

Abstract
Background
Macrophages of the reticuloendothelial system play a key role in recycling iron from hemoglobin of senescent or damaged erythrocytes. Heme oxygenase 1 degrades the heme moiety and releases inorganic iron that is stored in ferritin or exported to the plasma via the iron export protein ferroportin. In the plasma, iron binds to transferrin and is made available for de novo red cell synthesis. The aim of this study was to gain insight into the regulatory mechanisms that control the transcriptional response of iron export protein ferroportin to hemoglobin in macrophages.
Design and Methods
Iron export protein ferroportin mRNA expression was analyzed in RAW264.7 mouse macrophages in response to hemoglobin, heme, ferric ammonium citrate or protoporphyrin treatment or to siRNA mediated knockdown or overexpression of Btb And Cnc Homology 1 or nuclear accumulation of Nuclear Factor Erythroid 2-like. Iron export protein ferroportin promoter activity was analyzed using reporter constructs that contain specific truncations of the iron export protein ferroportin promoter or mutations in a newly identified MARE/ARE element.
Results
We show that iron export protein ferroportin is transcriptionally co-regulated with heme oxygenase 1 by heme, a degradation product of hemoglobin. The protoporphyrin ring of heme is sufficient to increase iron export protein ferroportin transcriptional activity while the iron released from the heme moiety controls iron export protein ferroportin translation involving the IRE in the 5′untranslated region. Transcription of iron export protein ferroportin is inhibited by Btb and Cnc Homology 1 and activated by Nuclear Factor Erythroid 2-like involving a MARE/ARE element located at position −7007/−7016 of the iron export protein ferroportin promoter.
Conclusions
This finding suggests that heme controls a macrophage iron recycling regulon involving Btb and Cnc Homology 1 and Nuclear Factor Erythroid 2-like to assure the coordinated degradation of heme by heme oxygenase 1, iron storage and detoxification by ferritin, and iron export by iron export protein ferroportin.
Keywords: reticuloendothelial system, FPN1, MARE/ARE, iron storage
Introduction
Approximately two-thirds of the total body iron content is bound to hemoglobin contained in mature erythrocytes and their precursors. To sustain hemoglobinization of new erythrocytes, approximately 20–25mg of iron is needed every day. Most of this iron is provided by specialized macrophages of the reticuloendothelial system (RES) that recycle iron from senescent or damaged erythrocytes. RES macrophages phagocytose and lyse effete erythrocytes and then catabolize the heme moiety with the help of heme oxygenase 1 to liberate inorganic iron, in addition to carbon monoxide (CO) and bilirubin.1
In conditions of severe intravascular hemolysis, such as in the thalassemias, but also in healthy individuals, intact hemoglobin is released into the plasma.2 Hemoglobin or free heme is then captured by haptoglobin or hemopexin and scavenged by macrophages via the surface receptor CD163 or the LDL receptor-related protein/CD91, respectively.3, 4 In addition, macrophages preserved the ability to acquire iron from transferrin via transferrin receptor 1 (TfR1).5 While multiple pathways control macrophage iron uptake, iron export appears to be the rate-limiting step that positively correlates with the need to sustain erythropoiesis. Targeted mutagenesis of the murine ferroportin1 (FPN1; also known as metal transport protein 1 [MTP1], iron-regulated transporter 1 [IREG1], or Slc11a3) locus (Slc40a1) shows that Fpn1 is the major iron export channel that mediates iron release from macrophages.6 Thus regulation of FPN1 expression is of major importance for the control of iron efflux. At the posttranslational level FPN1 expression is regulated via the small hepatic peptide hormone hepcidin that binds FPN1 to cause its internalization and proteolysis.7,8 In accordance with this mechanism, chronically elevated hepcidin levels [e.g. in the anemia of chronic diseases (ACD) or iron refractory iron deficiency anemia (IRIDA)] cause iron retention in macrophages and decreased intestinal iron absorption. By contrast, inappropriately low hepcidin levels (e.g. in hereditary hemochromatosis (HH) or disorders with inefficient erythropoiesis like the thalassemias) result in iron overload.9,10 In addition, FPN1 translation is controlled by the iron regulatory proteins (IRPs) 1 and 2 that bind to the iron-responsive element (IRE) located within the FPN1 5′untranslated region.11 Binding of IRPs to the FPN1 IRE in iron deficiency represses FPN1 translation, while FPN1 translation is activated in iron replete cells. Consistent with this mechanism, bone marrow derived macrophages (BMDM) lacking both IRP1 and IRP2 up-regulate FPN1 expression.12
While translational and posttranslational control mechanisms of FPN1 are increasingly well understood, comparatively little is known about how FPN1 expression is controlled at the transcriptional level. Transcriptional regulation of FPN1 was demonstrated in J774 cells13 and in bone marrow derived macrophages14,15 following erythrocytosis. In addition, FPN1 mRNA expression is also increased in the duodenum and spleen of haptoglobin-null mice which show increased plasma hemoglobin levels and unchanged hepcidin expression.16 These findings suggest that increased hemoglobin levels contribute to the regulation of FPN1 mRNA expression.
Heme regulates gene transcription of HO1,17 the globin genes,18,19 the heavy (H) and light (L) ferritin chains,20,21 thioredoxin reductase 120 and the NAD(P)H: quinone oxidoreductase 1 (qr)22 through the transcriptional repressor Btb And Cnc Homology 1 (Bach1). Bach1 is a basic leucine zipper (bZip) transcriptional repressor and a sensor of cellular heme levels.23 It antagonizes the activity of Maf-related oncogenes or small Maf proteins (sMaf) that bind Maf recognition elements (MAREs) to activate transcription of, for example, HO1.24 Heme binding to the Bach1 C-terminal domain induces its dissociation from small Maf proteins (sMAF) and triggers its ubiquitination, degradation and Crm1-dependent export from the nucleus.25–27 In addition, heme stabilizes the Nuclear Factor Erythroid 2-like (NRF2) that binds to small (s)MAF proteins to enhance gene transcription utilizing specific Antioxidant Response Elements (ARE) which are specific forms of MAREs.28 If heme levels are low, Kelch-like ECH-associated protein 1 (Keap1) sequesters NRF2 in the cytoplasm by binding to its Neh2 domain.29 Inducers of oxidative stress, such as sulforaphane or heme react with specific cystein residues in Keap1 causing dissociation of the Keap1-Nrf2 complex and Nrf2 nuclear accumulation.30,31
In this study we show that heme controls the transcription of the iron exporter FPN1 involving Bach1 activity, Nrf2 nuclear accumulation and a highly conserved MARE/ARE enhancer element located at position −7007/−7016 of the FPN1 promoter. This finding suggests that iron recycling from heme involves a single transcription control mechanism that regulates heme catabolism, iron storage and detoxification, as well as iron export in a coordinated manner.
Design and Methods
Cell culture
The murine macrophage RAW 264.7 cell line (ATCC number TIB-71) was cultured in Dulbecco modified Eagle medium (DMEM; high glucose; Invitrogen, Carlsbad, CA, USA). The medium was supplemented with 10% heat-inactivated low-endotoxin fetal bovine serum (FBS; Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were maintained at 37°C under 5% C02 atmosphere.
Human hemoglobin (hHb) (H7379; Sigma-Aldrich), sulforaphane (SF) (S6317; Sigma-Aldrich), hemin chloride (H9039; Sigma-Aldrich), protoporphyrin IX (PPIX) (P8293; Sigma-Aldrich), ferric ammonium citrate (FAC) (F5879; Sigma-Aldrich) and DMSO (Sigma) were used to treat the cells. SF, hemin and PPIX were dissolved in DMSO. hHb and FAC were dissolved in water. To treat RAW264.7 cells (2×106 cells/6-well plate) with the indicated substances, cells were initially maintained in DMEM without FCS at 37°C in 5% CO2 for one hour; the medium was then removed and substituted by DMEM supplemented with the reagents described above and the concentrations indicated in the text. After 8h the cells were washed twice with PBS and total RNA was extracted. To block RNA polymerase-dependent RNA synthesis, cells were treated with 0.5 μg/mL actinomycin D (ActD) (A1410; Sigma–Aldrich) for 8h.
RNA isolation
Total RNA was isolated using the Qiagen RNAeasy kit according to the manufacturer’s instructions (Qiagen). Concentration and purity of the RNA was determined by OD260/280 reading. Quality of the RNA was assessed by gel electrophoresis and ethidium bromide staining.
Reverse transcription and quantitative real-time PCR analysis
Two micrograms of total RNA were reverse transcribed using 10 μM each of dCTP, dGTP, dATP, and dTTP, 100 ng random primers, 1 x first-strand buffer (Gibco BRL, Carlsbad, CA, USA), 0.01 M DTT, and 200 units of SuperScript II reverse transcriptase (Gibco BRL) in a 20 μL reaction for 90 min at 42°C. Real-time polymerase chain reaction (PCR) was performed using the ABI Prism 7500 Applied Biosystems (Applera Deutschland, Darmstadt, Germany). Amplification reactions were carried out in a 20 μL volume using SYBR Green I dye and the following amplification conditions: 50°C for 2 min and 95°C for 10 min (95°C, 15 sec; 60°C, 15 sec) for 45 cycles. Primers were designed to specifically amplify 72 bp of mouse FPN1 cDNA (forward 5′-TGTTGTTGTGGCAGGAGAAA -3′, reverse 5′-AGCTGGTCAATCCTTCTAATGG -3′); 108 bp of mouse GAPDH cDNA (forward 5′-TCCACTCATGGCAAATTCAA -3′, reverse 5′-TTTGATGTTAGTGGGGTCTCG -3′);
77 bp of mouse HO1 cDNA (forward 5′-GTCAAGCACAGGGTGACAGA -3′, reverse 5′-ATCACCTGCAGCTCCTCAAA -3′); 68 bp of mouse QR cDNA (forward 5′-AGCGTTCGGTATTACGATCC -3′, reverse 5′-AGTACAATCAGGGCTCTTCTCG -3′); 62 bp of mouse TRR cDNA (forward 5′-TTTTGTCACACCGACTCCTCT -3′, reverse 5′-CCACATTCACACACGTTCCT -3′);
The mRNA/cDNA abundance of each gene was calculated relative to the expression of a housekeeping gene, GAPDH (glyceraldehyde-3-phosphate-dehydrogenase).
Promoter analysis
A -8949 nucleotide fragment containing the 5′-flanking genomic region as well as the 5′ untranslated region (UTR) of the murine FPN1 gene was subcloned by homologous recombination of the chromosome 1 Bacterial Artificial Chromosome (BAC; GenBank accession no. AC026803.7) into the pBluescript SK vector. This plasmid was digested with XhoI/EcoRV and the resulting fragment inserted into the promoterless luciferase reporter vector pGL3-Basic (Promega, Madison, WI, USA) previously digested with SmaI/XhoI. The resulting plasmid was named 8kb-Luc. To obtain the truncated forms of the FPN1 promoter, 6.8kb-Luc, 4.8kb-Luc and 2.4kb-Luc, the 8kb-Luc was cut with KpnI, PmlI or HindIII, respectively, and ligated using T4 ligase. The 8kbΔIRE-Luc, 6.8kbΔIRE-Luc, 4.8kbΔIRE-Luc and 2.4kb ΔIRE plasmids were constructed by deletion of the BamHI/SmaI segment of the FPN1 5′-UTR as described by Abboud S et al.11 The ARE/MARE (position -7007/-7016) binding site was mutated by site-directed mutagenesis (Invitrogen) using the following primer pairs: 5′-TGTCAGGCTCCTCGAAGGCCGCGGCCGCTGAGCTAAAGTTGA -3′ and 5′-GGCCTTCGAGGAGCCTGACACACAGTTAGTCATTA -3′.
Details of the constructs are available upon request. All constructs were confirmed by DNA sequencing. Search for putative transcription factor binding sites was performed using MatInspector24 (Genomatrix Software, München, Germany) and the position weight matrix (PWM) constructed by Wang X et al.32 The conservation analysis of genomic Slc40a1 locus was performed by PhastCons Placental Mammal Conserved 30-way Multiz Alignment UCSC Genome Browser.33
Cell transfection and luciferase assay
RAW264.7 cells were plated at 70% confluency in 24-well plates and incubated overnight. A total of 2 μg of pGL3 reporter vectors described above was co-transfected with 100 ng of a control plasmid containing the Renilla gene under the control of the CMV promoter. Transfections were performed using jetPEI-Macrophage (PolyPlus Transfection) according to the manufacturer’s instructions. Four hours after transfection the cells were harvested or treated with the indicated stimuli. At the indicated time points cells were lysed in passive lysis buffer (Promega), and cellular extracts were analyzed for luciferase activity using the Dual-Luciferase-Reporter assay system (Promega) and a Centro LB 960 luminometer (Berthold Technologies, Bad Wildbad, Germany).
The mouse Bach1 full-length containing plasmid was obtained from Open Biosystems (MMM1013-9201026); RAW264.7 cells were transfected with 2 μg of the plasmid DNA 24h after seeding, using jetPEI-Macrophage (PolyPlus Transfection) according to the manufacturer’s instructions. Control cells were transfected with pCMV-Luciferase vector. RNA was extracted and analyzed as described above.
siRNA-mediated knockdown of murine Bach1
RAW264.7 cells were plated at 70% confluency in DMEM supplemented with 10% FBS. After 24h, cells were transfected using INTERFERin (PolyPlus Transfection) and 100 nM pool of an siRNA-pool directed against mouse Bach1 (Dharmacon, Lafayette, CO, USA). As a control, siRNA directed against luciferase (Dharmacon) was transfected. The efficiency of the knockdown was analyzed at the protein level 72h later.
Western blotting
Fifty micrograms of total protein extracts were separated on 8% SDS-PAGE and analyzed by Western blotting using antibodies against mouse Bach1 and actin (Santa Cruz Biothecnology Inc, Santa Cruz, California, USA).
Statistics
Results were expressed as mean plus or minus SD. Student’s t-test was used for estimation of statistical significance.
Results
Heme activates FPN1 transcription in an iron-independent manner
To understand the molecular mechanisms that control FPN1 transcriptional activation in response to phagocytosis of senescent erythrocytes or hemoglobin scavenging by macrophages we asked ourselves how hemoglobin regulates FPN1 mRNA expression. Hemoglobin consists of several biologically active components such as globin chains, the protoporphyrin ring and atomic iron. Since each mol of hemoglobin contains 4 mols of heme and 4 mols of iron, we treated RAW264.7 cells with either 0.5 μM hemoglobin, 2 μM hemin (a chemical compound analogous to physiological heme), 2 μM protoporphyrin IX (PPIX) or 2 μM ferric ammonium citrate (FAC) for 8h and analyzed FPN1 mRNA expression. To assess whether FPN1 mRNA expression is controlled at the transcriptional level, we additionally treated cells with the RNA polymerase II inhibitor actinomycin D. Consistent with our previous observations, treatment of RAW264.7 cells with hemoglobin or hemin up-regulates FPN1 and HO1 mRNA expression. This finding suggests that heme rather than the globin chains mediate the FPN1 response to hemoglobin. Furthermore, actinomycin D completely inhibits the activation of FPN1, indicating that FPN1 is regulated at the transcriptional level. Interestingly, treatment with identical concentrations of PPIX, a heme precursor that does not contain iron atoms, shows comparable effects to hemoglobin, suggesting that the transcriptional FPN1 response is independent of iron release from hemoglobin by the HO1 catalytic activity. Consistent with this interpretation FAC treatment has no effect on FPN1 mRNA expression (Figure 1A). Taken together these results suggest an active role of the protoporphyrin ring in the hemoglobin-dependent transcription control of FPN1. As HO1 and FPN1 show similar regulatory responses (Figure 1A), our data further suggest that these two genes are co-regulated in macrophages by hemoglobin.
Figure 1.
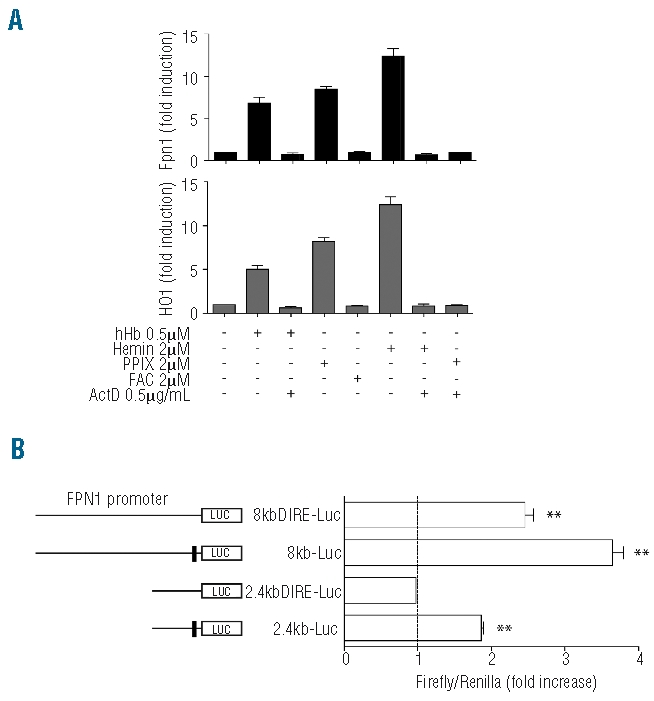
Heme activates FPN1 transcription in an iron independent manner. (A) FPN1 and HO1 mRNA levels were measured by qPCR in the RAW264.7 macrophage cell line following 8 h treatments with either human hemoglobin (hHb), hemin, protoporphyrin IX (PPIX), ferric ammonium citrate (FAC) and/or actinomycin D (ActD). Data were normalized to mRNA expression of GADPH and presented as fold change whereby the untreated control was set to 1. (B) Luciferase reporter assay. RAW264.7 macrophages were transfected with the 2.4kb, 2.4kb_IRE, 8kb and 8kb_IRE luciferase reporter vectors, treated with hHb or the solvent control after 6 h and luciferase activity was measured after 18 h. Transfections were performed in triplicates, and results are presented as fold change ± SEM of Renilla/Firefly. ** represents P<0.01.
To investigate how hemoglobin regulates FPN1 transcription and how the IRE within the FPN1 5′UTR may contribute to FPN1 expression, we generated two types of luciferase reporter vectors: 1) the 8kb-Luc construct, which contains the murine FPN1 5′untranslated region (5′UTR) and 8071 bps upstream of the transcription start site; and 2) the 2.4kb-Luc construct, which contains the 5′UTR of murine FPN1 and 2482 bps upstream of the transcription start site. Further constructs contain deletions of the iron responsive element (IRE) located in the 5′UTR region of the 8kb-Luc and 2.4kb-Luc constructs (8kbΔIRE-Luc and 2.4kbΔIRE-Luc, respectively; Figure 1B). RAW264.7 cells were transfected with the luciferase reporter vectors, treated with hemoglobin and luciferase activity was measured 18 h later. Luciferase activity increased approximately 2-fold (P<0.01) in response to Hb treatment in cells transfected with the 2.4Kb-Luc construct. Deletion of the IRE element (2.4kbΔIRE-Luc) completely abrogated this effect, suggesting that increased luciferase activity from this construct depends on the FPN1 IRE sequence. As FPN1 mRNA expression does not change in response to FAC treatment the alteration in luciferase activity likely is explained by iron regulatory protein-mediated translational derepression of FPN1 by the iron released from hemoglobin due to HO1 activity. Hemoglobin-dependent activation of luciferase activity was significantly higher in cells transfected with the 8Kb-Luc construct and 30% reduced upon the deletion of the IRE in 8KbΔIRE-Luc, while a highly significant induction by hemoglobin is maintained. These data suggest that FPN1 is transcriptionally activated by hemoglobin and that the promoter elements responsible for the hemoglobin-mediated transcriptional control are located between 2.4 and 8kb upstream of the transcription start site.
Bach1 and Nrf2 control FPN1 mRNA accumulation
Coregulation of HO1 and FPN1 mRNA expression in response to hemoglobin (Figure 1A) suggests the involvement of similar control mechanisms. Transcriptional regulation of HO1 by heme involves the transcription-repressor Bach117 and the transcription activator Nrf2.31,34 To test whether Bach1 mediates the hemoglobin response of FPN1 we reduced cellular Bach1 expression by siRNA-mediated gene knockdown and increased cellular Bach1 expression by protein overexpression in the RAW 264.7 cell line. A decrease in Bach1 protein levels by approximately 50% caused a 17-fold and 13-fold increase of endogenous FPN1 and HO1 mRNA expression, respectively, compared to a control siRNA (Figure 2A). Additional treatment of cells subjected to Bach1 silencing with heme was unable to further increase FPN1 mRNA levels (Online Supplementary Figure S1), suggesting that the majority of heme-dependent FPN1 activation is mediated by inactivation of the repressor Bach1. By contrast, Bach1 overexpression (by 6-fold) decreased FPN1 and HO1 mRNA expression 3-fold and 4.5-fold, respectively, compared to the respective control vector (Figure 2B).
Figure 2.

Bach1 and Nrf2 modulate FPN1 transcription in RAW264.7 cells. (A) siRNA mediated knockdown of Bach1. FPN1 and HO1 expression was analyzed by qPCR 72 h after transfection with specific siRNAs directed against Bach1 or luciferase as a control. (B) Bach1 overexpression. FPN1 and HO1 mRNA expression was analyzed by qPCR 72 h after transfection with pCMV-Bach1 or pCMV-luciferase as a control. Knockdown and overexpression of Bach1 was determined by Western Blot (bottom panels). Equal loading was controlled by the detection of Vinculin. Experiments were performed in triplicates. **represents P<0.01 and ***P<0.001. (C) Nrf2 nuclear accumulation mediated by sulforaphane (SF) treatment. Ferroportin (FPN1), NADH:quinone oxidoreductase (QR), TTR and HO1 expression was analyzed by qPCR after 8 h of treatment with 2 mM SF or 1%DMSO as a control. Data were normalized to mRNA expression of GADPH and presented as fold changes whereby the untreated control was set to 1.
To investigate whether Nrf2 is involved in the FPN1 transcription control we induced Nrf2 nuclear accumulation by sulforaphane treatment, a chemical inhibitor of KEAP1.30, 31 Following 8h of sulforaphane exposure, we analyzed the mRNA abundance of FPN1, as well as of NADP (H) quinone (oxido) reductase (QR), thioredoxin reductase1 (TRR) and HO1 as controls. Interestingly, FPN1 mRNA expression increases in sulforaphane treated cells to a similar extent as QR and even stronger than HO1, suggesting that the Nrf2/KEAP1 pathway and Bach1 regulate FPN1 transcription (Figure 2C).
Heme dependent FPN1 activation is mediated by an MARE/ARE element at position −7007/− 7016 of the FPN1 promoter
Both Bach1 and Nrf2 form heterodimers with transcription factors such as sMaf to bind to enhancer sequences within the HO1 promoters, the MAf Recognition Element/Antioxidant Responsive Elements (MARE/ARE). Bioinformatic analysis of 9kb genomic DNA upstream of the FPN1 transcription start site (see Design and Methods section) revealed 4 putative MARE/ARE sequence motifs at positions −1848, −3105, −5385 and −7007 (Figure 3; Online Supplementary Figure S2). Interestingly the MARE/ARE sequence motif at position −7007 is located within a highly conserved FPN1 promoter region (Online Supplementary Figure S3) suggesting a functional role of this sequence element in regulating FPN1 transcription. To assess whether the computationally identified MARE/ARE motifs are functional we truncated the 8.0kbΔIRE-luciferase construct in such a way that one putative transcription factor binding site was removed at a time within the reporter vectors tested.
Figure 3.

Bioinformatic analysis of genomic DNA upstream of the FPN1 coding region recognizes 4 putative Maf Recognition Element/Antioxidant Responsive Elements (MARE/ARE). The genomic position of the FPN1 locus (top bar) and the base count of approximately 8000bp upstream of the FPN1 translation start site are shown. The restriction sites used to generate the truncated FPN1 reporter constructs, as well as the putative MARE/ARE (gray box) are indicated, whereby the numbers relate to the 3′ position of the element. The mammalian conservation plot is shown. The big arrow marks the transcription start site. The IRE and the transcription start site (TSS) is indicated.
Hb treatment of cells for 18h only activates luciferase activity from the 8.0 ΔIRE reporter construct and not from the 2.4kbΔIRE, 4.8kbΔIRE and 6.8kb ΔIRE constructs (Figure 4A). This suggests that a putative MARE/ARE element must be located between 6.8kb and 8kb upstream of the FPN1 transcription start site. To determine whether the MARE/ARE element identified at position −7007/−7016 is involved in hemoglobin mediated FPN1 activation we mutated this sequence by inserting a NotI site (8.0kbDIRE-MARE/ARE*-luc; Figure 4A). Hb activation of luciferase activity from 8.0kbDIRE-MARE/ARE*-luc transfected cells was almost completely abolished compared to the 8.0KbDIRE-luc transfected cells, suggesting that the MARE/ARE element at position −7007/−7016 is critical to regulate FPN1 transcription in response to hemoglobin.
Figure 4.

A MAf Recognition Element/Antioxidant Responsive Elements (MARE/ARE) at position −7007/−7016 of the FPN1 promoter confers heme responsiveness. (A) Luciferase reporter assay. RAW264.7 cells were transfected with the 2.4kb_IRE, 4.8kb_IRE, 6.8kb_IRE 8kb_IRE and 8kb_IRE-MARE/ARE* luciferase reporter vectors, treated with hHb or the solvent control after 6 h and luciferase activity was measured after 18 h. Transfections were performed in triplicates, and results are presented as fold change ± SEM of Renilla/Firefly. **represents P<0.01. (B) MARE/ARE consensus sequence. The sequence AGATCAT at position −7010/−7016 upstream of the TSS was mutated to GCGGCCG creating a new NotI restriction site; Consensus ARE/MARE, wild-type and mutated site are shown.
Discussion
Macrophages of the human reticuloendothelial system recycle approximately 25mg iron from hemoglobin every day. Most of the iron is exported from macrophages by the iron exporter FPN1 to sustain de novo erythropoiesis. Here we show that hemoglobin activates transcription of FPN1 and HO1 with similar kinetics and that the coordinated expression of these two important proteins for macrophage iron recycling involves the transcriptional regulators Bach1 and Nrf2 as well as a MARE/ARE response element in the FPN1 promoter.
The transcription of FPN1 is activated by either hemoglobin, hemin or the protoporphyrin ring alone, suggesting that the iron released from the hemoglobin by HO1 activity is unlikely to be involved in this process. Consistently, treatment of macrophages with an equal molarity of iron as contained in the hemoglobin did not activate FPN1 transcription (Figure 1). This finding contrasts with previous studies that reported iron-controlled FPN1 transcriptional activation.13,35 The discrepant data may be explained by the fact that more than 10-fold higher iron concentrations were applied in the previous studies. Interestingly, hemoglobin and heme are both able to increase luciferase activity in cells transfected with the 2.4Kb-Luc reporter. This response is fully dependent on the IRE sequence in the FPN1 5′UTR, whereas deletion of the FPN1 5′UTR from the 8Kb-Luc construct only partially reduced luciferase activity. Moreover PPIX alone is able to increase the luciferase activity of the 8kb-Luc promoter but not of the 2.4kb (data not shown).
These data show that two overlapping regulatory mechanisms control FPN1 expression in macrophages in response to hemoglobin treatment: 1) transcriptional control mediated by the FPN1 promoter; and 2) translational control mediated by the IRE sequence within the FPN1 5′UTR. As iron is the major regulator of IRP1 and IRP2 activity, we believe that iron released from hemoglobin by the activity of HO1 is responsible for IRE/IRP mediated FPN1 regulation. Alternatively, regulation of IRP2 levels by heme36 may play a role in this process. A role for the 5′UTR IRE in the iron and/or heme-response of FPN1 is consistent with the finding that bone marrow derived macrophages lacking both IRP1 and IRP2 up-regulate FPN1 protein levels.12 Expression of FPN1 is thus controlled by multiple mechanisms underlining the importance of precise regulation of iron export: 1) transcriptional control by hemoglobin; 2) alternative promoter usage in duodenal enterocytes and erythroid precursor cells;37 3) translational control by the IRE/IRP system in response to iron levels; as well as 4) posttranslational control by hepcidin and systemic iron requirements.38 How the regulation of FPN1 by the different stimuli is prioritized will be the subject of further investigation.
Detailed analysis of the molecular mechanisms underlying the hemoglobin-mediated FPN1 transcriptional activation revealed that Bach1, Keap1 and Nrf2 as well as an MARE/ARE element at position bp−7007/bp− 7016 of the FPN1 promoter are important for this process. Together with previous reports this finding suggests that heme released from senescent or damaged red blood cells poses an oxidative stimulus to inhibit the enzyme Keap1.31 Keap1 inhibition prevents Nrf2 degradation and results in subsequent Nrf2 nuclear accumulation.29,39 Increased Nrf2 levels in the nucleus will displace the Bach1 repressor from the ARE/MARE sequence on the FPN1 enhancer to activate FPN1 transcription (Figure 5) by a similar mechanism reported for HO1.40, 41 Bach1 itself can be directly inhibited by heme23,26–27 and this mechanism seems not to require iron.39 Activation of HO1 by hemoglobin24 (Figure 1A) will result in iron release from heme which additionally activates FPN1 translation by preventing IRP1 and/or IRP2 binding to the FPN1 IRE. It is of note that Bach1 silencing increases FPN1 mRNA expression by a factor of 17 whereas Bach1 overexpression reduces FPN1 mRNA levels by a factor of three (Figure 2A and B). This finding suggests that in steady state conditions (e.g. in the absence of a heme stimulus), FPN1 expression in macrophages is strongly repressed.
Figure 5.

Regulation of iron recycling by a Bach1/Nrf2 dependent transcriptional control mechanism. Erythrophagocytosis or uptake of extracellular hemoglobin via the hemoglobin scavenging receptor CD163 cause increased intracellular heme levels (1). Heme accumulation leads to the release of the transcriptional repressor Bach1 from the ARE/MARE enhancer, KEAP1 degradation and subsequent nuclear accumulation of the transcriptional activator Nrf2. Nrf2 binding to small MAFs (SM) enhances the transcription of heme-oxygenase 1 (HO1), the ferritins and FPN1 (2). HO1 releases inorganic iron from the heme moiety (3) which inhibits IRP1 and IRP2 binding to the FPN1 and ferritin IREs to promote mRNA translation. Free iron is stored in ferritin or exported to the plasma via FPN1 (4). Plasma iron binds to transferrin and is made available for de novo red cell synthesis in the bone marrow (5).
The same mechanism involved in the FPN1 transcriptional response to hemoglobin in macrophages may also be operational in haptoglobin-null mice16 which are hallmarked by increased FPN1 expression in duodenal enterocytes and spleen macrophages together with chronic systemic hemoglobin overload. We speculate that in this mouse model, hemoglobin released as a consequence of intravascular hemolysis acts as a systemic signal to regulate FPN1 transcription. Consistently, FPN1 mRNA expression is also strongly increased in murine models of intra and extra-vascular hemolysis associated with thalassemia,42 phenylhydrazine treatment43 or lack of superoxide dismutase.44 Interestingly, transcription control of FPN1 by Bach1 and Nrf2 is also operational in a mouse hepatoma cell line (HepA1–6; data not shown) suggesting that this mechanism may be conserved in different cell types. It is, thus, of note that haptoglobin-null mice up-regulate FPN1 mRNA expression also in the liver. Heme and/or hemoglobin induced FPN1 transcription may thus enhance systemic iron availability to sustain erythropoiesis under conditions when intact erythrocytes are limiting.
In summary, our data show that FPN1 is an important novel player in a gene regulon controlled by Bach1, Nrf2 and MAF transcription factors that regulates both iron recycling from hemoglobin as well as the cellular antioxidant response.40 Phagocytosis of senescent or damaged erythrocytes releases hemoglobin which induces HO1 to trigger heme degradation, the heavy (H) and light (L) ferritin chains20–21 (Online Supplementary Figure S4) to store and detoxify the iron released from the heme moiety and FPN1 to pump iron out of the cell for transport in the serum. In other cell types, coregulation of FPN1 with antioxidant enzymes like thioredoxin reductase 1 and NAD(P)H: quinone oxidoreductase 1 (qr)22 by the same mechanism may contribute to the antioxidant defense by exporting iron out of the cell to avoid an aggravation of oxidative stress.
Footnotes
The Online version of this article has a Supplementary Appendix.
Authorship and Disclosures
SM, DC and JS performed experiments; EM cloned the FPN1 promoter; SM analyzed results and made the figures; SM, ET and MM designed the research and wrote the paper.
The authors reported no potential conflicts of interest.
References
- 1.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117(3):285–97. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 2.Kino K, Tsunoo H, Higa Y, Takami M, Nakajima H. Kinetic aspects of hemoglobin.haptoglobin-receptor interaction in rat liver plasma membranes, isolated liver cells, and liver cells in primary culture. J Biol Chem. 1982;257(9):4828–33. [PubMed] [Google Scholar]
- 3.Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 4.Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106(7):2572–9. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- 5.Hamilton TA, Weiel JE, Adams DO. Expression of the transferrin receptor in murine peritoneal macrophages is modulated in the different stages of activation. J Immunol. 1984;132(5):2285–90. [PubMed] [Google Scholar]
- 6.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 7.De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 2007;18(7):2569–78. doi: 10.1091/mbc.E07-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 9.Fleming MD. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Hematology Am Soc Hematol Educ Program. 2008:151–8. doi: 10.1182/asheducation-2008.1.151. [DOI] [PubMed] [Google Scholar]
- 10.Muckenthaler MU. Fine tuning of hepcidin expression by positive and negative regulators. Cell Metab. 2008;8(1):1–3. doi: 10.1016/j.cmet.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275(26):19906–12. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 12.Galy B, Ferring-Appel D, Kaden S, Grone HJ, Hentze MW. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab. 2008;7(1):79–85. doi: 10.1016/j.cmet.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Knutson MD, Vafa MR, Haile DJ, Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood. 2003;102(12):4191–7. doi: 10.1182/blood-2003-04-1250. [DOI] [PubMed] [Google Scholar]
- 14.Delaby C, Pilard N, Puy H, Canonne-Hergaux F. Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: early mRNA induction by haem, followed by iron-dependent protein expression. Biochem J. 2008;411(1):123–31. doi: 10.1042/BJ20071474. [DOI] [PubMed] [Google Scholar]
- 15.Delaby C, Pilard N, Hetet G, Driss F, Grandchamp B, Beaumont C, et al. A physiological model to study iron recycling in macrophages. Exp Cell Res. 2005;310(1):43–53. doi: 10.1016/j.yexcr.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Marro S, Barisani D, Chiabrando D, Fagoonee S, Muckenthaler MU, Stolte J, et al. Lack of haptoglobin affects iron transport across duodenum by modulating ferroportin expression. Gastroenterology. 2007;133(4):1261–71. doi: 10.1053/j.gastro.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21(19):5216–24. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun. 2004;324(1):77–85. doi: 10.1016/j.bbrc.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 19.Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, et al. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. 2004;279(7):5480–7. doi: 10.1074/jbc.M302733200. [DOI] [PubMed] [Google Scholar]
- 20.Hintze KJ, Katoh Y, Igarashi K, Theil EC. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, beta-globin, and NADP(H) quinone (oxido) reductase1. J Biol Chem. 2007;282(47):34365–71. doi: 10.1074/jbc.M700254200. [DOI] [PubMed] [Google Scholar]
- 21.Hintze KJ, Theil EC. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc Natl Acad Sci USA. 2005;102(42):15048–52. doi: 10.1073/pnas.0505148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280(17):16891–900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 23.Igarashi K, Hoshino H, Muto A, Suwabe N, Nishikawa S, Nakauchi H, et al. Multivalent DNA binding complex generated by small Maf and Bach1 as a possible biochemical basis for beta-globin locus control region complex. J Biol Chem. 1998;273(19):11783–90. doi: 10.1074/jbc.273.19.11783. [DOI] [PubMed] [Google Scholar]
- 24.Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol. 1996;16(11):6083–95. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001;20(11):2835–43. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H, Tashiro S, Hira S, Sun J, Yamazaki C, Zenke Y, et al. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J. 2004;23(13):2544–53. doi: 10.1038/sj.emboj.7600248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol. 2007;27(19):6962–71. doi: 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med. 2004;37(4):433–41. doi: 10.1016/j.freeradbiomed.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 29.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci USA. 2005;102(29):10070–5. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keum YS, Yu S, Chang PP, Yuan X, Kim JH, Xu C, et al. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006;66(17):8804–13. doi: 10.1158/0008-5472.CAN-05-3513. [DOI] [PubMed] [Google Scholar]
- 31.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Tomso DJ, Chorley BN, Cho HY, Cheung VG, Kleeberger SR, et al. Identification of polymorphic antioxidant response elements in the human genome. Hum Mol Genet. 2007;16(10):1188–200. doi: 10.1093/hmg/ddm066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kataoka K, Handa H, Nishizawa M. Induction of cellular antioxidative stress genes through heterodimeric transcription factor Nrf2/small Maf by antirheumatic gold(I) compounds. J Biol Chem. 2001;276(36):34074–81. doi: 10.1074/jbc.M105383200. [DOI] [PubMed] [Google Scholar]
- 35.Yang F, Wang X, Haile DJ, Piantadosi CA, Ghio AJ. Iron increases expression of iron-export protein MTP1 in lung cells. Am J Physiol Lung Cell Mol Physiol. 2002;283(5):L932–9. doi: 10.1152/ajplung.00114.2002. [DOI] [PubMed] [Google Scholar]
- 36.Goessling LS, Mascotti DP, Thach RE. Involvement of heme in the degradation of iron-regulatory protein 2. J Biol Chem. 1998;273(20):12555–7. doi: 10.1074/jbc.273.20.12555. [DOI] [PubMed] [Google Scholar]
- 37.Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009;9(5):461–73. doi: 10.1016/j.cmet.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci USA. 2005;102(5):1324–8. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan Y, Lambrecht RW, Donohue SE, Bonkovsky HL. Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J. 2006;20(14):2651–3. doi: 10.1096/fj.06-6346fje. [DOI] [PubMed] [Google Scholar]
- 40.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA. 2004;101(6):1461–6. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007;35(21):7074–86. doi: 10.1093/nar/gkm638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109(11):5027–35. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Latunde-Dada GO, Vulpe CD, Anderson GJ, Simpson RJ, McKie AT. Tissue-specific changes in iron metabolism genes in mice following phenylhydrazine-induced haemolysis. Biochim Biophys Acta. 2004;1690(2):169–76. doi: 10.1016/j.bbadis.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 44.Starzynski RR, Canonne-Hergaux F, Willemetz A, Gralak MA, Wolinski J, Stys A, et al. Haemolytic anaemia and alterations in hepatic iron metabolism in aged mice lacking Cu,Zn-superoxide dismutase. Biochem J. 2009;420(3):383–90. doi: 10.1042/BJ20082137. [DOI] [PubMed] [Google Scholar]