

Abstract
Background
Identification of mutations in the SERPINC1 gene has revealed different mechanisms responsible for antithrombin deficiency. Deletions and nonsense mutations associate with type I deficiency. Certain missense mutations cause type II deficiency by affecting the heparin binding site or the reactive center loop, while others result in type I deficiency by intracellular retention or RNA instability.
Design and Methods
We studied the molecular, biochemical, proteomic and glycomic characterization of a new natural mutant (K241E) that may be classified as pleiotropic.
Results
The mutation caused a significant decrease in the anticoagulant activity mainly due to a reduced heparin affinity and a modification of the electrostatic potential that might explain the impaired ability of the mutant protein to form complexes with the target protease in the absence of heparin. Mass spectrometry and glycomic analyses confirmed an increased molecular weight of 800 Da in the mutant protein possibly due to core-fucosylation, provoking the loss of heparin affinity. Additionally, carriers of this mutation also have a minor mutant isoform that still followed normal glycosylation, retaining similar heparin affinity to wild-type α-antithrombin, and certain anticoagulant activity, which may explain the milder thrombotic risk of patients carrying this mutation. Similar results were observed using recombinant K241E antithrombin molecules.
Conclusions
Our data suggest a new mechanism involved in antithrombin type II deficiency by indirectly affecting the glycosylation of a natural variant. Additional studies are required to confirm this hypothesis.
Keywords: antithrombin, glycosylation, heparin
Introduction
Antithrombin is a serpin that plays a key anticoagulant role by correctly controlling and preventing inappropriate, excessive or mislocalized clotting of blood which may cause thrombotic disorders.1 Indeed, this was the first hemostatic element shown to be involved in venous thrombosis. Since then, up to 228 distinct mutations have been described in the SERPINC1 gene associated with two types of deficiency:2 I and II, distinguished by the presence of a variant protein in the latter. Type II deficiency, with significant clinical heterogeneity, is further sub-classified on the basis of mutations that either alter the function of the reactive site, the heparin-binding site or have multiple or pleiotropic effects.3 Research has provided information concerning the mechanisms responsible for antithrombin deficiency. Thus, the identification of missense mutations affecting mobile regions (mainly the hinges of the reactive center loop (RCL) or the region involved in the shutter-like opening of the main β-sheet) that caused oligomer formation and intracellular retention, revealed a new pathological mechanism causing antithrombin deficiency, including some cases of thrombosis within the group of conformational diseases.4 Moreover, characterization of other missense mutations has been decisive in identifying key structural and functional domains, such as the heparin-binding domain, which is crucial for the control of its anticoagulant activity.5 Indeed, most circulating antithrombin has a native conformation with low anticoagulant properties that allows thrombin to form the clot.6 The binding of glycosaminoglycans to the heparin-binding site causes a conformational transition in the antithrombin that closes the β-sheet A, which expels the RCL, activating the molecule.7 This control allows a slight delay in the antithrombin anticoagulant function and ensures it remains localized at the site of vascular injury. Therefore, alteration of heparin affinity results in a defective inhibitory function of this serpin.
We present here the occurrence of a new pleiotropic natural mutant (K241E) that may induce alteration of the glycosylation which would result in a novel mechanism of antithrombin deficiency by an impaired heparin binding and thrombin inhibition. The impairment of antithrombin is confirmed through the recombinant expression of the mutant.
Design and Methods
Functional and genetic analysis
Anti-FXa activity, antigen levels and heparin affinity of plasma antithrombin from plasma of the patient and family members, and PCR amplification and sequencing of the SER-PINC1 gene was performed essentially as reported.8 Thrombophilic tests including protein C, protein S, antiphospholipid antibodies, FV Leiden and prothrombin G20210A were also performed. Crossed immunoelectrophoresis of antithrombin with unfractionated sodium heparin (Laboratorios Rovi SA, Madrid, Spain) was performed as previously reported.9
Protein purification
Antithrombin of the proband and his affected daughter were purified by heparin affinity chromatography at pH 7.4 on HiTrap Heparin columns (GE Healthcare, Barcelona, Spain) using a Biologic LP system (Bio-Rad, Madrid, Spain). Fractions with variant antithrombins were applied to a HiTrap Q affinity column (GE Healthcare, Barcelona, Spain). Purity and separation of proteins were evaluated by 8% SDS-PAGE, non-denaturing PAGE, and immunoblotting, as indicated elsewhere.10
Antithrombin activity
Formation of thrombin-antithrombin complex (TAT) was evaluated by incubation of 150 ng antithrombin with 0.25 U thrombin at 37ºC. Aliquots were withdrawn at different time intervals. The reaction was carried out with and without previous incubation of antithrombin with 0.6 U heparin for 30 min. These samples were evaluated by SDS-PAGE as indicated previously.
Fluorescence studies
Equilibrium dissociation constants (Kd) for the AT-heparin interaction were determined essentially as described previously.7 Briefly, change in intrinsic fluorescence of AT (25–50 nM) upon titration of the unfractionated heparin was monitored at 340 nm on a PerkinElmer Life Sciences 50B spectrofluorometer, with excitation at 280 nm and using bandwidths of 3.5 nm for both excitation and emission. All titrations were carried out at room temperature under physiological ionic strength (I = 0.15) in 20 mM NaPO4, 100 mM NaCl, 0.1 mM EDTA, 0.1% polyethylene glycol 8000, pH 7.4. Fluorescence emission intensity was taken as the average of 100 measurements recorded at one second intervals for each addition of heparin. Data were fitted as previously described.7
Endo and exoglycosidase digestions
Purified antithrombins (1.50 μg) were treated with 2 U α2–3,6,8,9 neuraminidase (sialidase) (N 3786, Sigma-Aldrich, Saint Louis, USA) at 37ºC for 18h in 50 mM sodium phosphate buffer, pH 6.0. Treatment with N-glycosidase F (Roche Diagnostics GmbH, Mannheim, Germany) required a previous denaturing step (5 min at 95ºC in 150 mM sodium phosphate buffer, pH 7.4). Afterwards, samples were chilled on ice and digested with 0.6 U N-glycosidase F by incubation at 37ºC for 15h. Samples were resolved by SDS-PAGE.
Plasma from the proband and healthy donors were treated with 0.01 U α-1,6-fucosidase (F 6272, Sigma-Aldrich, Saint Louis, USA) for 24h at 37ºC.
MALDI-TOF-MS analysis
A solution of 3,5-dimethoxy-4-hydroxycinnamic acid (10 g/L) in acetonitrile (ACN)/water/trifluoroacetic acid (TFA) (50:50:0.1 by vol.) was chosen as matrix for protein analysis. Experiments were carried out on a Voyager-DE™ STR Biospectrometry workstation (Applied Biosystems), equipped with a N2 laser (337 nm). Recorded data were processed with Data Explorer™ Software (Applied Biosystems).
Antithrombin samples were also digested in 100 mM NH4HCO3 (pH 7.8) containing trypsin (enzyme/substrate ratio 1:50) at 37ºC for 16h. Peptide mixtures from in situ digestion of proteins were desalted in a GELoader tip packed with 0.5 μL POROS-10 R2 (PerSeptive Biosystem) slurry.
Glycan profiling
Weak anion-exchange HPLC of 2-aminobenzamide (2-AB) N-linked glycans were carried on a VYDAC 301 VHP column (7.5 × 50 mm) using an Agilent 1090 HPLC equipped with a fluorescence detector (1100 Agilent fluorescence module): excitation λ=330 nm and emission λ=420 nm. The column was calibrated with 2-AB labeled N-glycans from bovine fetuin.11
Normal-phase high performance liquid chromatography (HPLC) analyses were performed on a capillary system used for SA analyses. Fluorescence detector parameters were those recommended for 2-AB tag-like in WAX analyses. Normal phase profiling was carried out on a TSK gel Amide-80 column (0.5 × 150 mm). The system was calibrated in glucose units using a 2-aminobenzamide-labeled dextran hydrolysate. The total running time was 125 min.
Recombinant antithrombin expression and purification
The K241E mutation was constructed on the recombinant β-glycoform S137A AT background in order to reduce glycosylation heterogeneity and facilitate purification, essentially as previously described.12 Site directed mutagenesis of the pCEP4-S137A/AT plasmid was performed using the Stratagene QuikChange Site-Directed Mutagenesis kit and the appropriate primers for the mutation. A second site directed mutagenesis was also performed to obtain the K241E mutant following the same strategy. Human Embryonic Kidney cells expressing the Epstein Barr Nuclear Antigen 1 (HEK-EBNA) were grown to 60% confluence at 37°C, 5% CO2, in DMEM with GlutaMAX-I medium (Invitrogen) supplemented with 5% fetal bovine serum (Sigma). Transfection was performed by addition of 1/10 volume of pCEP4/AT plasmid (3μg/mL) preincubated for 15 min in DMEM with polyethylenimine (15μg/mL). After 24h the cells were washed with PBS and exchanged into CD-CHO medium (Invitrogen) supplemented with 4mM L-glutamine and 0.25mg/mL Geneticin (Invitrogen). Cells were grown to confluence and media was harvested after 72h. The variant expressed at levels similar to wild-type, although the level of expression was low. Purification was achieved by affinity chromatography on heparin-Sepharose. Purified material was evaluated by SDS-PAGE and Western blot as described before.
Structural representation
Structural representation was performed by using SWISS-MODEL and the Swiss-PdbViewer programs13 (http://www.expasy.ch/spdbv) using native (PDB accession number: 1t1fA) conformational states as template. Electrostatic potential was studied using both Poisson-Boltzmann Solver (APBS) program14 and a Coulomb computation method. Python Molecule Viewer15 was used as the graphic interface for the calculated Poisson-Boltzmann electrostatic potential.
Results
Clinical data
The proband, a 44-year old Caucasian male, developed a deep venous thrombosis (popliteal venous of the right leg) at the age of 37 after traumatism and immobilization. Since then, he has been under stable oral anticoagulation with coumarins without recurrence. Plasma and DNA from all family members were obtained. All included subjects gave their informed consent to enter the study.
Identification of antithrombin Murcia, K241E
Thrombophilic test revealed that the proband had two prothrombotic defects, both in heterozygosis: Factor V (FV) Leiden, and antithrombin deficiency (60% anti-FXa levels). The deficiency of antithrombin should be a spontaneous mutation, as it was transmitted to one of his daughters, but was absent in his parents (Figure 1A).
Figure 1.

Characterization of the antithrombin K241E mutation (antithrombin Murcia). (A) Pedigree of proband with ages in brackets. The proband is indicated by the arrow, family members documented with antithrombin deficiency are in black, and individuals affected with Factor V Leiden are typed in gray. Anti-FXa activities (first row) and antigen levels (second row) are represented for each family member in percentage rates. (B) Crossed immunoelectrophoresis in the presence of heparin: (1) proband, (2) control. The antithrombin forms of high and low heparin affinity are indicated. (C) Electropherogram of the proband PCR-amplified product of exon 4, showing a heterozygous base C→T base change in exon 4 at codon 241. (D) Structural modeling of antithrombin Murcia. Strands are displayed in blue and helices in red. Mutation at residue 241 is represented as yellow CPK spheres. (E) Western blotting of antithrombin from plasma. Lane 1, proband; lane 2, control; run on native PAGE. The wild-type (WT) and the mutant variants (V1 and V2) identified in the proband are marked. (F) Western blotting of antithrombin from plasma run on native PAGE with urea. Lane 1, proband; lane 2, control. Native (N) and latent (L) antithrombins are indicated (wild-type-wt and mutant-mut).
Antithrombin levels of 77% in carriers and an increased fraction with low heparin affinity (Figure 1B) suggested a type II deficiency with heparin binding defect. Sequencing of the SERPINC1 gene in the proband and his affected daughter revealed a single base pair mutation responsible for the K241E change (Figure 1C). This mutation, which we called antithrombin Murcia, had not been described previously, and affected a non-conserved residue located at β-strand C4 (Figure 1D).
Western-blot analysis of native gels revealed two extra bands with increased mobility compared to wild-type antithrombin in the proband (Figure 1E). Native gels with urea showed two latent forms (wild-type and mutant) that shared a similar proportion compared with the native forms (~5%) (Figure 1F). The same results were observed in the proband’s daughter (data not shown).
Purification of wild-type and mutant antithrombins
Purification of antithrombins from plasma of the proband and his affected daughter was performed by heparin-agarose chromatography. Normal profiles were obtained by eluting with a 0.15–2 M NaCl gradient. Native-PAGE revealed two bands in the peaks containing wild-type α and latent forms (Figure 2). The main mutant variant observed in plasma, Variant 1 (V1), eluted in the flow-through fraction (Figure 2, lane 3) with trace amounts in the peak of the wild-type latent antithrombin (data not shown). A 0–2M NaCl gradient resulted in three peaks, with mutant V1 and wild-type latent form eluting in the first peak (Figure 2, lane 6). The second peak contained α-wild type and the minor mutant isoform, Variant 2 (V2) (Figure 2, lane 7). Since the mutation incorporates an extra negative charge, mutant variants were further purified by anion exchange chromatography at pH 6.0. After the second purification step, both V1 and V2 were pure (>90%), without contamination from wild-type latent and α isoforms (Figure 2, lanes 5 and 8). However, this step confirmed a different pI between both variants since variant 1 eluted before the wild-type latent and V2 after the wild-type α isoform (Online Supplementary Figure S1). Finally, wild-type β-antithrombin was purified from the fraction with the highest heparin affinity (Figure 2, lane 10).
Figure 2.

Western blot (native gel electrophoresis) of different fractions from purification steps of the antithrombin Murcia variants. Lane 1, plasma control; lane 2, plasma proband; lane 3, flow-through fraction when heparin affinity chromatography is performed using 100 mM Tris-HCl, 10 mM citric acid, 150 mM NaCl as start buffer; lane 4, flow-through fraction when heparin affinity chromatography is performed with the same start buffer but with no NaCl; lane 5, antithrombin Murcia V1 purified by ion exchange; lane 6, fraction eluted at 180–200 mM NaCl from heparin affinity chromatography, containing wild-type latent antithrombin and mutant V1; lane 7, fraction eluted at 0.98–1 M NaCl from heparin affinity chromatography, containing wild-type a-antithrombin and mutant V2; lane 8, mutant V2 purified by ion exchange; lanes 9 and 10, wild-type α- and β-antithrombin forms purified from proband.
Inhibitory activity and heparin affinity
Mutant V2 displayed similar inhibition of thrombin to wild-type α-antithrombin in the presence of heparin (Figure 3A). However, such inhibition was impaired without heparin, although it was still higher than that of V1 (Figure 3B). In contrast, mutant V1 showed very low thrombin inhibitory activity and long incubation periods were needed to establish the complex with the protease (Figure 3C). These results could explain why plasma anticoagulant activity in carriers of antithrombin Murcia was over 50%, probing the functionality of part of the mutant antithrombin.
Figure 3.

(A) Thrombin-antithrombin (TAT) complexes formed by wild-type a-antithrombin, mutant V1 and V2 in presence (+) or absence (−) of heparin and identified by SDS-PAGE and Western blotting after 5 min incubation. (B) Thrombin-antithrombin (TAT) complexes formed by V2 in presence (+) or absence (−) of heparin after different intervals of incubation. (C) Thrombin-antithrombin (TAT) complexes formed by V1 in presence of heparin after different intervals of incubation in comparison with wild type α and latent antithrombin. (D) Heparin binding titrations. The intrinsic fluorescence change upon unfractionated heparin addition was followed for antithrombin wild type (black circles) and for V1 (white circles) (Antithrombin-AT).
The heparin affinity of the variants was initially observed during purification with the heparin-Sepharose column, and was confirmed by determining the equilibrium dissociation constants of the antithrombin variants for the heparin by following changes in intrinsic fluorescence (Figure 3D). The dissociation constants (Kd values) were 551±0.3, and 43.9±0.4 nM for V1 and wild-type α-antithrombin, respectively. Unfortunately, we did not have enough V2 protein to determine its heparin affinity by this method.
Biochemical characterization
SDS-PAGE analysis suggested that mutant V1 isoform had slightly higher molecular weight than mutant V2 isoform (Figure 4A). This could be due to a differential glycosylation pattern, which was examined by treating the mutant variants with sialidase and N-glycosidase F, plus subsequent evaluation by electrophoresis and mass spectrometry. Removing sialic acid residues with sialidase slightly reduced the size of both mutant variants, but did not diminish the difference in molecular weight between V1 and V2 (Figure 4A). MALDI-TOF analysis confirmed the mutation in the peptides obtained from both variants and showed an approximately 800 Da mass increase for V1 in comparison to wild-type α-antithrombin and V2. As wild-type β-antithrombin is physiologically produced due to the absence of glycosylation at N135,16,17 we suspected that V2 could represent the mutant β-isoform. However, the MALDI-TOF spectrum analysis of V2 after treatment with N-glycosidase F18 and partial de-N-glycosylation confirmed the presence of five different centroids in the mutant V2 (Figure 4B). These correspond to the presence of up to four residual N-glycans in the treated protein, the same pattern observed in wild-type α-antithrombin (Figure 4B). These data were corroborated at the glycoprotein level through treatment with several exoglycosidases and eventually N-glycosidase F yielding the completely de-N-glycosylated antithrombin at approximately 49 kDa (data not shown). Furthermore, glycan profiling analysis showed the same glycosylation pattern for V2 as found in wild-type α-antithrombin (Figure 4C). Nevertheless, the mutant V2 was not identical to wild-type α-antithrombin, as it showed a slightly faster electrophoretic mobility on native gels (Figure 2). Moreover, V2 was less efficient than wild-type α-antithrombin in forming complexes with thrombin when heparin was absent (Figure 3), besides which it displayed a different isoelectric point.
Figure 4.
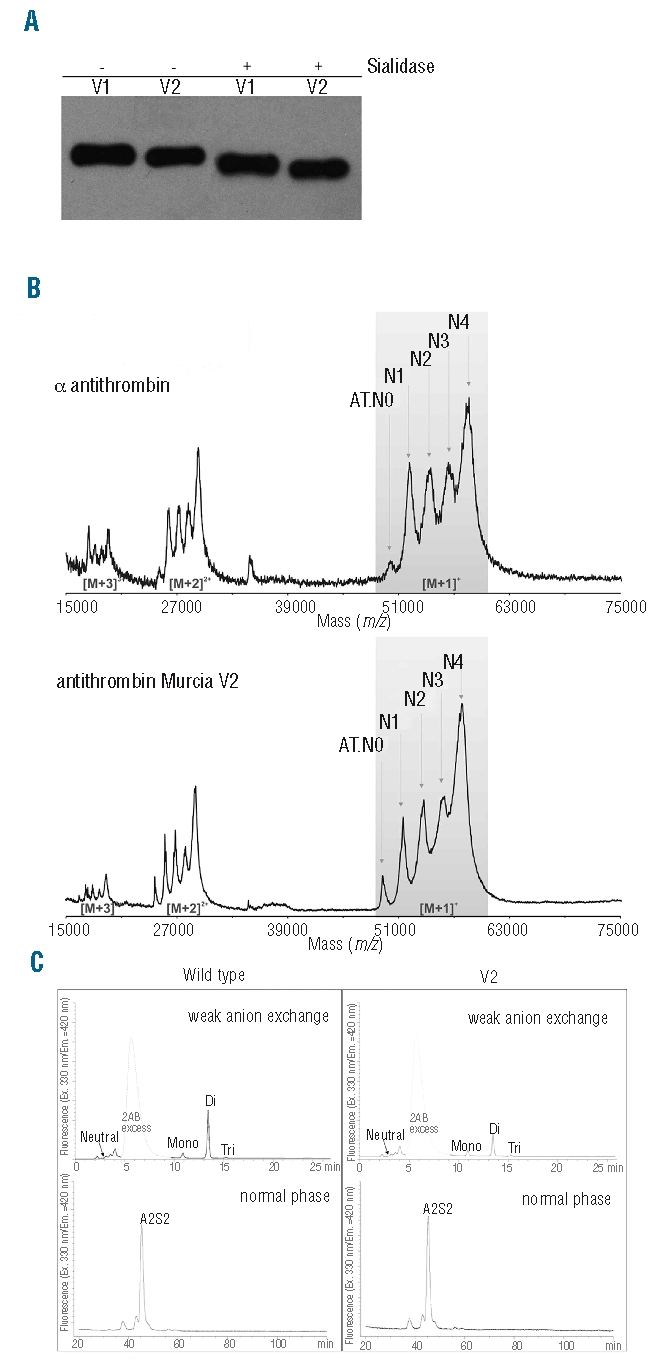
Analysis of the glycosylation of antithrombin Murcia. (A) Western blot of desialylated antithrombin run on SDS-PAGE. (B) Mass spectra of partially deglycosylated wild-type α-antithrombin and mutant V2 using N-glycosidase F. (C) Glycan profiling of wild-type α-antithrombin and mutant V2. Glycan structures were determined by normal phase and anion exchange in an HPLC system. AB: aminobenzamide; Mono: monoantennary; Di: diantennary; Tri: tri-antennary; A2S2: diantennary and desialylated.
Since fucosylation of antithrombin has been reported to reduce the heparin affinity of the protein,19–21 treatment of the plasma from proband and a control pool of citrated plasma from 100 healthy subjects with α-1,6-fucosidase was carried out. Figure 5 shows that this enzyme only provoked a change in the mobility of the main mutant form, probing the presence of core-fucosylation in V1.
Figure 5.
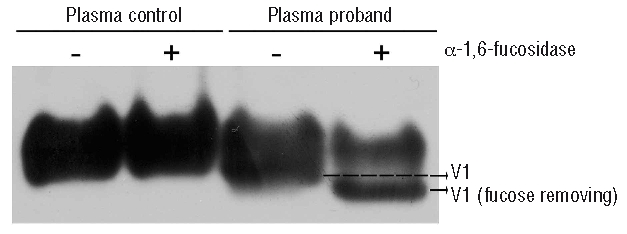
Determination of the presence of core-fucosylation in the V1 isoform of antithrombin Murcia. Western blot of plasma antithrombin under non-denaturing PAGE.
Recombinant K241E
Cells transfected with wild-type cDNA only produced recombinant antithrombin with high heparin affinity. In contrast, cells transfected with mutant cDNA produced two isoforms with different heparin affinity and size (Figure 6). In accordance with data from the patient, the mutant isoform with low heparin affinity had higher molecular weight than the isoform with high heparin affinity, which had a similar size to the wild-type recombinant antithrombin (Figure 6). Thus, recombinant V1 seemed to have an altered glycosylation that did not affect N135. Unfortunately, the level of expression was too low to perform additional biochemical, functional or glycomic analysis.
Figure 6.

Western blot (SDS-PAGE) of fractions of wild-type and antithrombin Murcia recombinants eluted with NaCl from the heparin affinity chromatography
Discussion
Natural variants of antithrombin have provided valuable insights into its anticoagulant function, its structural features, the mechanisms leading to its deficiency and the associated thrombotic risk. The clinical, biochemical, proteomic, glycomic and electrostatic potential analysis of a new pleiotropic mutation (K241E, antithrombin Murcia) located in the β-strand C4 have now widened our understanding (Figure 7A).
Figure 7.
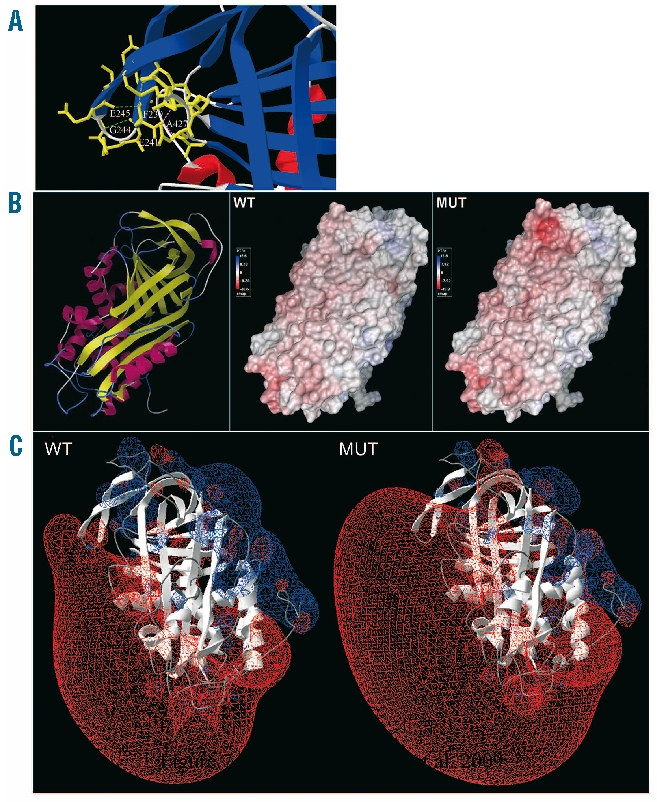
Structural representation of antithrombin Murcia. (A) Molecular representation of the residues involved in the interaction with the mutated residue at position 241. Hydrogen bonds at a distance from 2.35 up to 3.2 Å are drawn as green dotted lines, whilst weaker hydrogen bonds appear in gray. Steric hindrances are shown in purple. (B) Solvent accessible surface of wild-type protein (WT) and antithrombin Murcia (MUT), displaying their electrostatic potentials by Poisson-Boltzmann method. A ribbon diagram of the protein (left) is drawn in yellow for β-strands and in pink for α-helices. (C) Electrostatic potential of wild-type protein (WT) and antithrombin Murcia (MUT) by Coulomb method. Charge distribution is represented as a gradient in which positive potentials are drawn in blue and negative in red.
The presence of different isoforms of antithrombin has been well defined for both wild-type16,17,22 and mutant antithrombin.23 However, our study revealed a new heterogeneity. Subjects carrying antithrombin Murcia had two circulating mutant isoforms that displayed different biochemical and functional features. V1, the main mutant form, exhibited the highest mobility in native gels (Figure 1E), a decreased heparin affinity (Figure 2), and very low anticoagulant function (Figure 3). Although this might suggest a latent conformation for mutant V1, such a variant was denatured in native gels with urea (Online Supplementary Figure S2) in concordance with the levels of mutant latent form identified in plasma (Figure 1F). Moreover, mutant V1 had higher molecular weight and a different pI than V2, which is not compatible with a latent form (Figure 4; Online Supplementary Figure S1) and it was able to inhibit thrombin when lengthy incubations were carried out (Figure 3C). On the contrary, V2, the minor mutant form, had the same heparin affinity and activity if activated by heparin than wild-type α-antithrombin (Figures 2 and 3) but it showed an impaired function in absence of heparin (Figure 3B). Similar results were observed with recombinant proteins (Figure 6).
Changes in the electrostatics of the surface of antithrombin just below the RCL (Figure 7) described in previous mutants at C-sheet might explain the reduced activity of mutant V2 in the absence of heparin.24 However, binding of heparin to the mutant protein, by activating the molecule, would overcome such limitation (Figure 3).
But how could the heparin affinity of V2 be explained if both variants have the same mutation? The answer could be found by analyzing the differences between V1 and V2. As they differ in circulating levels, size and heparin affinity, our first hypothesis was to consider V1 and V2 to be the α and β mutant isoforms, respectively. Indeed, two isoforms of wild-type antithrombin are present in plasma: the main α form (90% of circulating antithrombin), has four N-glycans and lower heparin affinity than the minor β form, smaller because of its three N-glycans.16,17 We speculated that if V2 were the β mutant isoform, the absence of N-glycosylation at position 135, by removing steric hindrance, might compensate the effects of the K241E mutation on the heparin affinity. However, MALDI-TOF spectrum of the N-glycosidase treated V2 revealed the presence of four N-glycans as in wild-type α-antithrombin (Figure 4B). Consequently, there was something else, affecting V1 but not V2, responsible for the loss in heparin affinity. As V1 was approximately 800 Da larger than V2, the only posttranslational modification affecting antithrombin is N-glycosylation,16,17 and since differences in glycosylation may affect the heparin affinity of this serpin,19–21,25 we tried to identify an abnormal glycosylation on V1. A relevant modification of the mobility of V1 was observed after treatment with α-1,6-fucosidase (Figure 5), supporting the hypothesis that V1 could present core-fucosylation. The election of this exoglycosidase was based on the severe effect that fucose has on the heparin affinity of antithrombin when it is expressed in recombinant systems.19–21,25 Unfortunately, despite being the main mutant form, the lower purification yield did not allow us to perform additional studies to define the structure of the N-linked oligosaccharides in the V1 mutant isoform. However, there might be additional glycosylation modifications in V1 since after treatment with α-1,6-fucosidase the mobility of the V1 in native gels increased (Figure 5). This could be explained by an increased sialylation that would explain the different pI between the two variants and that has been reported to occur in transgenic mouse models from hepatocellular carcinoma.26 For such a reason, the difference in sialylation would explain the different pI between the two variants. To determine if the difference in sialylation between V1 and V2 could be responsible for the loss of activity in V1, sialic acid in such a form was removed. However, negligible anti-FIIa and FXa activity in the presence of heparin was detected for treated V1 whereas the same activity was obtained with the treated wild-type protein (data not shown). Moreover, after sialidase treatment V1 was evaluated in native gel pointing out the presence of an altered glycosylation (Figure 8). Therefore, the presence of core-fucose in V1 would explain the increased size of this isoform and its lower heparin affinity. To complete the puzzle, we had to explain why a mutation located in the C-sheet allowed an altered glycosylation. This is merely speculative, but the answer could be related to an improved accessibility of the Asn-linked GlcNAc to the fucosyltransferase-VIII after folding or an impaired secretion during Golgi secretion.27 The importance of this result lies in the functional consequences of this posttranslational modification and its pathological implication. The different glycosylation pattern reduced the heparin affinity and caused the loss of activity. This is, as far as we know, a novel mechanism involved in the deficiency of this key anticoagulant and it is described for the first time for a natural mutation.
Figure 8.
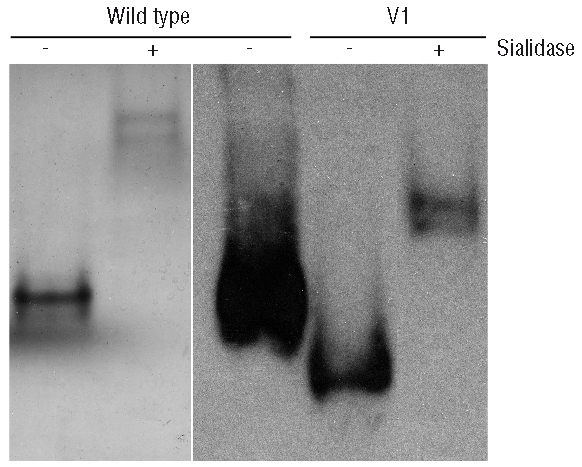
Sialidase treatment of mutant V1 and wild-type antithrombin evaluated in native gels by Western blot.
It only remained then to provide an explanation for V2. We suggest that some mutant molecules (V2) will not be core-fucosylated resulting in the glycosylation pattern of wild-type α-antithrombin with the same size and glycomic features (Figure 4B and C). Exactly this heterogeneity has already been described in recombinant wild-type antithrombin produced by baby hamster kidney or Chinese hamster ovary cells, which produced a low-heparin affinity form with increased carbohydrate content (with fucose) and other smaller forms with normal affinity to heparin,18–20,25 proving the incomplete nature of the glycosylation machinery.27 The clinical consequences of this heterogeneity may be relevant, as a small but significant proportion of the mutant protein present in plasma has heparin affinity and functional activity, which is consistent with the reduced anticoagulant activity observed in carriers of this mutation (60–65%). These results also help to explain the relatively moderate thrombotic profile of the patient who had additional thrombotic risk factors, both genetic (FV Leiden) and environmental (traumatism and immobilization).
In conclusion, our studies on a new natural pleiotropic mutant (antithrombin Murcia, K241E) and the concordant results obtained with the recombinant molecule underline the relevance that not only the primary sequence, but also the three-dimensional structure and posttranslational modifications, have on the affinity of antithrombin for heparin. Our results suggest a new mechanism leading to deficiency of antithrombin since this mutation might facilitate an altered glycosylation, probably involving fucosylation that damages the heparin affinity and causes the deficiency. However, mutant molecules that still follow a standard glycosylation pattern retain the heparin affinity and some functionality that could contribute to reduce the thrombotic risk. Despite the coherent and solid evidence presented, we are fully aware of the main limitation of this study. The small amounts of natural protein available, purified from plasma, or recombinantly expressed mutant protein, did not allow us to perform confirmative analysis, such as an exhaustive glycomic analysis. Therefore, further studies are required to sustain the hypothesis suggested by our results.
Acknowledgments
the authors wish to acknowledge the invaluable technical support of Antonia Miñano. We also want to thank Dr Rocio González-Conejero, Dr Maria Luisa Lozano, Dr Fernando Corrales and Dr Santiago Nieto for their helpful discussion and comments and management of patients, and to José Padilla and Nuria García Barberá for their technical help. Moreover, we thank Dr JA Huntington for providing the HEK-EBNA cells and the plasmid containing the wild-type cDNA of human antitrombin. Finally, we are deeply acknowledged to the proband and his family for their cooperation.
Footnotes
Funding: this work was partially supported by 04515/GERM/06 from Fundación Séneca, SAF2006-06212 and SAF2009-08993 (MICINN & FEDER), RETICS (RECAVA RD06/0014/0039) from ISCIII, Fundación Mutua Madrileña, and BFU2006-00494 (MCyT). IMM and CM are researchers from Fundación para la Formación e Investigación Sanitarias (FFIS), Spain. AO is a holder of a predoctoral research grant from FFIS, Spain.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
IMM, VV and JC, project planning, design of the study, analysis and interpretation of data; IMM and JC, wrote the manuscript; IMM, AO and JC, molecular analysis; IMM, AO and CM, biochemical and functional analysis; IMM, JNF, APL and RG, structural studies; RGG and EL, proteomic and glycomic analysis.
The authors reported no potential conflicts of interest.
References
- 1.Quinsey NS, Greedy AL, Bottomley SP, Whisstock JC, Pike RN. Antithrombin: in control of coagulation. Int J Biochem Cell Biol. 2004;36(3):386–9. doi: 10.1016/s1357-2725(03)00244-9. [DOI] [PubMed] [Google Scholar]
- 2.Cooper DN, Ball EV, Stenson PD, Phillips AD, Howells K, Mort ME, Thomas NST. The Human Gene Mutation Database. http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SERPINC1.2009.
- 3.Lane DA, Bayston T, Olds RJ, Fitches AC, Cooper DN, Miller DS, Jochmans K, Perry DJ, Okajima K, Thein SL, Emmerich J. Antithrombin mutation database: 2nd (1997) update. Thromb Haemost. 1997;77(1):197–211. [PubMed] [Google Scholar]
- 4.Corral J, Vicente V, Carrell RW. Thrombosis as a conformational disease. Haematologica. 2005;90(2):238–46. [PubMed] [Google Scholar]
- 5.Olson ST, Björk I, Bock SC. Identification of critical molecular interactions mediating heparin activation of antithrombin. Trends Cardiovasc Med. 2002;12(5):198–205. doi: 10.1016/s1050-1738(02)00160-3. [DOI] [PubMed] [Google Scholar]
- 6.Johnson DJ, Langdown J, Li W, Luis SA, Baglin TP, Huntington JA. Crystal structure of monomeric native antithrombin reveals a novel reactive center loop conformation. J Biol Chem. 2006;281(46):35478–86. doi: 10.1074/jbc.M607204200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langdown J, Belzar KJ, Savoir WJ, Baglin TP, Huntington JA. The critical role of hinge-region expulsion in the induced-fit heparin binding mechanism of antithrombin. J Mol Biol. 2009;386(5):1278–89. doi: 10.1016/j.jmb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corral J, Huntington JA, Gonzalez-Conejero R, Mushunje A, Navarro M, Marco P, et al. Mutations in the shutter region of antithrombin result in formation of disulfide-linked dimers and severe venous thrombosis. J Thromb Haemost. 2004;2(6):931–9. doi: 10.1111/j.1538-7836.2004.00749.x. [DOI] [PubMed] [Google Scholar]
- 9.Corral J, Rivera J, Martínez C, Gónzalez-Conejero R, Miñano A, Vicente V. Detection of conformational transformation of antithrombin in blood with crossed immunoelectrophoresis: new application for a classical method. J Lab Clin Med. 2003;142(5):298–305. doi: 10.1016/S0022-2143(03)00136-7. [DOI] [PubMed] [Google Scholar]
- 10.Mushunje A, Evans G, Brennan SO, Carrell RW, Zhou A. Latent antithrombin and its detection, formation and turnover in the circulation. J Thromb Haemost. 2004;2(12):2170–7. doi: 10.1111/j.1538-7836.2004.01047.x. [DOI] [PubMed] [Google Scholar]
- 11.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 12.Mushunje A, Zhou A, Carrell RW, Huntington JA. Heparin-induced substrate behavior of antithrombin Cambridge II. Blood. 2003;102(12):4028–34. doi: 10.1182/blood-2003-05-1560. [DOI] [PubMed] [Google Scholar]
- 13.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis. 1997;18(15):2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 14.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98(8):10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanner MF. Python: a programming language for software integration and development. J Mol Graphics Mod. 1999;17(1):57–61. [PubMed] [Google Scholar]
- 16.Picard V, Ersdal-Badju E, Bock SC. Partial glycosylation of antithrombin III asparagine-135 is caused by the serine in the third position of its N-glycosylation consensus sequence and is responsible for production of the b-antithrombin III isoform with enhanced heparin affinity. Biochemistry. 1995;34(26):8433–40. doi: 10.1021/bi00026a026. [DOI] [PubMed] [Google Scholar]
- 17.McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW. Structure of beta-antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J Mol Biol. 2003;326(3):823–33. doi: 10.1016/s0022-2836(02)01382-7. [DOI] [PubMed] [Google Scholar]
- 18.Muller R, Marchetti M, Kratzmeier M, Elgass H, Kuschell M, Zenker A, Allmaier G. Comparison of planar SDS-PAGE, CGE-on-the-chip and MALDI-TOF mass spectrometry for the analysis of the enzymatic de-N-glycosylation of antithrombin III and coagulation factor IX with PNGase F. Anal Bioanal Chem. 2007;389(6):1859–68. doi: 10.1007/s00216-007-1586-3. [DOI] [PubMed] [Google Scholar]
- 19.Garone L, Edmunds T, Hanson E, Bernasconi R, Huntington JA, Meagher JL, et al. Antithrombin-heparin affinity reduced by fucosylation of carbohydrate at asparagine 155. Biochemistry. 1996;35(27):8881–9. doi: 10.1021/bi960542m. [DOI] [PubMed] [Google Scholar]
- 20.Björk I, Ylinenjärvi K, Olson ST, Hermentin P, Conradt HS, Zettlmeissl G. Decreased affinity of recombinant antithrombin for heparin due to increased glycosylation. Biochem J. 1992;286(Pt 3):793–800. doi: 10.1042/bj2860793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olson ST, Frances-Chmura AM, Swanson R, Björk I, Zettlmeissl G. Effect of individual carbohydrate chains of recombinant antithrombin on heparin affinity and on the generation of glycoforms differing in heparin affinity. Arch Biochem Biophys. 1997;341(2):212–21. doi: 10.1006/abbi.1997.9973. [DOI] [PubMed] [Google Scholar]
- 22.Demelbauer UM, Plematl A, Kremser L, Allmaier G, Josic D, Rizzi A. Characterization of glyco isoforms in plasma-derived human antithrombin by online capillary zone electrophoresis-electro-spray ionization-quadrupole ion trap-mass spectrometry of the intact glycoproteins. Electrophoresis. 2004;25(13):2026–32. doi: 10.1002/elps.200305936. [DOI] [PubMed] [Google Scholar]
- 23.Olds RJ, Lane DA, Boisclair M, Sas G, Bock SC, Thein SL. Antithrombin Budapest 3. An antithrombin variant with reduced heparin affinity resulting from the substitution L99F. FEBS Lett. 1992;300(3):241–6. doi: 10.1016/0014-5793(92)80854-a. [DOI] [PubMed] [Google Scholar]
- 24.Lane DA, Olds RJ, Conard J, Boisclair M, Bock SC, Hultin M, et al. Pleiotropic effects of antithrombin strand 1C substitution mutations. J Clin Invest. 1992;90(6):2422–33. doi: 10.1172/JCI116133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan B, Crews BC, Turko IV, Choay J, Zettlmeissl G, Gettins P. Heterogeneity of recombinant human antithrombin III expressed in baby hamster kidney cells. Effect of glycosylation differences on heparin binding and structure. J Biol Chem. 1993;268(23):17588–96. [PubMed] [Google Scholar]
- 26.Pousset D, Piller V, Bureaud N, Monsigny M, Piller F. Increased a2,6 Sialylation of N-Glycans in a Transgenic Mouse Model of Hepatocellular Carcinoma. Cancer Res. 1997;57(19):4249–56. [PubMed] [Google Scholar]
- 27.Schachter H. Biosynthetic controls that determine the branching and microheterogeneity of protein-bound oligosaccharides. Biochem Cell Biol. 1986;64(3):163–81. doi: 10.1139/o86-026. [DOI] [PubMed] [Google Scholar]