in shape and dimensions at the expected position of subunit Isol (Fig. 5).
Figure 5.

Docking of the E–G peripheral stalk complex into the 23-Å EM density of the intact T. thermophilus A-ATPase/synthase. (a–c) The EM reconstruction with only the EG peripheral stalk docked in is shown from the side (a), top (b) and a vertical cross-section just below the A3B3 head group (c). (d–f) EM reconstruction plus a model (in gray) of the X-ray structures of the T. thermophilus A1 complex10 (A3B3DF (PDB accession code 3a5c), T. thermophilus subunit C46 (PDB accession code 1r5z) and a model of 12 E. coli subunit c protomers based on NMR data47 (PDB accession code 1c17). Residue Lys25 from subunit B is indicated by red spheres. The coordinates of the composite model of these structures are available in the Supplementary Data. (d) The eukaryotic V-ATPase subunit C41 (Vma5p; PDB accession code 1u7l) is docked (in purple) to demonstrate its complementarity in size and shape to the soluble part of subunit I. (e,f) Close-up view of the distal (e) and proximal (f) E–G stator head group fitted into the EM density. Gray arrowheads indicate deviations of the EG structure from the EM density.
The residual EM density highlights the position of two peripheral stalks, which are attached asymmetrically to the complex such that one binds to the B subunit directly above the ion channel (proximal) and the other binds to the B subunit on the adjacent A–B dimer (distal)11–14,38,39,42 (Fig. 1a). The coiled coil of E–G is consistent with the rod-shaped density that extends from Isol to the top of subunit B, where there is a mass of density corresponding to the globular head group. Thus, in the intact complex, the peripheral stalks are oriented with their C-terminal head groups above the nucleotide-binding domain pointing toward each other and the N terminus of the RHCC in contact with Isol (Fig. 5). In this orientation, the interface between the peripheral stalk and the nucleotide-binding domain (Fig. 1a) consists mainly of subunit E, which forms a concave surface that complements the size and shape of subunit B (Fig. 5d). This is consistent with previous cross-linking studies in yeast, which indicated that the residue equivalent to yeast Lys45 (T. thermophilus Lys25) is in contact with subunit E43. Residue Lys25 is situated at the top of subunit B in close proximity to subunit E (Fig. 5d,e).
Surprisingly, docking of the peripheral stalks highlighted differences in the EM density between the proximal and distal stalks. There is significant surface correlation between the EM density of the distal peripheral stalk and the entire crystal structure of the E–G heterodimer (Fig. 5). However, although there is also reasonable complementarity between the long helices of the E–G heterodimer and the EM density in the proximal stalk, the density deviates from the structure toward the globular domain (Fig. 5b,f).
DISCUSSION
This study has provided the most complete composite model of any A- or V-ATPase so far and the first glimpse of a heterodimeric RHCC structure, to our knowledge. In addition, it constitutes direct experimental evidence of right-handed coiling of the peripheral stalk in H+-ATPases and extends the prediction of RHCCs in the peripheral stalks of bacterial F-type ATP synthases to A- and V-ATPases, which suggests that opposite coiling in the central and peripheral stalks may be a conserved feature in most subtypes of H+-ATPases. This, as proposed earlier, might provide a second avenue for the storage of transient elastic energy during the rotary catalytic cycle36. Docking of the E–G heterodimer into the EM density revealed the structural orientation of the peripheral stalk, which indicates that subunit E but not subunit G functions to tether the peripheral stalk to the A3B3 nucleotide-binding domain (Fig. 5). This has important implications for right-handed coiling of the peripheral stalk, at least during ATP synthesis, during which the central stalk rotates counter-clockwise when viewed from above. A consequence of the right-handed coiling of the peripheral stalks is that during ATP synthesis, subunit E is driven into the short helix of subunit G (Fig. 1b). In contrast, left-handed coiling in the peripheral stalk would position the C terminus of subunit G on the opposite side of subunit E such that during ATP synthesis, subunit E is pulled away from subunit G. Thus, right-handed coiling puts subunit G in a mechanically superior position to act as an obstruction restricting bowing of the peripheral stalk during ATP synthesis. Interestingly, Fourier analysis of a selection of homologous sequences suggests that the peripheral stalks of eukaryotic V-ATPases also contain RHCCs (Supplementary Fig. 3), although during proton pumping the mechanical implications of right-handed coiling of the peripheral stalk are less clear. Unlike ATP synthesis, during which the E–G complex counteracts rotation of the A3B3 nucleotide-binding domain, during proton pumping, the E–G complex counteracts rotation of subunit I relative to the proteolipid ring. Hence, subunit I rather than A3B3 imposes rotational strain on the E–G complex, and the implications of the RHCC during proton pumping will depend on its interaction with Isol. However, without the structure of the EGI or equivalent complexes, the necessity of RHCCs for proton pumping remains uncertain and their presence in V-ATPases may be purely an evolutionary legacy.
In contrast to the peripheral stalk of A-ATPases, the peripheral stalk of eukaryotic F-type ATP synthase does not form a RHCC. The N-terminal helix of subunit G shows limited sequence homology to the peripheral stalk subunit b from F-type ATP synthase, and similarly its soluble part forms a 160-Å long helix in the crystal structure of the mitochondrial peripheral stalk25. Rather than forming a RHCC, the single mitochondrial subunit b is flanked by shorter fragments of helices from subunits d and F6 that are connected by extended loops. The C-terminal kink in subunit G is reminiscent of subunit F6 in the bovine peripheral stalk structure, which wraps around subunit b in a right-handed fashion and may have a similar role in mechanically stabilizing subunit b. F-type ATP synthases do not have an amphipathic channel-forming subunit. Instead, the ion channel–forming subunit a is entirely integrated into the membrane. Hence, the interaction between stator and channel is very different. Although neither subunit E nor subunit G extends into the membrane, F-type ATP synthase subunit b is amphipathic and forms the membrane anchor, a role taken by the ion channel–forming amphipathic subunit I or a in A-ATPases or V-ATPases, respectively.
Docking of two E–G complexes into the EM density revealed significant asymmetry in the two peripheral stalks, which suggests conformational differences in the intact complex. Several lines of evidence indicate that conformational changes in the peripheral stalks are required for the assembly of A- and V-ATPases and possibly also for their function. First, all H+-ATPases contain a set of three chemically identical B subunits or their homologs. In eukaryotic V-ATPases, these are matched with three peripheral stalks that each bind to one B subunit at the top of the complex and are linked at their bases, one pair by subunit a (equivalent to subunit I) and the other by Vma5p. As A-ATPases do not have a subunit equivalent to Vma5p, they can contain only two peripheral stalks. Thus, the E–G heterodimer does not per se bind to the B subunit, as only two of the three B subunits in A-ATPases are occupied with peripheral stalks12,13. Second, although the two peripheral stalks in the complex are cross-linked by Isol, resulting in a stoichiometry of 2:2:1, in isolation the E–G–Isol complex assembles with a stoichiometry of 1:1:1 (refs. 20,40). In the intact complex, an E–G–Isol (1:1:1) complex forms a C-shape with the globular domain of EG pointing in the same direction as Isol (Fig. 5d). Small-angle X-ray scattering of the E–G–Isol complex40 indicates that in isolation, the subcomplex forms a structure in which the globular head group of EG points in a different direction to Isol. Similarly, the isolated yeast E–G–Vma5p complex has a stoichiometry of 1:1:1, and small-angle X-ray scattering experiments have shown that the globular head group of EG does not point in the same direction as Vma5p20,24. Thus, significant structural rearrangement of both the E–G–Isol complex and E–G–Vma5p complex is required for these structures to be consistent with the shape of the intact A- and V-ATPases. The native C-shape and the binding site for the second E–G complex may form via movement in the head domain, perhaps via the three-proline hinge loop and kink in chain G described above (Fig. 2a) or through a rotation in the coiled coil domain. Finally, the inability to spontaneously form the in vivo E–G–Isol conformation suggests that the assembly processes may require a source of energy input. In support of this idea, it has been suggested that Vma5p can bind to ATP at low affinity and become phosphorylated44, which could lead to the conformational changes needed to trigger its binding to two peripheral stalks and into the V-ATPase complex.
When expressed alone, subunit G from several organisms can form homodimers14,40,45, and purified subunit E has the tendency to oligomerize14,40,45. Furthermore, on the basis of the crystal packing of the P. horikoshii subunit E C-terminal domain, it has been suggested that this subunit may form a homodimer in intact A- or V-ATPase27. This homodimer, however, cannot be fitted into the EM density of the intact A-ATPase without significantly altering the conformation of the protein, as the N-terminal helices (which are truncated in the P. horikoshii structure) would point in opposite directions, thus spanning a total length of more than 250 Å, consequently exceeding the longest dimension of the A-ATPase complex. Furthermore, when co-expressed, subunits E and G from T. thermophilus favor the formation of heterodimers over homodimers. Both subunits E and G from T. thermophilus are fragile when expressed on their own but are stable when coexpressed. The size and congruency of the intersubunit interface in the EG structure explains the preferred formation of EG heterodimers when both subunits are present.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/nsmb/.
Supplementary Material
Table 1.
Data collection and refinement statistics
| SeMet crystala | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 75.8, 79.8, 75.8 |
| a, b, g (°) | 90.0, 97.6, 90.0 |
| Wavelength (Å) | 0.97957 |
| Resolution (Å) | 30-3.1 (3.26-3.10)b |
| Rmerge | 9.0 (52.2) |
| I / sI | 11.9 (1.8) |
| Completeness (%) [Anomalous] | 95.3 (95.3) [89.7] |
| Redundancy | 4.1 (2.7) |
| Refinement | |
| Resolution (Å) | 3.10 |
| No. reflections | 14,648 |
| Rwork / Rfree | 23.2/28.7 |
| No. atoms | |
| Protein | 4,056c |
| B-factors | |
| Protein | 48.5 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.91 |
The working data set was obtained by merging of data from two isomorphous selenomethionine (SeMet)-containing crystals.
Values in parentheses are for the shell of highest resolution.
Two E–G complexes per asymmetric unit.
ACKNOWLEDGMENTS
We thank the staff at beamline ID14-4 at the European Synchrotron Research Facility, Grenoble, France, at beamline 14-ID (BioCARS) and 23-ID (General Medicine and Cancer Institutes Collaborative Access Team (GM/CA-CAT)), at the Advanced Photon Source, Chicago, U.S.A and at beamline 3-BM1 at the Australian Synchrotron, Victoria, for support; K. Miki and colleagues are acknowledged for providing coordinates of the T. thermophilus A1 structure before release from the protein data bank. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science (contract DEAC02-06CH11357), and use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources (grant RR007707). GM/CA-CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). This work was supported by the Australian Synchrotron Research Program of the Australian Nuclear Science Technology Organization and Australian National Health & Medical Research Council (grant DP573712).
Footnotes
Accession codes. Protein Data Bank: atomic coordinates and structure factors for the peripheral stalk subunits have been deposited in the under accession code 3k5b.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
AUTHOR CONTRIBUTIONS
L.K.L. collected X-ray diffraction data, solved, built and refined the crystal structure, performed the Fourier analysis of coiled coils and wrote the manuscript; A.G.S. expressed, purified and crystallized the E-G complex, solved, built and refined the crystal structure and wrote the manuscript; M.D. cloned, expressed, purified and crystallized the E-G complex; R.B. docked subunits into EM density; D.S. conceived the project, collected X-ray diffraction data, built and refined the structure and wrote the manuscript.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
Published online at http://www.nature.com/nsmb/.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 2.Toei M, et al. Dodecamer rotor ring defines H+/ATP ratio for ATP synthesis of prokaryotic V-ATPase from Thermus thermophilus. Proc. Natl. Acad. Sci. USA. 2007;104:20256–20261. doi: 10.1073/pnas.0706914105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida M, Muneyuki E, Hisabori T. ATP synthase-a marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2001;2:669–677. doi: 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- 4.Cross RL, Muller V. The evolution of A-, F-, and V-type ATP synthases and ATPases: reversals in function and changes in the H+ATP coupling ratio. FEBS Lett. 2004;576:1–4. doi: 10.1016/j.febslet.2004.08.065. [DOI] [PubMed] [Google Scholar]
- 5.Nakano M, et al. ATP hydrolysis and synthesis of a rotary motor V-ATPase from Thermus thermophilus. J. Biol. Chem. 2008;283:20789–20796. doi: 10.1074/jbc.M801276200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feniouk BA, Suzuki T, Yoshida M. Regulatory interplay between proton motive force, ADP, phosphate, and subunit epsilon in bacterial ATP synthase. J. Biol. Chem. 2007;282:764–772. doi: 10.1074/jbc.M606321200. [DOI] [PubMed] [Google Scholar]
- 7.Gruber G, Wieczorek H, Harvey WR, Muller V. Structure-function relationships of A-, F- and V-ATPases. J. Exp. Biol. 2001;204:2597–2605. doi: 10.1242/jeb.204.15.2597. [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama K, et al. Subunit arrangement in V-ATPase from Thermus thermophilus. J. Biol. Chem. 2003;278:42686–42691. doi: 10.1074/jbc.M305853200. [DOI] [PubMed] [Google Scholar]
- 9.Makyio H, et al. Structure of a central stalk subunit F of prokaryotic V-type ATPase/synthase from Thermus thermophilus. EMBO J. 2005;24:3974–3983. doi: 10.1038/sj.emboj.7600859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Numoto N, Hasegawa Y, Takeda K, Miki K. Inter-subunit interaction and quaternary rearrangement defined by the central stalk of prokaryotic V1-ATPase. EMBO Rep. 2009;10:1228–1234. doi: 10.1038/embor.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernal RA, Stock D. Three-dimensional structure of the intact Thermus thermophilus H+-ATPase/synthase by electron microscopy. Structure. 2004;12:1789–1798. doi: 10.1016/j.str.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Kish-Trier E, Briere LK, Dunn SD, Wilkens S. The stator complex of the A1A0-ATP synthase-structural characterization of the E and H subunits. J. Mol. Biol. 2008;375:673–685. doi: 10.1016/j.jmb.2007.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esteban O, et al. Stoichiometry and localization of the stator subunits E and G in Thermus thermophilus H+-ATPase/synthase. J. Biol. Chem. 2008;283:2595–2603. doi: 10.1074/jbc.M704941200. [DOI] [PubMed] [Google Scholar]
- 14.Kish-Trier E, Wilkens S. Domain architecture of the stator complex of the A1A0-ATP synthase from Thermoplasma acidophilum. J. Biol. Chem. 2009;284:12031–12040. doi: 10.1074/jbc.M808962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karrasch S, Walker JE. Novel features in the structure of bovine ATP synthase. J. Mol. Biol. 1999;290:379–384. doi: 10.1006/jmbi.1999.2897. [DOI] [PubMed] [Google Scholar]
- 16.Bottcher B, Bertsche I, Reuter R, Graber P. Direct visualisation of conformational changes in EF0F1 by electron microscopy. J. Mol. Biol. 2000;296:449–457. doi: 10.1006/jmbi.1999.3435. [DOI] [PubMed] [Google Scholar]
- 17.Lau WC, Baker LA, Rubinstein JL. Cryo-EM structure of the yeast ATP synthase. J. Mol. Biol. 2008;382:1256–1264. doi: 10.1016/j.jmb.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 18.Rubinstein JL, Walker JE, Henderson R. Structure of the mitochondrial ATP synthase by electron cryomicroscopy. EMBO J. 2003;22:6182–6192. doi: 10.1093/emboj/cdg608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkens S, Capaldi RA. ATP synthase' second stalk comes into focus. Nature. 1998;393:29. doi: 10.1038/29908. [DOI] [PubMed] [Google Scholar]
- 20.Diepholz M, et al. A different conformation for EGC stator subcomplex in solution and in the assembled yeast V-ATPase: possible implications for regulatory disassembly. Structure. 2008;16:1789–1798. doi: 10.1016/j.str.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa N, Mazon H, Heck AJ, Wilkens S. Stoichiometry of the peripheral stalk subunits E and G of yeast V1-ATPase determined by mass spectrometry. J. Biol. Chem. 2008;283:3329–3337. doi: 10.1074/jbc.M707924200. [DOI] [PubMed] [Google Scholar]
- 22.Muench SP, et al. Cryo-electron microscopy of the vacuolar ATPase motor reveals its mechanical and regulatory complexity. J. Mol. Biol. 2009;386:989–999. doi: 10.1016/j.jmb.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, et al. Structure of the yeast vacuolar ATPase. J. Biol. Chem. 2008;283:35983–35995. doi: 10.1074/jbc.M805345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vonck J, Pisa KY, Morgner N, Brutschy B, Muller V. Three-dimensional structure of A1A0 ATP synthase from the hyperthermophilic archaeon Pyrococcus furiosus by electron microscopy. J. Biol. Chem. 2009;284:10110–10119. doi: 10.1074/jbc.M808498200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickson VK, Silvester JA, Fearnley IM, Leslie AG, Walker JE. On the structure of the stator of the mitochondrial ATP synthase. EMBO J. 2006;25:2911–2918. doi: 10.1038/sj.emboj.7601177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carbajo RJ, et al. How the N-terminal domain of the OSCP subunit of bovine F1Fo-ATP synthase interacts with the N-terminal region of an a subunit. J. Mol. Biol. 2007;368:310–318. doi: 10.1016/j.jmb.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 27.Lokanath NK, Matsuura Y, Kuroishi C, Takahashi N, Kunishima N. Dimeric core structure of modular stator subunit E of archaeal H+-ATPase. J. Mol. Biol. 2007;366:933–944. doi: 10.1016/j.jmb.2006.11.088. [DOI] [PubMed] [Google Scholar]
- 28.Parry DA, Fraser RD, Squire JM. Fifty years of coiled-coils and a-helical bundles: a close relationship between sequence and structure. J. Struct. Biol. 2008;163:258–269. doi: 10.1016/j.jsb.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 29.Stetefeld J, et al. Crystal structure of a naturally occurring parallel right-handed coiled coil tetramer. Nat. Struct. Biol. 2000;7:772–776. doi: 10.1038/79006. [DOI] [PubMed] [Google Scholar]
- 30.Kuhnel K, et al. The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc. Natl. Acad. Sci. USA. 2004;101:17027–17032. doi: 10.1073/pnas.0403069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 32.Strelkov SV, Burkhard P. Analysis of a-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J. Struct. Biol. 2002;137:54–64. doi: 10.1006/jsbi.2002.4454. [DOI] [PubMed] [Google Scholar]
- 33.Cherepanov DA, Mulkidjanian AY, Junge W. Transient accumulation of elastic energy in proton translocating ATP synthase. FEBS Lett. 1999;449:1–6. doi: 10.1016/s0014-5793(99)00386-5. [DOI] [PubMed] [Google Scholar]
- 34.Oster G, Wang H. Reverse engineering a protein: the mechanochemistry of ATP synthase. Biochim. Biophys. Acta. 2000;1458:482–510. doi: 10.1016/s0005-2728(00)00096-7. [DOI] [PubMed] [Google Scholar]
- 35.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 36.Del Rizzo PA, Bi Y, Dunn SD. ATP synthase b subunit dimerization domain: a right-handed coiled coil with offset helices. J. Mol. Biol. 2006;364:735–746. doi: 10.1016/j.jmb.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 37.Gruber M, Soding J, Lupas AN. REPPER-repeats and their periodicities in fibrous proteins. Nucleic Acids Res. 2005;33:W239–W243. doi: 10.1093/nar/gki405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boekema EJ, et al. Connecting stalks in V-type ATPase. Nature. 1999;401:37–38. doi: 10.1038/43369. [DOI] [PubMed] [Google Scholar]
- 39.Schafer IB, et al. Crystal Structure of the Archaeal A1AO ATP synthase subunit B from Methanosarcina mazei Gö1: implications of nucleotide-binding differences in the major A1AO subunits A and B. J. Mol. Biol. 2006;358:725–740. doi: 10.1016/j.jmb.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto M, et al. Interaction and stoichiometry of the peripheral stalk subunits NtpE and NtpF and the N-terminal hydrophilic domain of NtpI of Enterococcus hirae V-ATPase. J. Biol. Chem. 2008;283:19422–19431. doi: 10.1074/jbc.M801772200. [DOI] [PubMed] [Google Scholar]
- 41.Drory O, Frolow F, Nelson N. Crystal structure of yeast V-ATPase subunit C reveals its stator function. EMBO Rep. 2004;5:1148–1152. doi: 10.1038/sj.embor.7400294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coskun U, et al. Structure and subunit arrangement of the A-type ATP synthase complex from the archaeon Methanococcus jannaschii visualized by electron microscopy. J. Biol. Chem. 2004;279:38644–38648. doi: 10.1074/jbc.M406196200. [DOI] [PubMed] [Google Scholar]
- 43.Arata Y, Baleja JD, Forgac M. Cysteine-directed cross-linking to subunit B suggests that subunit E forms part of the peripheral stalk of the vacuolar H+-ATPase. J. Biol. Chem. 2002;277:3357–3363. doi: 10.1074/jbc.M109967200. [DOI] [PubMed] [Google Scholar]
- 44.Armbruster A, et al. Evidence for major structural changes in subunit C of the vacuolar ATPase due to nucleotide binding. FEBS Lett. 2005;579:1961–1967. doi: 10.1016/j.febslet.2005.02.042. [DOI] [PubMed] [Google Scholar]
- 45.Biukovic G, Rossle M, Gayen S, Mu Y, Gruber G. Small-angle X-ray scattering reveals the solution structure of the peripheral stalk subunit H of the A1AO ATP synthase from Methanocaldococcus jannaschii and its binding to the catalytic A subunit. Biochemistry. 2007;46:2070–2078. doi: 10.1021/bi062123n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.