

Abstract
Background
Type 2B von Willebrand factor (VWF) is characterized by gain of function mutations in the A1 domain inducing a greater affinity for platelet GPIb, possibly associated with the disappearance of large VWF multimers and thrombocytopenia.
Design and Methods
VWF survival was explored using 1-desamino-8-D-arginine vasopressin (DDAVP) in 18 patients with type 2B von Willebrand disease (VWD) and compared with their platelet count and large VWF multimer representation.
Results
A similarly significant shorter VWF survival, expressed as T1/2elimination (T1/2el), was observed in patients lacking large VWF multimers (type 2B) and in those with a normal multimer pattern (atypical type 2B) (4.47±0.41 h and 4.87±0.9 h, respectively, vs. normal 15.53±2.17 h) due mainly to a greater VWF clearance. The half-life of large VWF multimers, explored by VWF collagen binding (VWF:CB) activity, was likewise reduced. The similarly reduced VWF half-life was also confirmed by the increase in the VWF propeptide ratio (a useful tool for exploring VWF survival) which was found to be the same in type 2B and atypical type 2B patients. The post-DDAVP drop in platelet count occurred in all patients lacking large multimers but not in those with a normal multimer pattern. A correlation was always found between pre- and/or post-DDAVP thrombocytopenia and the lack of large VWF multimers in type 2B VWD while these were unrelated to the reduced VWF half-life.
Conclusions
In addition to demonstrating that a shorter VWF survival contributes to the type 2B and atypical type 2B VWD phenotype, our findings suggest that VWF clearance and proteolysis are independent phenomena.
Keywords: von Willebrand factor, type 2B VWD, VWF half-life, type 2B thrombocytopenia, large VWF multimers
Introduction
Von Willebrand factor (VWF) is a multimeric glycoprotein consisting of linear arrangements of disulfide-linked 275 kDa subunits, ranging from 0.5 to more than 10 million Dalton in size.1 The mature VWF subunit contains functional domains, including the A1 domain that is the VWF binding site for platelet GPIb.2 High molecular weight multimers contain many platelet-binding sites and are also recognized as being biologically the most active.3 VWF multimer size is regulated by the specific cleaving protease ADAMTS134 whose proteolytic action gives each VWF oligomer its typical triplet structure.5 ADAMTS13 deficiency gives rise to ultralarge VWF multimers in the circulation which are responsible for thrombotic thrombocytopenic purpura,6 whereas an increased VWF proteolysis leads to the loss of large VWF multimers.7
Reduced VWF levels and/or anomalies cause von Willebrand disease (VWD), one of the most common inherited bleeding disorders.1,8 According to the current classification of VWD, types 1 and 3 are quantitative VWF defects while type 2 refers to abnormal VWF functions.9 Type 2B is an unusual variant of VWD, characterized by an enhanced platelet GPIb and VWF interaction10,11 due to gain of function mutations in the A1 domain of VWF,12,13 one of the consequences of which is the disappearance of large VWF multimers from the plasma due to their spontaneous binding to platelet GPIb. Large multimers are synthesized normally, as confirmed by the normal platelet VWF content seen in type 2B VWD, but currently cannot be detected in vivo for their removal. Enhanced ristocetin-induced platelet aggregation (RIPA), spontaneous platelet aggregation (SPA)14–16 and sometimes thrombocytopenia are also part of the type 2B VWD phenotype.17 Thrombocytopenia may be persistent or transient but is commonly worse after DDAVP infusion, pregnancy, exercise or surgery.17–21 Giant platelets may also be associated with thrombocytopenia. Variants of type 2B VWD have been described, however, that have a normal multimer pattern in spite of an enhanced GPIb-VWF interaction, with no thrombocytopenia.16,22,23
Mean plasma VWF concentrations depend partly on the balance between the release of VWF from endothelial cells and its removal from the circulation. Many factors affect VWF survival, such as ABO blood group and mutations in specific VWF domains.24–26 Healthy individuals with the O blood group have a shorter VWF survival than those with other blood groups.24 VWF mutations, such as R1205H and C1130F in the D3 domain, are also associated with a shorter VWF half-life.25–27
Here, we investigated the survival of type 2B VWF after DDAVP with a view to clarifying the contribution made by the disappearance of large VWF multimers.
Design and Methods
Patients and healthy subjects were enrolled for the study after obtaining their written informed consent in accordance with the Helsinki Declaration and the study was approved by the University of Padua’s institutional review board.
Blood was drawn from the antecubital vein and anticoagulated using 3.8% sodium citrate (1:10, vol/vol); samples for platelet preparations also contained 50 mM EDTA, 50 IU/mL Trasylol, 10 mM leupeptin, and 60 mM N-ethylmaleimide as protease inhibitors. The platelet-rich and platelet-poor plasma was prepared as described elsewhere.28 For platelet VWF:Ag assay, platelet-rich plasma was sedimented at 12,000 x g for 1 min and resuspended in phosphate-buffered saline (PBS) containing 3% EDTA. The procedure was repeated three times; platelets were finally adjusted to a count of 106/μL in PBS containing protease inhibitors and were lysed by adding 1% Triton X-100.
RIPA was determined at ristocetin concentrations from 1.2 mg/mL to 0.3 mg/mL.28 VWF ristocetin cofactor activity (VWF:RCo) was assessed with normal washed, formalin-fixed platelets and 1.0 mg/mL ristocetin in a Chronolog aggregometer.28 Plasma and platelet VWF antigen (VWF:Ag) was obtained with a home-made enzyme-linked immunosorbent assay (ELISA), using a horseradish peroxidase (HRP)-conjugated anti-VWF antibody (Dako, Netherlands). VWF collagen binding (VWF:CB) activity was assessed by ELISA using type III collagen (Sigma, Milan, Italy) diluted in acetic acid. Factor VIII (FVIII) coagulant was measured using a one-stage method, with cephaloplastin as activated cephalin. The above FVIII and VWF assays were conducted using a pool of normal plasma samples for reference; for platelet VWF:Ag the reference curve was constructed using a pool of washed normal platelets, taking the first dilution as 100 U/dL. VWF propeptide (VWFpp) was determined using an ELISA (GTI Diagnostics, Waukesha, WI, USA). Briefly, prediluted calibrators and diluted plasma samples were added to microwells coated with monoclonal antibodies specific for VWFpp; bound VWFpp was assessed with biotinylated anti-VWFpp monoclonal antibody and streptavidin-labeled HRP. The results are given in U/dL taking the first reference curve dilution as 100 U/dL. VWF multimer analysis was performed on high gelling temperature agarose containing 0.1% sodium dodecyl sulphate (SDS), and using 1.2% and 2.2% agarose to obtain a low and high resolution, respectively.28 Multimers were detected by autoradiography after reaction with a purified anti-VWF 125I-labeled antibody.
DDAVP (1-desamino-8-D-arginine vasopressin, Emosint, Sclavo, Italy) was administered subcutaneously at a dose of 0.3 μg/kg. Blood samples were collected before and then 15, 30, 60, 120, 180, 240, 360 and 480 min, and 24 hours after administering DDAVP. The time courses of the VWF:Ag and VWF:CB plasma concentrations after DDAVP were analyzed using a one-compartment model with first-order input and output kinetics, including baseline concentrations, B and a time lag between DDAVP administration and the increase in plasma concentration, t’, as follows:
where A is the Y-axis intercept, Kre is the release rate constant, Kel is the elimination rate constant, and t is the time. The model was fitted to each set of concentration-time data using the Prism statistical package (GraphPad Software, San Diego, CA, USA). The goodness of fit was evaluated by r2. The elimination half-live (T1/2el) was calculated using the standard formula, i.e. T1/2el=0.693/kel.
Using this kinetic model,29 the amount of VWF:Ag released by DDAVP (Q) is: Q=A × VD × (Kre-Kel)/Kre, where VD is the volume of VWF:Ag distribution. Likewise, plasma clearance (CL) is: CL = Kel × VD and the baseline rate of VWF:Ag release (Vre) is: Vre = B × CL. The VD of VWF could not be calculated from our data, so the VD reported by Menache et al.30 after intravenous VWF administration (40 mL/kg) was considered to obtain an approximate estimate of Q, CL and Vre in our patients. The kinetic parameters were compared with those obtained previously in a group of normal subjects.24
Human genomic DNA was extracted from peripheral blood leukocytes using the QIAmp DNA blood Mini Kit (QIAGEN, Hilden, Germany). VWF cDNA nucleotides were numbered in agreement with the latest recommendations of the ISTH Scientific Subcommittee on VWD, establishing the “A” of the initiation codon ATG as +1.31
Laboratory data and pharmacokinetic parameters were expressed as means ± standard errors (SE).
Student’s t-test was used to compare the pharmacokinetic parameters of type 2B and atypical type 2B VWF patients with those of normal subjects. P values below 0.05 were considered statistically significant. Welch’s correction was applied when variances were not equal.
Results
Patients
Fifteen type 2B patients (from 7 unrelated families) and 3 atypical type 2B VWD patients (from 2 unrelated families) were considered. Forty-seven healthy subjects served as controls. The criteria for diagnosing type 2B VWD were: a greater interaction between VWF and platelet GPIb as documented by lower minimal aggregating doses of ristocetin (MADR), the presence of mutations in the A1 domain of VWF, and the absence of large VWF multimers. Atypical type 2B VWD was defined as being characterized by hyper-responsiveness to ristocetin, mutations in the A1 domain, and a full complement of VWF multimers. Thirteen of the type 2B patients carried the R1308C mutation; one had the V1316M mutation and one the R1306W mutation. Among the atypical type 2B VWD cases, one patient was carrying the P1266L mutation (previously called type New York/type Malmo VWD) and 2 had the R1379C mutation.
Hemostatic findings
Patients’ main hemostatic findings are shown in Table 1. MADR ranged between 0.3 and 0.5 mg/mL ristocetin (vs. normal ≥1.0 mg/mL) in both type 2B and atypical type 2B VWD; SPA was seen, to different degrees, in all patients. Five type 2B VWD patients had thrombocytopenia, while the platelet count was normal in the atypical type 2B patients. Type 2B VWD patients had lower plasma VWF:Ag levels associated with a pronounced decrease in VWF:CB (mean VWF:CB/VWF:Ag ratio 0.18±0.02 vs. normal 1.12±0.06), while an intermediate decrease was seen for VWF:RCo (VWF:RCo/VWF:Ag ratio was 0.48±0.02 vs. normal 0.98±0.17) (Table 1). Atypical type 2B VWD cases had lower VWF:Ag, VWF:CB and VWF:RCo levels, but normal VWF:CB and VWF:RCo ratios (0.85±0.07 and 0.86±0.22, respectively). All type 2B VWD patients had a normal platelet VWF content, while this was lower in atypical type 2B VWD (Table 1).
Table 1.
Patients’ main hemostatic findings.

The plasma VWF multimer pattern was normal in atypical type 2B patients, while the large and intermediate VWF multimers were lacking in type 2B VWD (Figure 1A). High-resolution gel (2.2% agarose) revealed a greater proportion of small type 2B VWF multimers than in normal individuals, with a more pronounced representation of the satellite bands of each oligomer; no difference in VWF oligomer pattern was observed in atypical type 2B patients (Figure 1B).
Figure 1.
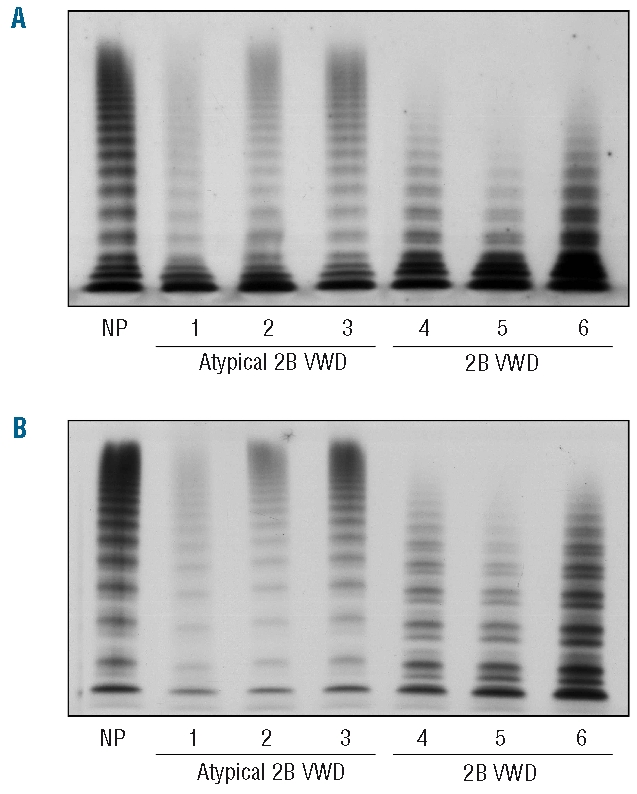
Plasma VWF multimer pattern observed in atypical type 2B and 2B VWD patients compared with normal plasma (NP). Electrophoresis was performed using 1.2% (A) and 2.2% (B) agarose gel containing 0.1% SDS. Multimers were detected using a 125I-labeled anti-VWF antibodies. Large VWF multimers are at the top, small multimers at the bottom.
Post-DDAVP hemostatic findings
DDAVP induced a dramatic drop in platelet count in type 2B VWD patients: a sudden decrease was seen after 15 min and the lowest platelet count was reached 30 min after DDAVP; platelets started to increase again after 120 min and had increased significantly after 360 min (Figure 2A). On the other hand, there were no changes in platelet count in patients with atypical type 2B VWD (Figure 2B). No large VWF multimers appeared in type 2B after DDAVP, while small and intermediate multimers and satellite bands were relatively more represented than before DDAVP (Figure 2A). Conversely, a significant increase in all VWF multimers was observed in atypical type 2B patients, with ultralarge VWF forms typical of a normal response to DDAVP being observed (Figure 2B). There were no adverse clinical effects after DDAVP in the patients studied.
Figure 2.
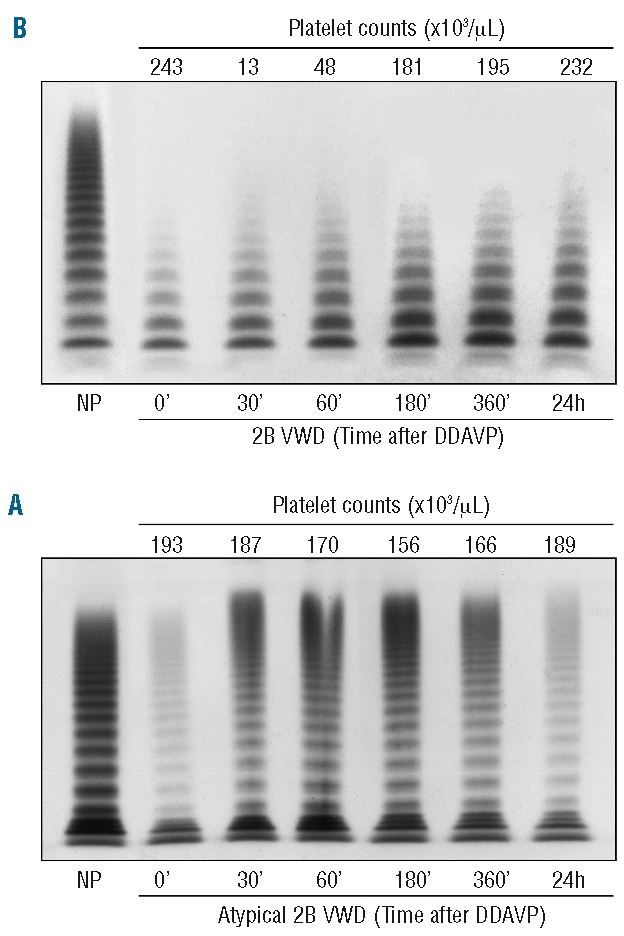
Time course of VWF multimers before and after DDAVP as observed in type 2B (A) and atypical type 2B (B) VWD patients. The analysis was performed using a 1.2% agarose gel electrophoresis. Similar findings were recorded in the other patients studied.
VWF pharmacokinetic parameters
The mean time courses of post-DDAVP VWF:Ag and VWF:CB plasma concentrations in type 2B VWD, atypical type 2B VWD, and normal subjects are shown in Figure 3. The associated pharmacokinetic parameters exploring VWF release (Q, Vre) and survival (T1/2el, CL) are shown in Table 2.
Figure 3.
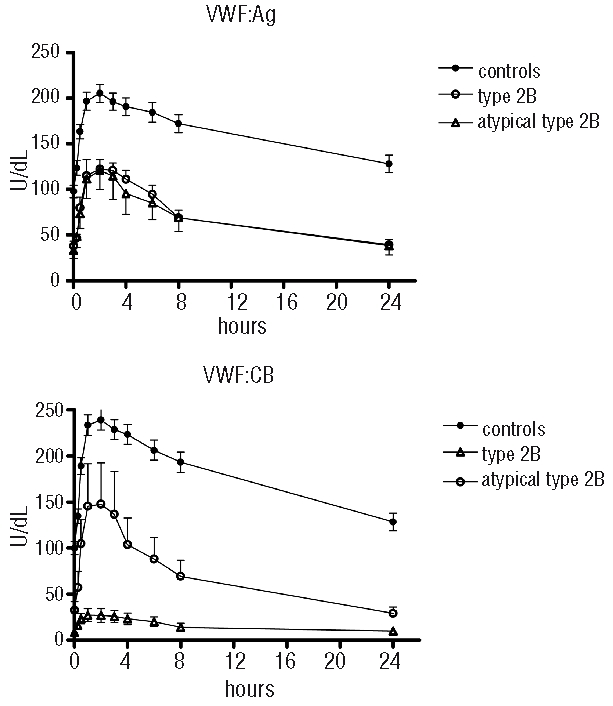
Mean VWF:Ag and VWF:CB survival observed in normal controls and in type 2B and atypical type 2B VWD patients.
Table 2.
Main VWF pharmacokinetic parameters observed in the VWD patients studied.

The amount of VWF:Ag released by DDAVP (Q) did not differ significantly between type 2B, atypical type 2B and normal cases (49.9±3.2 U/h/kg, 43.6±7.7 U/h/kg, and 48.5±2.5 U/h/kg, respectively), but the Q of VWF:CB was much lower in type 2B VWD patients (12.3±2.5 U/h/kg vs. normal 66.6±3.5 U/h/kg (Table 2). The rate of release (Vre) of VWF:Ag under steady state conditions was normal in type 2B and atypical type 2B patients (Table 2) suggestive of a normal release from endothelial cells, whereas the baseline release of VWF:CB was much slower in type 2B patients (Vre: 0.52±0.07 U/h/kg vs. normal 2.82±0.23 U/h/kg) (Table 2).
As for VWF survival, VWF:Ag T1/2el was shorter in the VWD patients (4.47±0.41 h in type 2B and 4.87±0.99 h in atypical type 2B) than in the normal controls (15.53±2.17 h, P<0.0001). VWF:CB half-life was also significantly reduced in type 2B and atypical type 2B patients (4.03±0.75 h and 4.23±1.22 h, respectively, vs. 12.67±1.88 h in controls). In both instances, the shorter T1/2el was due to a greater clearance of VWF:Ag and VWF:CB which was much the same in type 2B and atypical type 2B VWD patients (Table 2).
VWFpp and VWFpp ratio
Further confirmation of a shorter type 2B VWF survival emerged on measuring the VWFpp and its ratio to VWF:Ag (VWFpp ratio). Mean VWFpp was normal in type 2B (111.0±13.4 U/dL vs. 101.8±0.1 U/dL in controls) but reduced in atypical type 2B VWD (46.3±5.2 U/dL). Much the same increase in VWFpp ratio was observed in the patients investigated: 2.62±0.14 in type 2B and 2.26±0.06 in atypical type 2B VWD vs. normal controls 1.41±0.07, P<0.0001.
Discussion
The survival of type 2B and atypical type 2B VWF was explored using DDAVP, mainly evaluating any relationship with the shortage of large VWF multimers and thrombocytopenia. A similarly reduced VWF half-life was identified in both types of VWD due entirely to an increased VWF clearance, irrespective of the proportion of large multimers or pre- and/or post-DDAVP thrombocytopenia. There was always a correlation between the disappearance of large VWF multimers and thrombocytopenia, while these were unrelated to VWF half-life.
Type 2B VWD represents a paradoxical hemostatic defect in which a greater affinity of the mutated VWF for platelet GPIb is responsible for bleeding rather than thrombotic symptoms.11 All the type 2B mutations reported to date have occurred in the A1 domain of VWF. Most of them are associated with a shortage of large VWF multimers (type 2B VWD) but some have a full complement of large VWF multimers.22,23,32 For the purpose of the study, and to emphasize the difference from and similarity to classic type 2B, these latter patients are identified as cases of atypical type 2B VWD.
The absence of circulating large VWF multimers in type 2B VWD is thought to be due to their removal as a result of the spontaneous binding of mutated VWF to platelet GPIb.11 Based on these findings, we advanced a working hypothesis that the half-life of 2B VWF is shorter than normal. Indeed, our data confirm a shorter survival of both type 2B and atypical type 2B VWF irrespective of large VWF multimer representation. The phenomenon appeared to depend on a similar increase in VWF:Ag and VWF:CB elimination rate (with a rise in CL and a decline in T1/2el) in both type 2B VWD subtypes, suggesting that large and small multimers are removed from the circulation at much the same rate. This confirms the findings already reported in a murine model.33
It is generally accepted that a molecule’s clearance is the result of its receptor-mediated removal or proteolysis, so theoretically three mechanisms may be advanced to explain the removal of VWF from the circulation: i) ADAMTS13-mediated proteolysis; ii) binding to platelet receptor, which is increased in both 2B subtypes; and iii) receptor-mediated liver uptake. We know that proteolysis has to be involved in the type 2B VWF picture because the absence of large multimers is associated with an increase in proteolytic fragments,34 mainly faster and slower bands of each oligomer organized as a triplet, as confirmed by the high-resolution multimers in our patients. Since the oligomer structure of VWF is due to the action of ADAMTS13,5 an increased representation of fragments is the result of a greater proteolytic action on 2B VWF. Atypical type 2B VWD behaves differently, however, given that the triplet organization of the VWF oligomers is not modified and the large multimers do not disappear. The fact that the mean clearance of VWF:Ag and VWF:CB was comparable in type 2B and atypical type 2B cases would rule out the possibility of ADAMTS13 activity inducing an accelerated VWF elimination. Similarly, if VWF removal were to occur via a greater platelet binding of the high molecular weight multimers, we ought to find a loss of large multimers in both 2B and atypical type 2B VWD, but this was not the case. The hypothesis of an increased liver uptake is consequently the most likely and, whatever the mechanisms involved, 2B and atypical type 2B mutations in the A1 domain are associated with a faster removal of VWF from the circulation.
The phase of VWF release from endothelial cells, that we explored by means of Q and Vre kinetic parameters, revealed a similarly normal behavior in type 2B and atypical type 2B VWD, while VWF:CB (used as a marker of large VWF multimer representation) was characterized by abnormalities in type 2B cases. Although post-DDAVP VWF:Ag release (Q) was normal in type 2B, the amount of VWF:CB released was considerably reduced. This divergent behavior may be explained by large VWF multimers being converted by ADAMTS13 into small multimers soon after their release without reducing the overall number of molecules, so that the total amount of VWF:Ag released remains the same. This would also explain why large multimers are never detectable in the circulation in cases of type 2B VWD. A rapid proteolysis during steady-state VWF release may also justify the decrease in the continuous release (Vre) of VWF:CB observed in type 2B patients.
Thrombocytopenia may complicate type 2B VWD, even though it is not a constant feature of the disorder, neither in a given individual, nor in patients from the same family or patients from different families carrying the same mutation.17,18, 35 Thrombocytopenia has been said to be due to the enhanced affinity of mutated VWF for platelet GPIb, which in turn triggers platelet aggregation due to the presence of hemostatically more efficient DDAVP-released VWF multimers.36 A greater interaction between mutant VWF and platelet GPIb is not enough, however, to justify the onset of thrombocytopenia and the associated disappearance of large VWF multimers, as seen in atypical type 2B VWD. In fact, no decline in platelet count before or after DDAVP was observed in the atypical type 2B VWD patients studied, while there was a marked drop in all type 2B VWD patients after DDAVP, even if this was not apparent under basal conditions. This post-DDAVP type 2B thrombocytopenia is not the result of a true platelet consumption, however, because the platelet count almost returns to the baseline within a few hours after DDAVP, confirming previous reports.17,18 After DDAVP, the transient fall in platelet count was paralleled by a shift from large to small type 2B VWF multimers with a consequent relatively stronger representation of the small forms and oligomer satellite bands. We can, therefore, infer that an enhanced VWF binding to platelet GPIb receptor, a transient drop in platelets and ADAMTS13 activation are pathogenically linked. It has been demonstrated36–39 that the platelet-VWF complexes activate ADAMTS13 under high shear stress conditions, and the latter cleaves the mutant VWF and dissolves the platelet aggregates, with the consequent disappearance of large VWF multimers. In type 2B VWD, the reduced platelet count may thus represent the prevalence of platelet aggregate formation over their ADAMTS13-dependent dissolution. The role of platelets in the loss of large VWF multimers is elegantly confirmed by a recent observation of the restoration of large VWF multimers in a patient with type 2B VWD after the onset of a severe immune thrombocytopenia.40
Finally, the shorter survival that we observed in type 2B and atypical type 2B VWD is not as severe as in type Vicenza VWD or type 1 VWD patients carrying the C1130F mutation, even though it is severe enough to raise the VWFpp ratio (recently proposed as a tool for investigating VWF survival).25,27 The VWFpp ratio ranges between 2 and 3 in type 2B and atypical type 2B VWD patients, with no significant differences emerging between the two forms. This confirms their comparable VWF half-life, but also means that the VWFpp ratio cannot be used to distinguish type 2B from atypical type 2B VWD.
In conclusion, a greater VWF clearance seems to contribute to the type 2B and atypical type 2B VWD phenotypes, down-regulating the circulating VWF levels, irrespective of large VWF multimers representation. A prolonged or abnormal VWF binding to platelet GPIb in type 2B VWD may give ADAMTS13 the chance to deplete the large VWF multimers, while this is not the case in atypical type 2B VWD. What causes bleeding in atypical type 2B VWD is still not clear. It also remains to be seen whether types 2B and atypical type 2B VWD should be considered as one and the same defect, or as distinct abnormalities.
Footnotes
Funding: this work was supported by grants from the Telethon Foundation and MURST 2006 (Rome, Italy). We thank Dr. Francesca Sartorello for her contribution to the patients’ genetic characterization. We are also grateful to Frances Coburn for editing the text.
Authorship and Disclosures
AC designed the research, analyzed the data and wrote the paper; LG monitored DDAVP administration and performed the genetic characterization; MGC took part in the hemostatic analysis; EP performed the multimeric analysis and VWF propeptide assays; RP performed the pharmacokinetic analysis and wrote the paper; AB monitored DDAVP administration; VD performed the statistical analysis; AP organized the study and discussed the results.
The authors reported no potential conflicts of interest.
References
- 1.Sadler JE. Biochemistry and genetics of von Willebrand factor. Ann Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 2.Ruggeri ZM. Von Willebrand factor. Curr Opin Hematol. 2003;10(2):142–9. doi: 10.1097/00062752-200303000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Gralnick HR, Williams SB, Morisato DK. Effect of multimeric structure of the factor VIII/von Willebrand factor protein on binding to platelets. Blood. 1981;58(2):387–97. [PubMed] [Google Scholar]
- 4.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276(44):41059–63. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 5.Dent JA, Galbusera M, Ruggeri ZM. Heterogeneity of plasma von Willebrand factor multimers resulting from proteolysis of the constituent subunit. J Clin Invest. 1991;88(33):774–82. doi: 10.1172/JCI115376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347(8):589–600. doi: 10.1056/NEJMra020528. [DOI] [PubMed] [Google Scholar]
- 7.Dent JA, Berkowitz SD, Ware J, Kasper CK, Ruggeri ZM. Identification of a cleavage site directing the immunochemical detection of molecular abnormalities in type IIA von Willebrand factor. Proc Natl Acad Sci USA. 1990;87(16):6306–10. doi: 10.1073/pnas.87.16.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruggeri ZM, Zimmerman TS. Von Willebrand factor and von Willebrand disease. Blood. 1987;70(4):895–904. [PubMed] [Google Scholar]
- 9.Sadler JE, Budde U, Eikenboom CJ, Favaloro EJ, Hill FG, Holmberg L, et al. Working Party on von Willebrand disease classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4(10):2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 10.Ruggeri ZM, Pareti FI, Mannucci PM, Ciavarella N, Zimmerman TS. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. N Engl J Med. 1980;302(19):1047–51. doi: 10.1056/NEJM198005083021902. [DOI] [PubMed] [Google Scholar]
- 11.Ruggeri ZM. Type IIB von Willebrand disease: a paradox explains how von Willebrand factor works. J Thromb Haemost. 2004;2(1):2–6. doi: 10.1111/j.1538-7836.2003.00523.x. [DOI] [PubMed] [Google Scholar]
- 12.Randi AM, Rabinowitz I, Mancuso DJ, Mannucci PM, Sadler JE. Molecular basis of von Willebrand disease type IIB. Candidate mutations cluster in one disulfide loop between proposed platelet glycoprotein Ib binding sequences. J Clin Invest. 1991;87(4):1220–6. doi: 10.1172/JCI115122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rendal E, Penas N, Larrabeiti B, Pérez A, Vale A, Lopez-Fernandez MF, et al. Type 2B von Willebrand’s disease due to Val1316Met mutation. Heterogeneity in the same sibship. Ann Hematol. 2001;80(6):354–60. doi: 10.1007/s002770100303. [DOI] [PubMed] [Google Scholar]
- 14.Gralnick HR, Williams SB, McKeown LP, Rick ME, Maisonneuve P, Jenneau C, et al. Von Willebrand’s disease with spontaneous platelet aggregation induced by an abnormal plasma von Willebrand factor. J Clin Invest. 1985;76(4):1522–9. doi: 10.1172/JCI112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Marco L, Mazzucato M, Grazia Del Ben M, Budde U, Federici AB, Girolami A, et al. Type IIB von Willebrand factor with normal sialic acid content induces platelet aggregation in the absence of ristocetin. Role of platelet activation, fibrinogen, and two distinct membrane receptors. J Clin Invest. 1987;80(2):475–82. doi: 10.1172/JCI113095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss HJ, Sussman II. A new von Willebrand factor variant (type I, New York): increased ristocetin-induced platelet aggregation and plasma von Willebrand factor containing the full complement of multimers. Blood. 1986;68(1):149–56. [PubMed] [Google Scholar]
- 17.Casonato A, Sartori MT, De Marco L, Girolami A. 1-Desamino-8-D-arginine vasopressin (DDAVP) infusion in type IIB von Willebrand's disease: shortening of bleeding time and induction of a variable pseudothrombocytopenia. Thromb Haemost. 1990;64(1):117–20. [PubMed] [Google Scholar]
- 18.Casonato A, Steffan A, Pontara E, Zucchetto A, Rossi C, De Marco L, et al. Post-DDAVP thrombocytopenia in type 2B von Willebrand disease is not associated with platelet consumption: failure to demonstrate glycocalicin increase or platelet activation. Thromb Haemost. 1999;81(2):224–8. [PubMed] [Google Scholar]
- 19.Casonato A, Sartori MT, Bertomoro A, Fede T, Vasoin F, Girolami A. Pregnancy-induced worsening of thrombocytopenia in a patient with type IIB von Willebrand’s disease. Blood Coagul Fibrinolysis. 1991;2(1):33–40. doi: 10.1097/00001721-199102000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Hultin MB, Sussman II. Postoperative thrombocytopenia in type IIB von Willebrand disease. Am J Hematol. 1990;33(1):64–8. doi: 10.1002/ajh.2830330113. [DOI] [PubMed] [Google Scholar]
- 21.Stakiw J, Bowman M, Hegadorn C, Pruss C, Notley C, Groot E, et al. The effect of exercise on von Willebrand factor and ADAMTS-13 in individuals with type 1 and type 2B von Willebrand disease. J Thromb Haemost. 2007;6(1):90–6. doi: 10.1111/j.1538-7836.2007.02790.x. [DOI] [PubMed] [Google Scholar]
- 22.Holmberg L, Berntorp E, Donner M, Nilsson IM. Von Willebrand’s disease characterized by increased ristocetin sensitivity and the presence of all von Willebrand factor multimers in plasma. Blood. 1986;68(3):668–72. [PubMed] [Google Scholar]
- 23.Casonato A, Sartorello F, Pontara E, Gallinaro L, Bertomoro A, Grazia Cattini M, et al. A novel von Willebrand factor mutation (I1372S) associated with type 2B-like von Willebrand disease: an elusive phenotype and a difficult diagnosis. Thromb Haemost. 2007;98(6):1182–7. doi: 10.1160/th07-05-0347. [DOI] [PubMed] [Google Scholar]
- 24.Gallinaro L, Cattini MG, Sztukowska M, Padrini R, Sartorello F, Pontara E, et al. A shorter von Willebrand factor survival in O blood group subjects explains how ABO determinants influence plasma von Willebrand factor. Blood. 2008;111(7):3540–5. doi: 10.1182/blood-2007-11-122945. [DOI] [PubMed] [Google Scholar]
- 25.Casonato A, Pontara E, Sartorello F, Cattini MG, Sartori MT, Padrini R, et al. Reduced von Willebrand factor survival in type Vicenza von Willebrand disease. Blood. 2002;99(1):180–4. doi: 10.1182/blood.v99.1.180. [DOI] [PubMed] [Google Scholar]
- 26.Sztukowska M, Gallinaro L, Cattini MG, Pontara E, Sartorello F, Daidone V, et al. Von Willebrand factor propeptide makes it easy to identify the shorter von Willebrand factor survival in patients with type 1 and type Vicenza von Willebrand disease. Brit J Haematol. 2008;143(1):107–14. doi: 10.1111/j.1365-2141.2008.07311.x. [DOI] [PubMed] [Google Scholar]
- 27.Haberichter SL, Castaman G, Budde U, Peake I, Goodeve A, Rodeghiero F, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD) Blood. 2008;111(10):4979–85. doi: 10.1182/blood-2007-09-110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casonato A, De Marco L, Mazzuccato M, De Angelis V, De Roia D, Fabris F, et al. A new congenital platelet abnormality characterized by spontaneous platelet aggregation, enhanced von Willebrand factor platelet interaction, and the presence of all von Willebrand factor multimers in plasma. Blood. 1989;74(6):2028–33. [PubMed] [Google Scholar]
- 29.Gibaldi M, Perrier D. Pharmacokinetics. New York, NY: Marcel Dekker; 1975. [Google Scholar]
- 30.Menache D, Aronson DL, Darr F, Montgomery RR, Gill JC, Kessler CM, et al. Pharmacokinetics of von Willebrand factor and factor VIIIC in patients with severe von Willebrand disease (type 3VWD): estimation of the rate of factor VIIIC synthesis. Cooperative Study. Br J Haematol. 1996;94(4):740–5. doi: 10.1046/j.1365-2141.1996.d01-1860.x. [DOI] [PubMed] [Google Scholar]
- 31.Goodeve AC, Eikenboom JCJ, Ginsburg D, Hilbert L, Mazurier C, Peake IR, et al. A standard nomenclature for von Willebrand factor gene mutations and polymorphisms. On behalf of the ISTH SSC Subcommittee on von Willebrand factor. Thromb Haemost. 2001;85(5):929–31. [PubMed] [Google Scholar]
- 32.Othman M, Favaloro EJ. Genetics of type 2B von Willebrand disease: “true 2B,” “tricky 2B,” or “not 2B.” What are the modifiers of the phenotype? Sem Thromb Hemost. 2008;34(6):520–31. doi: 10.1055/s-0028-1103363. [DOI] [PubMed] [Google Scholar]
- 33.Lenting PJ, Westein E, Terraube V, Ribba AS, Huizinga EG, Meyer D, et al. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J Biol Chem. 2004;279(13):12102–9. doi: 10.1074/jbc.M310436200. [DOI] [PubMed] [Google Scholar]
- 34.Zimmerman TS, Dent JA, Ruggeri ZM, Nannini LH. Subunit composition of plasma von Willebrand factor. Cleavage is present in normal individuals, increased in IIA and IIB von Willebrand disease, but minimal in variants with aberrant structure of individual oligomers (types IIC, IID, and IIE) J Clin Invest. 1986;77(3):947–51. doi: 10.1172/JCI112394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Federici AB, Mannucci PM, Castaman G, Baronciani L, Bucciarelli P, Canciani MT, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort sturdy of 67 patients. Blood. 2009;113(3):526–34. doi: 10.1182/blood-2008-04-152280. [DOI] [PubMed] [Google Scholar]
- 36.Shim K, Anderson PJ, Tuley EA, Wiswall E, Sadler JE. Platelet-VWF complexes are preferred substrate of ADAMTS13 under fluid shear stress. Blood. 2008;111(2):651–7. doi: 10.1182/blood-2007-05-093021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rayes J, Hommais A, Legendre P, Tout H, Veyradier A, Obert B, et al. Effect of von Willebrand disease type 2B and type 2M mutations on the susceptibility of von Willebrand to ADAMTS-13. J Thromb Haemost. 2006;5(2):321–8. doi: 10.1111/j.1538-7836.2007.02296.x. [DOI] [PubMed] [Google Scholar]
- 38.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–9. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 39.Nishio K, Anderson PJ, Zheng XL, Sadler JE. Binding of platelet glycoprotein Ibalpha to von Willebrand factor domain A1 stimulates the cleavage of the adjacent domain A2 by ADAMTS13. Proc Natl Acad Sci USA. 2004;101(29):10578–83. doi: 10.1073/pnas.0402041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang ZJ, Onwuzurike N, Callaghan MU, Rajpurkar M, Chitlur M, Lusher JM. Decreased clearance of von Willebrand factor in a patient with type 2B von Willebrand disease following development of immune thrombocytopenia. Pediatr Blood Cancer. 2008;51(3):416–8. doi: 10.1002/pbc.21593. [DOI] [PubMed] [Google Scholar]