

Abstract
Background and purpose:
Excessive production of nitric oxide (NO) by inducible NO synthase (iNOS) is thought to underlie the vascular dysfunction, systemic hypotension and organ failure that characterize endotoxic shock. Plasma levels of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and C-type natriuretic peptide (CNP) are raised in animal models and humans with endotoxic shock and correlate with the associated cardiovascular dysfunction. Since both NO and natriuretic peptides play important roles in cardiovascular homeostasis via activation of guanylate cyclase-linked receptors, we used mice lacking natriuretic peptide receptor (NPR)-A (NPR1) to establish if natriuretic peptides contribute to the cardiovascular dysfunction present in endotoxic shock.
Experimental approach:
Wild-type (WT) and NPR-A knockout (KO) mice were exposed to lipopolysaccharide (LPS) and vascular dysfunction (in vitro and in vivo), production of pro-inflammatory cytokines, and iNOS expression and activity were evaluated.
Key results:
LPS-treated WT animals exhibited a marked fall in mean arterial blood pressure (MABP) whereas NPR-A KO mice maintained MABP throughout. LPS administration caused a greater suppression of vascular responses to the thromboxane-mimetic U46619, ANP, acetylcholine and the NO-donor spermine-NONOate in WT versus NPR-A KO mice. This differential effect on vascular function was paralleled by reduced pro-inflammatory cytokine production, iNOS expression and activity (plasma [NOx] and cyclic GMP).
Conclusions and implications:
These observations suggest that NPR-A activation by natriuretic peptides facilitates iNOS expression and contributes to the vascular dysfunction characteristic of endotoxic shock. Pharmacological interventions that target the natriuretic peptide system may represent a novel approach to treat this life-threatening condition.
Keywords: atrial natriuretic peptide, inducible nitric oxide synthase, cyclic GMP, endotoxaemia, hypotension, vascular smooth muscle
Introduction
Endotoxic shock is a systemic inflammatory syndrome in which vascular smooth muscle and endothelial dysfunction contribute to a progressive failure of the circulation to provide blood to vital organs with consequent hypotension, multiple organ failure and death (Hotchkiss and Karl, 2003). The condition remains a major cause of death in intensive care patients with 30–60% of individuals failing to recover (Singer, 2006). Whilst considerable progress has been made in understanding the molecular and cellular mechanisms underlying the pathogenesis of endotoxic shock, little advance has been achieved in terms of therapy (Singer, 2006). Thus, identification and characterization of novel pathways contributing to disease progression could prove significant in reducing the associated mortality and morbidity.
Animal models and patient studies have confirmed a central role for excessive nitric oxide (NO) production by inducible nitric oxide synthase (iNOS) in the development of endotoxic shock. Under physiological conditions iNOS is effectively absent; however, in response to pro-inflammatory cytokines and bacterial lipopolysaccharide (LPS, endotoxin), iNOS is expressed in several cell types resulting in a ‘high-output’ NO production. Although NO is essential for combating infection, excessive NO production during inflammatory episodes results in NO-mediated host damage (MacMicking et al., 1997). In particular, endothelial and smooth muscle dysfunction result in a dramatic decrease in blood pressure due to increased NO-mediated dilatation and hyporeactivity to catecholamines (Julou-Schaeffer et al., 1990; Fleming et al., 1992). In accord, selective iNOS inhibitors restore blood pressure and reduce mortality in experimental animal models of sepsis (Kilbourn et al., 1990; Petros et al., 1994; Rees et al., 1998) and iNOS knockout (KO) mice are resistant to LPS-induced vascular dysfunction (Hollenberg et al., 2000; Chauhan et al., 2003). A role for iNOS in the pathogenesis of endotoxic shock has also been established in humans. For instance, total plasma nitrite (NO2-) and nitrate (NO3-; NOx) levels (an index of NO production) and iNOS expression are significantly elevated in endotoxic patients compared to healthy individuals (Ochoa et al., 1991; Annane et al., 2000). Despite these animal and human data advocating the use of iNOS inhibitors in the treatment of endotoxic shock, clinical trials with the non-selective NOS inhibitor NG-methyl-L-arginine (L-NMA) have shown no beneficial effect on survival (Lopez et al., 2004). Therefore, new therapeutic targets that can reverse the cardiovascular dysfunction associated with endotoxic shock may offer a valuable alternative.
Atrial natriuretic peptide (ANP), a key cardiovascular homeostatic hormone (Ahluwalia et al., 2004), plays a role in regulating inflammation and modulates iNOS expression in a number of cell types in vitro(Marumo et al., 1995; Vollmar and Schulz, 1995). Several studies have alluded to a potential role for natriuretic peptides (particularly ANP and B-type natriuretic peptide, BNP) in the pathogenesis of endotoxic shock, although a definitive function has yet to be established. Intravenous injection of LPS in rodents causes a rise in plasma ANP concentrations and a reduction in plasma volume (Aiura et al., 1995). This is also true in humans with endotoxic shock, in whom plasma ANP and BNP levels are significantly elevated (Witthaut et al., 2003). Furthermore, ANP and BNP concentrations in the circulation are increased in cardiovascular diseases such as heart failure, which is a common complication of sepsis (Potter et al., 2006). Indeed, in an experimental model of ovine sepsis, selective blockade of the natriuretic peptide receptor A and B (NPR-A/B) by HS-142-1 reverses many indices of myocardial depression associated with the condition (Hinder et al., 1997; Stubbe et al., 2004). Hence, it has been proposed that ANP and cyclic GMP (cGMP) (the intracellular second messenger responsible for mediating the biological activity of ANP) could be markers of severe and fatal myocardial depression early in the course of human endotoxic shock (Hartemink et al., 2001). In combination, these in vitro and in vivo data hint at a role for natriuretic peptides in the vascular abnormalities associated with endotoxic shock. However, little is known about the relationship between natriuretic peptides, iNOS and the vascular dysfunction characteristic of this condition.
To address this deficit, we used NPR-A (NPR1) KO mice (the principal receptor mediating the biological actions of ANP and BNP) to explore the role of natriuretic peptides in regulating iNOS expression and activity in the context of (cardio)vascular dysfunction during endotoxic shock in vitro and in vivo.
Methods
Reagents were obtained from Sigma (St. Louis, MO, USA) unless stated otherwise. All experiments were performed in accordance with the Animals (Scientific procedures) Act 1986. Drug and receptor nomenclature conforms to the ‘Guide to receptors and ion channels’ (Alexander et al., 2008).
LPS treatment and tissue homogenization
Salmonella typhimuriumLPS (Poole, Dorset, UK; 12.5 mg·kg−1, i.v.) or vehicle (physiological saline, sodium chloride 0.9% w/v, Ivex Pharmaceuticals, Antrim, Northern Ireland) were administered to male NPR-A KO mice [kind gift of Prof O. Smithies, University of North Carolina, Chapel Hill, NC, USA (Oliver et al., 1997)] or littermate controls (25–30 g) via the tail vein. After 16 h animals were killed by cervical dislocation and tissues and blood collected. Tissues were snap frozen in liquid nitrogen and stored at –80°C. They were then homogenized using an ice-cold mortar and pestle, transferred to whole cell homogenization buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 8 mM EGTA, protease inhibitor cocktail tablet; Roche, Burgess Hill, UK) and incubated on ice with vortexing every 10 min for a minimum of 30 min. Samples were centrifuged at 16 060×g for 15 min at 4°C and the supernatant was retained for subsequent analysis. Blood samples were centrifuged at 16 060×g for 5 min at room temperature and plasma retained for nitrate/nitrite, cGMP and cytokine determination.
Measurement of mean arterial blood pressure
Mean arterial blood pressure (MABP) was measured in chronically instrumented, conscious male NPR-A KO or WT mice as we have described previously (Connelly et al., 2005). After the mice had recovered from surgery (24 h), resting MABP was measured for 24 h on a pre-calibrated PowerLab system (ADInstruments, Castle Hill, New South Wales, Australia); 24 h later, LPS (12.5 mg·kg−1) was administered via the jugular vein and the MABP monitored over a further 16 h period.
Functional reactivity
Male NPR-A KO or WT mice were treated with LPS or vehicle as described above. After 16 h the animals were killed by cervical dislocation and the thoracic aortae were carefully excised, cleaned of connective tissue and mounted in 10 mL organ baths for isometric tension recordings, as described previously (Hussain et al., 1999). Following equilibration, an EC80 concentration of the thromboxane-mimetic, U46619 (Biomol International, Exeter, UK) was added to the organ bath and once a stable contraction was attained, cumulative concentration-response curves to ACh (1 nM–10 µM), ANP (0.01 nM–1 µM), the NO donor SPER-NO (Calbiochem, Nottingham, UK; 1 nM–100 µM) and the adenylyl cyclase activator forskolin (Calbiochem; 1 nM–100 µM) were obtained. The concentration of U46619 used to pre-contract the vessels from NPR-A KO and LPS-treated animals was carefully titrated to match the contractile response in control tissues. Data were captured using PowerLab and Chart version 5 (AD Instruments, Oxfordshire, UK).
Western blot analysis
Determination of iNOS expression by immunoblot was achieved as we have described previously (Connelly et al., 2005), using a primary anti-iNOS antibody (BD Biosciences, Cowley, Oxford UK; 1:2000 dilution) and a secondary horseradish peroxidise-conjugated goat anti-rabbit IgG antibody (Dako, Cambridge UK; 1:1000 dilution). Bands were quantified by densitometry using AlphaImager (Alpha Innotech, San Leandro, CA, USA).
Plasma NOx, cytokine and cGMP measurements
Plasma samples were analysed for NO2- and NO3- using chemiluminesence as we have described previously (Connelly et al., 2005). A BioPlex® (Bio-Rad, Hemel Hempstead, UK) array was used to determine cytokine production in response to LPS in the plasma of WT and NPR-A KO mice according to the manufacturer's instructions. Plasma cGMP concentrations were measured using a specific ELISA (R & D Systems, Abingdon, UK).
Data analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Densitometric analyses were performed using AlphaEaseFC (Alpha Innotech). All data are plotted graphically as mean values, with vertical bars representing standard error of the mean. Student's t-test or one-way analysis of variance (anova) followed by Bonferroni's multiple comparison test was used to assess differences between individual experimental conditions. Two-way anova was used to compare temporal changes in MABP in WT and NPR-A KO animals. A probability (P) value of <0.05 was taken as an appropriate level of significance.
Results
Haemodynamic profile in WT and NPR-A KO mice in response to LPS
The resting MABP in the WT and NPR-A KO animals was 113.9 ± 2.9 and 131.6 ± 2.5 mmHg, respectively. These values are consistent with previous studies reporting increased blood pressure in NPR-A-deficient mice (Oliver et al., 1997). Administration of LPS (12.5 mg·kg−1; i.v.) caused a slow decline in the MABP in WT animals such that there was a mean reduction of 11.5 ± 0.8 mmHg over 16 h (Figure 1), in accord with previous findings (Connelly et al., 2005; Vo et al., 2005). In NPR-A KO animals, following administration of LPS (12.5 mg·kg−1; i.v.) there was little or no deviation in blood pressure over a 16 h period (Figure 1), such that the MABP was not significantly different from the initial value (Δ2.6 ± 0.9 mmHg). Interestingly, following administration of LPS there was a small, rapid decrease in MABP in both WT and NPR-A KO animals (Figure 1), consistent with endothelial NOS (eNOS) activation, as we and others have reported (Connelly et al., 2005; Vo et al., 2005).
Figure 1.
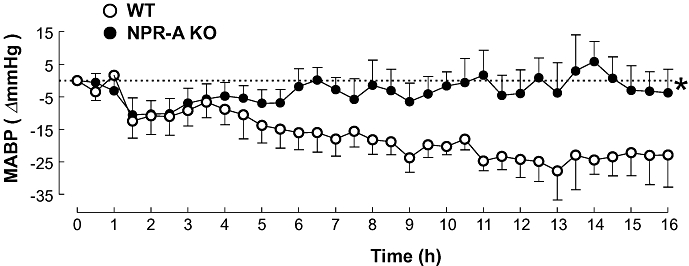
Change in mean arterial blood pressure (MABP) in WT and NPR-A KO mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Data are represented as mean ± standard error of the mean, n = 9; *P < 0.05 versus WT (across the whole time period). WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
Effect of NPR-A gene deletion on plasma NOx and cGMP in response to LPS
Administration of LPS (12.5 mg·kg−1; i.v.) led to an increase in plasma NOx concentration, as expected in animals with endotoxic shock (Connelly et al., 2005; Vo et al., 2005); however, levels were significantly lower in NPR-A KO mice in comparison to WT animals (Figure 2). NOx levels were indistinguishable between saline-treated NPR-A KO and WT animals (Figure 2). Accordingly, plasma cGMP levels were raised in response to LPS, but this increase was significantly smaller in NPR-A KO animals compared to WT controls (Figure 2).
Figure 2.
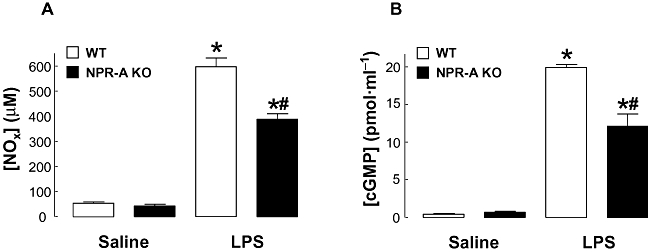
Plasma NOx (A) and cGMP (B) levels in WT and NPR-A KO mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Data are represented as mean ± standard error of the mean, n = 7; *P < 0.05 versus corresponding saline control, #P < 0.05 versus LPS-treated WT. cGMP, cyclic GMP; WT, wild-type; KO, knockout; LPS, lipopolysaccharide; NPR-A, natriuretic peptide receptor-A.
Effect of NPR-A gene deletion of plasma cytokine levels in response to LPS
Administration of LPS (12.5 mg·kg−1; i.v.) led to an increase in the plasma concentrations of interleukin (IL)-1β, IL-10, interferon (IFN)γ and tumour necrosis factor (TNF)α (Figure 3), but not IL-2, IL-4 and IL-5 (data not shown). However, plasma levels of IL-1α, IFNγ and TNFα were all significantly lower in NPR-A KO animals, whereas the plasma concentration of IL-10 was markedly increased in these mice (Figure 3).
Figure 3.
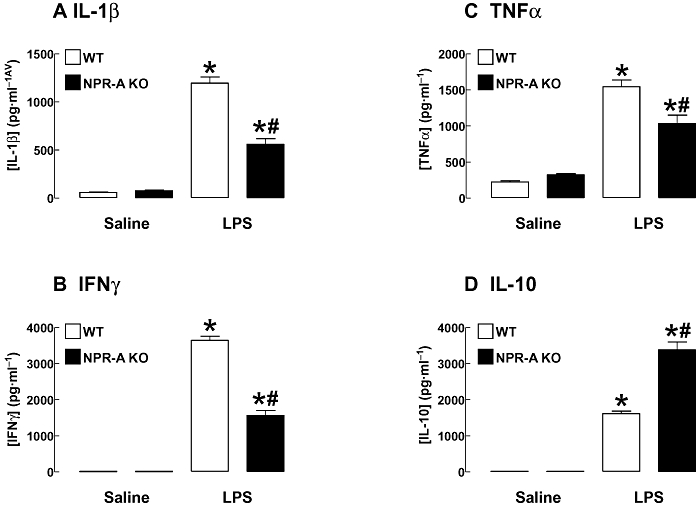
Plasma IL-1β (A), IFNγ (B), TNFα (C) and IL-10 (D) in WT and NPR-A KO mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Data are represented as mean ± standard error of the mean, n = 6–7; *P < 0.05 versus corresponding saline control, #P < 0.05 versus LPS-treated WT. IL, interleukin; IFNγ, interferon- γ; TNF-α, tumour necrosis factor-α; WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
Effect of NPR-A deletion on iNOS expression in response to LPS
There was no detectable iNOS expression in tissues from saline-treated WT or NPR-A KO mice. Administration of LPS (12.5 mg·kg−1; i.v.) for 16 h led to an increase in the expression of iNOS protein in lung and aorta of WT and NPR-A KO mice. However, in tissues from NPR-A KO mice there was a significantly lower level of iNOS protein detected (lung: –38.5 ± 6.3%, aorta: –80.3 ± 10.2%; Figure 4), that paralleled the reduced levels of plasma NOx, cGMP and pro-inflammatory cytokines (Figures 2 and 3).
Figure 4.
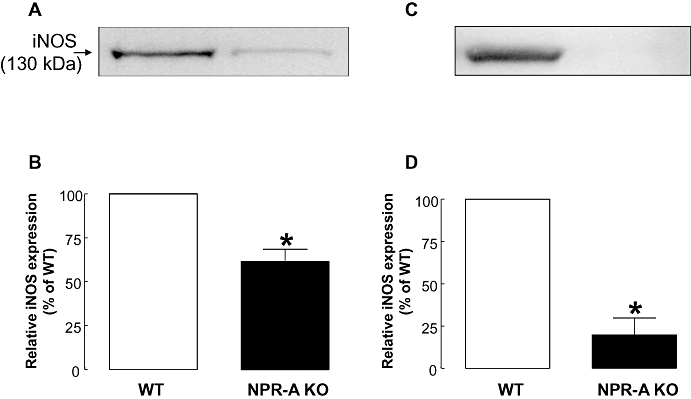
Expression of iNOS protein in lung (A and B) and aorta (C and D) from WT and NPR-A KO mice treated with LPS (12.5 mg·kg−1; i.v.) for 16 h. Protein expression was analysed by Western blot (A and C) and quantified by densitometry (B and D). Data are represented as mean ± standard error of the mean, n = 5 (lung); n = 4 (aorta); *P < 0.05 versus WT. iNOS, inducible nitric oxide synthase; WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
Ex vivo pharmacology
Since treatment of NPR-A KO mice with LPS resulted in dramatically reduced iNOS expression and plasma NOx, cGMP and pro-inflammatory cytokine levels in comparison to WT animals, functional pharmacological studies were conducted using thoracic aortae from saline (volume-matched) or LPS (12.5 mg·kg−1; both i.v.,16 h)-treated NPR-A KO and WT mice to assess the effect of NPR-A deficiency on the vascular dysfunction associated with endotoxaemia.
(1) Effect of NPR-A deletion on vascular responses to U46619 following LPS.
U46619 caused a concentration-dependent contraction in tissues from both WT and NPR-A KO mice. In WT aortae, in vivo LPS treatment caused a small but significant rightward shift in the concentration-response curve to U46619 (pEC50: 7.69 ± 0.12 and 7.94 ± 0.05; P < 0.05; LPS and saline, respectively; Figure 5). In contrast, in tissues from NPR-A KO mice LPS treatment had no significant effect on responses to U46619 compared to control (pEC50: 6.41 ± 0.16 and 7.12 ± 0.18; P > 0.05; LPS and saline, respectively; Figure 5).
Figure 5.
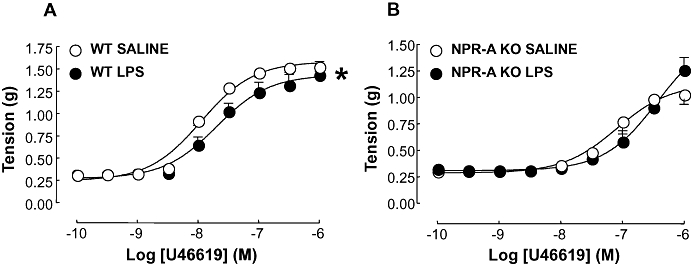
Concentration-response curves to U46619 in aortic rings from WT (A) and NPR-A KO (B) mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Contraction is expressed as mean ± standard error of the mean tension in g; *P < 0.05 versus saline-treated animals (across the entire curve), n = 7. WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
(2) Effect of NPR-A deletion on vascular responses to ANP following LPS.
In order to explore the effects of LPS on relaxant responses, tissues were pre-contracted with an approximate EC80 of U46619 that was titrated to account for the altered potency between aortae from WT and NPR-A KO animals. Tissues from WT mice treated with LPS exhibited a significant rightward shift in the concentration-response curve to ANP (pEC50: 7.89 ± 0.20 and 8.13 ± 0.13; P < 0.05; LPS and saline, respectively; Figure 6) accompanied by a significant decrease in Emax (Emax: 53.61 ± 4.13 and 76.16 ± 3.58; P < 0.05; LPS and saline, respectively; Figure 6). Aortae from NPR-A KO animals did not exhibit vasorelaxation in response to ANP, confirming the lack of NPR-A in these animals (Figure 6).
Figure 6.
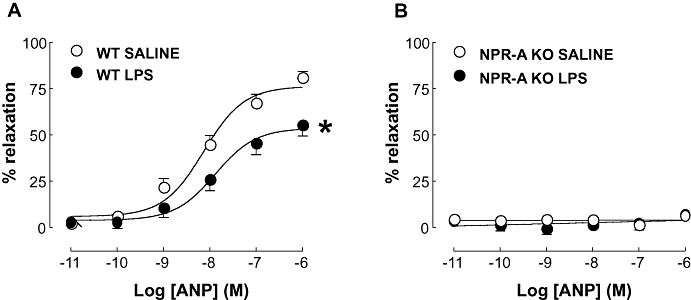
Concentration-response curves to ANP in aortic rings from WT (A) and NPR-A KO (B) mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Relaxation is expressed as mean ± standard error of the mean percentage reversal of U46619-induced tone; *P < 0.05 versus saline-treated animals (across the entire curve), n = 9. ANP, atrial natriuretic peptide; WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
(3) Effect of NPR-A deletion on vascular responses to ACh following LPS
Treatment of WT animals with LPS caused a significant rightward shift in the concentration-response curve to the endothelium-dependent relaxant ACh (pEC50: 6.69 ± 0.21 and 7.00 ± 0.08; P < 0.05; LPS and saline, respectively; Figure 7) and a significant decrease in the maximum (Emax: 25.46 ± 2.11 and 66.36 ± 2.22; P < 0.05; LPS and saline, respectively; Figure 7). Vessels from LPS-treated NPR-A KO mice showed almost complete restoration of the responsiveness to ACh when compared to WT animals treated with LPS (pEC50: 6.70 ± 0.24 and 7.30 ± 0.28; LPS and saline, respectively; Figure 7). Furthermore, the Emax was indistinguishable in LPS and saline-treated NPR-A KO mice (Emax: 77.31 ± 6.92 and 79.58 ± 6.93; P > 0.05; LPS and saline, respectively; Figure 7).
Figure 7.
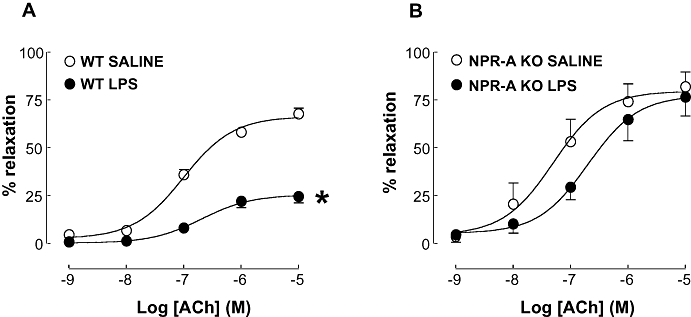
Concentration-response curve to ACh in aortic rings from WT (A) and NPR-A KO (B) mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Relaxation is expressed as mean ± standard error of the mean percentage reversal of U46619-induced tone; *P < 0.05 versus saline-treated animals (across the entire curve), n = 5. WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
(4) Effect of NPR-A deletion on vascular responses to SPER-NO following LPS
Treatment of WT mice with LPS caused asignificant rightward shift in the concentration response curve to the NO donor SPER-NO (pEC50: 5.08 ± 0.16 and 5.86 ± 0.07; P < 0.05; LPS and saline, respectively; Figure 8). However, the Emax remained unchanged (Emax: 101.4 ± 12.13 and 94.40 ± 3.01; P > 0.05; LPS and saline, respectively; Figure 8). This reduction in potency following LPS administration was not observed in NPR-A KO animals where the curves were superimposable (pEC50: 5.25 ± 0.12 and 5.42 ± 0.18; Emax: 95.73 ± 7.09 and 91.64 ± 8.83; both P > 0.05; LPS and saline, respectively; Figure 8).
Figure 8.
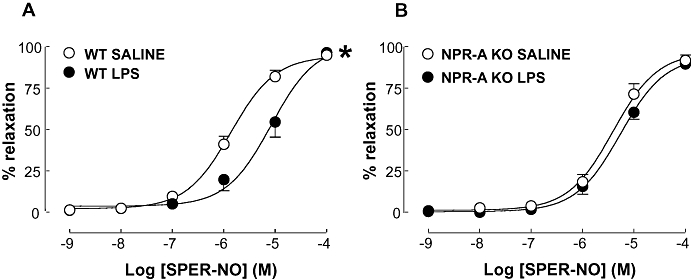
Concentration-response curve to SPER-NO in aortic rings from WT (A) and NPR-A KO (B) mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Relaxation is expressed as mean ± standard error of the mean percentage reversal of U46619-induced tone; *P < 0.05 versus saline-treated animals (across the entire curve), n = 5. SPER-NO, spermine-NONOate; WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
(5) Effect of NPR-A deletion on vascular responses to forskolin following LPS.
In order to confirm that the vascular dysfunction observed in WT and NPR-A KO mice following LPS treatment was restricted to cGMP (and not cAMP)-dependent pathways, concentration-response curves to the adenylate cyclase activator, forskolin, were constructed. As expected from previous studies (Hussain et al., 1999; Sabrane et al., 2003; Madhani et al., 2006) the vasorelaxant potency to forskolin was identical in LPS and saline-treated WT and NPR-A KO animals (Figure 9).
Figure 9.
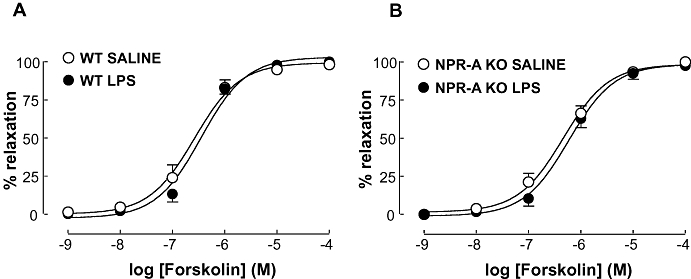
Concentration-response curve to forskolin in aortic rings from WT (A) and NPR-A KO (B) mice treated with LPS (12.5 mg·kg−1) or saline (both i.v.) for 16 h. Relaxation is expressed as mean ± standard error of the mean percentage reversal of U46619-induced tone; *P < 0.05 versus saline-treated animals (across the entire curve), n = 5. WT, wild-type; KO, knockout; NPR-A, natriuretic peptide receptor-A; LPS, lipopolysaccharide.
Discussion
This study demonstrates that NPR-A activation (by ANP and/or BNP) plays a key role in facilitating pro-inflammatory cytokine production, iNOS expression/activity and the subsequent development of vascular hyporeactivity in LPS-induced murine shock. In NPR-A KO animals treated with LPS in vivo, circulating levels of the pro-inflammatory cytokines IL-1β, IFNγ and TNFα, iNOS expression and activity (as assessed by plasma NOx and cGMP accumulation), endothelial and vascular smooth muscle dysfunction, and systemic hypotension are all significantly reduced in comparison to WT animals; in contrast, plasma concentrations of the anti-inflammatory cytokine IL-10 are significantly elevated. These findings give rise to the thesis that blockade of natriuretic peptide signalling may represent a novel therapeutic target that would prevent the (cardio)vascular hyporeactivity that underlies the high mortality associated with this condition.
Endotoxic shock remains a major cause of death worldwide. Despite a more detailed understanding of many aspects of disease progression, this has not yet translated into improved therapy and reductions in mortality (Singer, 2006). As such, identification of novel pathways contributing to the development of this condition may offer potential for superior treatment regimens. Inducible NOS is well-established to play a pivotal role in the development of the (cardio)vascular hyporeactivity, which characterizes endotoxic shock and is a major cause of death in patients with this condition (Petros et al., 1994; Chauhan et al., 2003). High-output NO production by this isoform of NOS results in both hypo-responsiveness to vasoconstrictors and endothelial dysfunction, which combine to elicit a marked systemic hypotension. In accord with this schema, iNOS KO mice and selective iNOS inhibitors have proven effective in restoring the vascular dysfunction associated with endotoxic shock (Julou-Schaeffer et al., 1990; Kilbourn et al., 1994; MacMicking et al., 1995; Chauhan et al., 2003). Disappointingly, however, clinical trials of a non-selective NOS inhibitor (L-NMA) in patients with septic shock have reported increased, rather than decreased, mortality (Lopez et al., 2004).
More recently, a key role for the endothelial isoform of NOS, eNOS, in regulating the inflammatory response, particularly iNOS expression and NO production during endotoxaemia, has been described by ourselves and others (Connelly et al., 2005; Vo et al., 2005). In this case, immediate activation of eNOS (via an Akt- and PI3K-dependent phosphorylation) by LPS results in a cGMP-mediated facilitation of iNOS expression and activity; accordingly, eNOS KO mice do not exhibit hypotension and are resistant to endotoxaemia. Since plasma levels of natriuretic peptides are raised in endotoxaemia (Aiura et al., 1995; Witthaut et al., 2003), and these vasoactive mediators play analogous cardiovascular homeostatic roles to NO, via production of cGMP, we hypothesized that natriuretic peptides might also play a key role in the pathogenesis of endotoxic shock. Thus, we assessed whether ANP and/or BNP signalling is important for the vascular dysfunction observed during endotoxic shock by comparing the haemodynamic profile of WT and NPR-A KO mice following exposure to LPS. Both NPR-A KO and WT animals showed a small reduction in systemic blood pressure within 1 h of exposure to LPS, which is consistent with a rapid, transient activation of eNOS (Connelly et al., 2005; Vo et al., 2005). The blood pressure rapidly recovered and was maintained at basal levels in NPR-A KO animals for the duration of the study. However, the blood pressure of WT mice continued to drop steadily throughout the remainder of the investigation; by the 18 h time point the MABP had dropped by almost 30 mmHg. This improved haemodynamic profile in NPR-A KO animals demonstrates that a lack of NPR-A conveys resistance to the vascular dysfunction characteristic of endotoxic shock. This phenomenon is likely to result from both direct and indirect consequences of NPR-A deficiency. Firstly, increased levels of natriuretic peptides (i.e. ANP and BNP) occurring during endotoxic shock would not be able to mediate their direct vasodilator activity via this NPR subtype nor stimulate excretion of fluid (and salt; i.e. natriuresis) to lower blood volume. Secondly, natriuretic peptides facilitate pro-inflammatory cytokine production and iNOS expression via a NPR-A- and cGMP- dependent mechanism (akin to NO). Indeed, the regulation of pro-inflammatory gene expression, particularly iNOS, by cGMP-dependent mechanisms has been previously documented (Choi et al., 1999; Perez-Sala et al., 2001; Connelly et al., 2003; Connelly et al., 2005; Gunnett et al., 2005; Vo et al., 2005) and a common pathway appears to be modulation of NFκB activity (Traenckner et al., 1995; Kalra et al., 2000). Thus, it is likely that similar downstream cGMP-triggered pathways underlie the pro-inflammatory role for natriuretic peptides (via NPR-A activation) in the pathogenesis of endotoxic shock. Whether the regulation of iNOS expression is a direct effect of natriuretic peptides, or secondary to a NPR-A-driven increase in pro-inflammatory cytokine production [e.g. IL-1β, TNFα IFNγ; each a potent stimulus for iNOS induction (Xie and Nathan, 1994)] remains to be elucidated. Regardless, this clear effect of NPR-A gene deletion on the production of, and balance between, pro- and anti-inflammatory cytokines suggests that the natriuretic peptide family underpin a novel mechanism governing host defence.
To examine this concept further, we investigated if the observed haemodynamic responses in NPR-A KO animals were paralleled by altered pro-inflammatory cytokine production, iNOS expression, NO production and cGMP generation. Plasma levels of the pro-inflammatory cytokines IL-1β, IFNγ and TNFα were all decreased in NPR-A KO versus WT animals treated with LPS. Conversely, plasma concentrations of the anti-inflammatory cytokine IL-10 were increased in NPR-A KO animals when compared to WT mice. The balance in favour of pro- versus anti-inflammatory cytokine production was mirrored by increased expression of iNOS protein, and enhanced production of NO and cGMP in WT versus NPR-A KO animals. The dramatic reduction in iNOS expression and activity correlated well with the haemodynamic changes following induction of endotoxic shock and confirm that NPR-A KO animals exhibit a considerably smaller pro-inflammatory response, paralleling the observations made in eNOS KO animals (Connelly et al., 2005). This observation also fits with a well-established role for IL-1β, IFNγ and TNFα in facilitating iNOS expression in numerous cell types (Xie and Nathan, 1994). Interestingly, there was almost complete abrogation of iNOS expression in the lungs and aorta of NPR-A KO mice treated with LPS, and vascular responsiveness to endothelium-dependent and independent vasorelaxants was normal. Yet, there was still a significant production of NOx and cGMP, suggesting that iNOS activity accounts for ∼50% of the total NO/cGMP production during endotoxaemia. Undoubtedly, eNOS activation in response to LPS (Connelly et al., 2005; Vo et al., 2005), will contribute to the NO/cGMP generation. However, NPR-A deficient animals also have an intact CNP-NPR-B-cGMP system that may account for a proportion of the remaining cGMP production; this is particularly relevant since sepsis and pro-inflammatory cytokines represent a strong stimulus for CNP release from the endothelium and sepsis is one of the few disorders in which plasma CNP levels are raised (Suga et al., 1993; Hama et al., 1994). Nonetheless, this ∼50% reduction in plasma NOx/cGMP levels observed in NPR-A KO mice in response to LPS is sufficient to permit normal vascular function (i.e. MABP in vivo and endothelial and smooth muscle function in vitro). This suggests that there is a threshold of NO/cGMP production within which vascular homeostasis, in particular endothelial function, can be maintained effectively. Outside that, endothelial dysfunction occurs and there is inappropriate control of local blood flow (and systemic blood pressure). In vivo, this manifests as the systemic hypotension, reduced tissue perfusion and end organ failure associated with sepsis.
Having established that NPR-A deletion alters the severity of endotoxic shock in vivo, we examined whether the well-established vascular dysfunction occurring in this condition was alleviated in animals lacking this NPR subtype in vitro. Contractile responses to the thromboxane mimetic U46619 were significantly (albeit marginally) reduced in tissues from WT animals treated with LPS compared to WT controls. This is consistent with an iNOS-dependent inhibition of the contractile apparatus (Chauhan et al., 2003). However, responsiveness to U46619 in aortae from NPR-A KO mice was equivalent in control and LPS-treated animals. Again, this fits with a much reduced iNOS expression and activity in these tissues. In aortae from WT animals, the potency of both ACh (endothelium-dependent) and SPER-NO (endothelium-independent) was significantly reduced following exposure to LPS, whereas in tissues from NPR-A KO animals there was no difference in dilator responsiveness in the absence or presence of LPS. Such observations provide further evidence that the diminished inflammatory response in NPR-A KO animals during endotoxic shock results in the restoration of the endothelial and smooth muscle dysfunction that lead to haemodynamic depression. It is also possible that endothelin (ET)-1 release is up-regulated in WT versus NPR-A KO animals challenged with LPS (Mansart et al., 2008; Scicluna et al., 2008) resulting in a greater functional antagonism of vasodilator activity. Notably, vascular responsiveness to the cAMP-dependent dilator forskolin was not impaired, confirming that vascular dysfunction is restricted to the cGMP signalling cascade. We and others have previously demonstrated (Papapetropoulos et al., 1996; Hussain et al., 1999; Madhani et al., 2003; Sabrane et al., 2003) that vascular responsiveness to NO is exquisitely sensitive to the ambient NO and natriuretic peptide concentrations. In the current study, the reduced potency of ACh and SPER-NO illustrates the impaired endothelial production of NO and smooth muscle responsiveness, respectively, during endotoxaemia. In tandem with the hyporesponsiveness to the vasoconstrictor U46619, this entails that not only is there a systemic hypotension, but that endothelial cells cannot regulate vascular tone and blood flow at a local level, perhaps explaining the aberrant organ perfusion that occurs in this condition. These impairments are not as severe in NPR-A KO animals, resulting in an improved haemodynamic profile and vascular function.
In summary, results from the current study provide convincing evidence that natriuretic peptides (principally ANP and BNP), via NPR-A activation, play a role in the pathogenesis of endotoxic shock. Specifically, these vasoactive mediators facilitate the expression of pro-inflammatory cytokines and iNOS expression/activity, and augment the vascular dysfunction and detrimental haemodynamic profile associated with this disease. Whilst these findings will need confirmation in longer-term, more clinically representative models of human sepsis, the data from the current model represent interesting proof of concept studies that warrant further investigation. Accordingly, inhibition of natriuretic peptide release or bioactivity may provide a novel approach to alleviating the haemodynamic problems associated with endotoxic shock and to reduce the associated mortality.
Acknowledgments
The authors wish to thank Nicholas Davies for his technical assistance and Sarah Yates for guidance with the cytokine arrays. This work was supported by the Wellcome Trust.
Glossary
Abbreviations:
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- CNP
C-type natriuretic peptide
- eNOS
endothelial nitric oxide synthase
- ET-1
endothelin-1
- IFNγ
interferon-γ
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- KO
knockout
- L-NMA
NG-methyl-L-arginine
- LPS
lipopolysaccharide
- MABP
mean arterial blood pressure
- NO
nitric oxide
- NOS
nitric oxide synthase
- [NOx]
total [NO2-] + [NO3-]
- NPR-A
natriuretic peptide receptor-A (NPR1)
- SPER-NO
spermine-NONOate
- TNFα
tumour necrosis factor-α
- U46619
9,11-dideoxy-9α,11α-methanoepoxy prostaglandin F2α
- WT
wild-type
Conflicts of interest
None.
References
- Ahluwalia A, MacAllister RJ, Hobbs AJ. Vascular actions of natriuretic peptides. Cyclic GMP-dependent and -independent mechanisms. Basic Res Cardiol. 2004;99:83–89. doi: 10.1007/s00395-004-0459-6. [DOI] [PubMed] [Google Scholar]
- Aiura K, Ueda M, Endo M, Kitajima M. Circulating concentrations and physiologic role of atrial natriuretic peptide during endotoxic shock in the rat. Crit Care Med. 1995;23:1898–1906. doi: 10.1097/00003246-199511000-00017. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–S254. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annane D, Sanquer S, Sebille V, Faye A, Djuranovic D, Raphael JC, et al. Compartmentalised inducible nitric-oxide synthase activity in septic shock. Lancet. 2000;355:1143–1148. doi: 10.1016/S0140-6736(00)02063-8. [DOI] [PubMed] [Google Scholar]
- Chauhan SD, Seggara G, Vo PA, MacAllister RJ, Hobbs AJ, Ahluwalia A. Protection against lipopolysaccharide-induced endothelial dysfunction in resistance and conduit vasculature of iNOS knockout mice. FASEB J. 2003;17:773–775. doi: 10.1096/fj.02-0668fje. [DOI] [PubMed] [Google Scholar]
- Choi SH, Shin KH, Kang SW, Chun YS, Chun BG. Guanosine 5’,3’-cyclic monophosphate enhances lipopolysaccharide- induced nitric oxide synthase expression in mixed glial cell cultures of rat. Neurosci Lett. 1999;276:29–32. doi: 10.1016/s0304-3940(99)00783-1. [DOI] [PubMed] [Google Scholar]
- Connelly L, Jacobs AT, Palacios-Callender M, Moncada S, Hobbs AJ. Macrophage endothelial nitric-oxide synthase autoregulates cellular activation and pro-inflammatory protein expression. J Biol Chem. 2003;278:26480–26487. doi: 10.1074/jbc.M302238200. [DOI] [PubMed] [Google Scholar]
- Connelly L, Madhani M, Hobbs AJ. Resistance to endotoxic shock in endothelial nitric-oxide synthase (eNOS) knock-out mice: a pro-inflammatory role for eNOS-derived no in vivo. J Biol Chem. 2005;280:10040–10046. doi: 10.1074/jbc.M411991200. [DOI] [PubMed] [Google Scholar]
- Fleming I, Dambacher T, Busse R. Endothelium-derived kinins account for the immediate response of endothelial cells to bacterial lipopolysaccharide. J Cardiovasc Pharmacol. 1992;20(Suppl 12):S135–S138. doi: 10.1097/00005344-199204002-00038. [DOI] [PubMed] [Google Scholar]
- Gunnett CA, Lund DD, McDowell AK, Faraci FM, Heistad DD. Mechanisms of inducible nitric oxide synthase-mediated vascular dysfunction. Arterioscler Thromb Vasc Biol. 2005;25:1617–1622. doi: 10.1161/01.ATV.0000172626.00296.ba. [DOI] [PubMed] [Google Scholar]
- Hama N, Itoh H, Shirakami G, Suga S, Komatsu Y, Yoshimasa T, et al. Detection of C-type natriuretic peptide in human circulation and marked increase of plasma CNP level in septic shock patients. Biochem Biophys Res Commun. 1994;198:1177–1182. doi: 10.1006/bbrc.1994.1166. [DOI] [PubMed] [Google Scholar]
- Hartemink KJ, Groeneveld AB, de Groot MC, Strack van Schijndel RJ, van KG, Thijs LG. Alpha-atrial natriuretic peptide, cyclic guanosine monophosphate, and endothelin in plasma as markers of myocardial depression in human septic shock. Crit Care Med. 2001;29:80–87. doi: 10.1097/00003246-200101000-00019. [DOI] [PubMed] [Google Scholar]
- Hinder F, Booke M, Traber LD, Traber DL. The atrial natriuretic peptide receptor antagonist HS 142-1 improves cardiovascular filling and mean arterial pressure in a hyperdynamic ovine model of sepsis. Crit Care Med. 1997;25:820–826. doi: 10.1097/00003246-199705000-00018. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Broussard M, Osman J, Parrillo JE. Increased microvascular reactivity and improved mortality in septic mice lacking inducible nitric oxide synthase. Circ Res. 2000;86:774–778. doi: 10.1161/01.res.86.7.774. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- Hussain MB, Hobbs AJ, MacAllister RJ. Autoregulation of nitric oxide-soluble guanylate cyclase-cyclic GMP signalling in mouse thoracic aorta. Br J Pharmacol. 1999;128:1082–1088. doi: 10.1038/sj.bjp.0702874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julou-Schaeffer G, Gray GA, Fleming I, Schott C, Parratt JR, Stoclet JC. Loss of vascular responsiveness induced by endotoxin involves L-arginine pathway. Am J Physiol. 1990;259:H1038–H1043. doi: 10.1152/ajpheart.1990.259.4.H1038. [DOI] [PubMed] [Google Scholar]
- Kalra D, Baumgarten G, Dibbs Z, Seta Y, Sivasubramanian N, Mann DL. Nitric oxide provokes tumor necrosis factor-alpha expression in adult feline myocardium through a cGMP-dependent pathway. Circulation. 2000;102:1302–1307. doi: 10.1161/01.cir.102.11.1302. [DOI] [PubMed] [Google Scholar]
- Kilbourn RG, Cromeens DM, Chelly FD, Griffith OW. NG-methyl-L-arginine, an inhibitor of nitric oxide formation, acts synergistically with dobutamine to improve cardiovascular performance in endotoxemic dogs. Crit Care Med. 1994;22:1835–1840. [PubMed] [Google Scholar]
- Kilbourn RG, Jubran A, Gross SS, Griffith OW, Levi R, Adams J, et al. Reversal of endotoxin-mediated shock by NG-methyl-L-arginine, an inhibitor of nitric oxide synthesis. Biochem Biophys Res Commun. 1990;172:1132–1138. doi: 10.1016/0006-291x(90)91565-a. [DOI] [PubMed] [Google Scholar]
- Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med. 2004;32:21–30. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Madhani M, Okorie M, Hobbs AJ, MacAllister RJ. Reciprocal regulation of human soluble and particulate guanylate cyclases in vivo. Br J Pharmacol. 2006;149:797–801. doi: 10.1038/sj.bjp.0706920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani M, Scotland RS, MacAllister RJ, Hobbs AJ. Vascular natriuretic peptide receptor-linked particulate guanylate cyclases are modulated by nitric oxide-cyclic GMP signalling. Br J Pharmacol. 2003;139:1289–1296. doi: 10.1038/sj.bjp.0705365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansart A, Ross JJ, Reilly CS, Brown NJ, Brookes ZL. LPS abolishes extrasplenic vasoconstriction to atrial natriuretic peptide: the role of NO and endothelin 1. Shock. 2008;29:675–680. doi: 10.1097/shk.0b013e31815811a3. [DOI] [PubMed] [Google Scholar]
- Marumo T, Nakaki T, Hishikawa K, Hirahashi J, Suzuki H, Kato R, et al. Natriuretic peptide-augmented induction of nitric oxide synthase through cyclic guanosine 3’,5’-monophosphate elevation in vascular smooth muscle cells. Endocrinology. 1995;136:2135–2142. doi: 10.1210/endo.136.5.7536663. [DOI] [PubMed] [Google Scholar]
- Ochoa JB, Udekwu AO, Billiar TR, Curran RD, Cerra FB, Simmons RL, et al. Nitrogen oxide levels in patients after trauma and during sepsis. Ann Surg. 1991;214:621–626. doi: 10.1097/00000658-199111000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A. 1997;94:14730–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Abou-Mohamed G, Marczin N, Murad F, Caldwell RW, Catravas JD. Downregulation of nitrovasodilator-induced cyclic GMP accumulation in cells exposed to endotoxin or interleukin-1 beta. Br J Pharmacol. 1996;118:1359–1366. doi: 10.1111/j.1476-5381.1996.tb15545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Sala D, Cernuda-Morollon E, Diaz-Cazorla M, Rodriguez-Pascual F, Lamas S. Posttranscriptional regulation of human iNOS by the NO/cGMP pathway. Am J Physiol Renal Physiol. 2001;280:F466–F473. doi: 10.1152/ajprenal.2001.280.3.F466. [DOI] [PubMed] [Google Scholar]
- Petros A, Lamb G, Leone A, Moncada S, Bennett D, Vallance P. Effects of a nitric oxide synthase inhibitor in humans with septic shock. Cardiovasc Res. 1994;28:34–39. doi: 10.1093/cvr/28.1.34. [DOI] [PubMed] [Google Scholar]
- Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27:47–72. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- Rees DD, Monkhouse JE, Cambridge D, Moncada S. Nitric oxide and the haemodynamic profile of endotoxin shock in the conscious mouse. Br J Pharmacol. 1998;124:540–546. doi: 10.1038/sj.bjp.0701815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabrane K, Gambaryan S, Brandes RP, Holtwick R, Voss M, Kuhn M. Increased sensitivity to endothelial nitric oxide (NO) contributes to arterial normotension in mice with vascular smooth muscle-selective deletion of the atrial natriuretic peptide (ANP) receptor. J Biol Chem. 2003;278:17963–17968. doi: 10.1074/jbc.M213113200. [DOI] [PubMed] [Google Scholar]
- Scicluna JK, Mansart A, Ross JJ, Reilly CS, Brown NJ, Brookes ZL. Reduced vascular response to phenylephrine during exposure to lipopolysaccharide in vitro involves nitric oxide and endothelin 1. Shock. 2008;29:417–421. doi: 10.1097/shk.0b013e318142c5df. [DOI] [PubMed] [Google Scholar]
- Singer M. The key advance in the treatment of sepsis in the last 10 years … doing less. Crit Care. 2006;10:122. doi: 10.1186/cc4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe HD, Traber DL, Booke M, Traber LD, Westphal M, Van AH, et al. Role of atrial natriuretic peptide in pulmonary permeability and vasoregulation in ovine sepsis. Crit Care Med. 2004;32:2491–2495. doi: 10.1097/01.ccm.0000147834.01191.4e. [DOI] [PubMed] [Google Scholar]
- Suga S, Itoh H, Komatsu Y, Ogawa Y, Hama N, Yoshimasa T, et al. Cytokine-induced C-type natriuretic peptide (CNP) secretion from vascular endothelial cells – evidence for CNP as a novel autocrine/paracrine regulator from endothelial cells. Endocrinology. 1993;133:3038–3041. doi: 10.1210/endo.133.6.8243333. [DOI] [PubMed] [Google Scholar]
- Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo PA, Lad B, Tomlinson JA, Francis S, Ahluwalia A. Autoregulatory role of endothelium-derived nitric oxide (NO) on Lipopolysaccharide-induced vascular inducible NO synthase expression and function. J Biol Chem. 2005;280:7236–7243. doi: 10.1074/jbc.M411317200. [DOI] [PubMed] [Google Scholar]
- Vollmar AM, Schulz R. Atrial natriuretic peptide inhibits nitric oxide synthesis in mouse macrophages. Life Sci. 1995;56:L149–L155. doi: 10.1016/0024-3205(94)00484-a. [DOI] [PubMed] [Google Scholar]
- Witthaut R, Busch C, Fraunberger P, Walli A, Seidel D, Pilz G, et al. Plasma atrial natriuretic peptide and brain natriuretic peptide are increased in septic shock: impact of interleukin-6 and sepsis-associated left ventricular dysfunction. Intensive Care Med. 2003;29:1696–1702. doi: 10.1007/s00134-003-1910-0. [DOI] [PubMed] [Google Scholar]
- Xie Q, Nathan C. The high-output nitric oxide pathway: role and regulation. J Leukoc Biol. 1994;56:576–582. doi: 10.1002/jlb.56.5.576. [DOI] [PubMed] [Google Scholar]