

Abstract
Familial breast and ovarian cancers are often defective in homologous recombination (HR) due to mutations in the BRCA1 or BRCA2 genes. Cisplatin chemotherapy or poly(ADP-ribose) polymerase (PARP) inhibitors are tested for these tumours in clinical trials. In a screen for novel drugs that selectively kill BRCA2-defective cells, we identified 6-thioguanine (6TG), which induces DNA double-strand breaks (DSBs) that we show are repaired by HR. Furthermore, we show that 6TG is as efficient as a PARP inhibitor in selectively killing BRCA2-defective tumours in a xenograft model. Spontaneous BRCA1 defective mammary tumours gain resistance to PARP inhibitors through increased p-glycoprotein expression. Here, we show that 6TG efficiently kills such BRCA1 defective PARP inhibitor resistant (PIR) tumours. We also show that 6TG can kill cells and tumours that have gained resistance to PARP inhibitors or cisplatin through genetic reversion of the BRCA2 gene. Although HR is reactivated in PIR BRCA2-defective cells, it is not fully restored for the repair of 6TG-induced lesions. This is likely to be due to several recombinogenic lesions being formed after 6TG. We show that BRCA2 is required for survival also to mismatch repair-independent lesions formed by 6TG, which do not include DSBs. This suggests that HR is involved in repair of 6TG-induced DSBs as well as mismatch repair-independent 6TG-induced DNA lesion. Altogether, our data show that 6TG efficiently kills BRCA2-defective tumours and suggest that 6TG may be effective in the treatment of advanced tumours that have developed resistance to PARP inhibitors or platinum-based chemotherapy.
Breast cancer is the most common cancer in women in the Western world today and in the UK breast cancer incidence rates have increased by more than 50% over the last 25 years. Familial mutations in the breast cancer susceptibility genes BRCA1 or BRCA2 are associated with an increased risk of several cancers, particularly breast, ovarian and prostate cancer (1). The proteins encoded by these genes both play important roles in homologous recombination (HR) repair (2-4) and it is likely that their tumour suppressor function is explained by their role in reducing mutation rates (5). This hypothesis is also supported by the observation that proteins with related functions have also been linked with a predisposition to developing breast cancer, i.e. CHEK2 (6), ATM (7), PALB2 (FANCN) (8-10) and BRIP1 (BACH1) (11).
HR-defective cells are characterised by hypersensitivity to crosslinking agents, which is thought to be related to a role of HR in bypassing inter-strand crosslinks during DNA replication (12). HR-defective cells are also hypersensitive to poly(ADP-ribose) polymerase (PARP) inhibitors (13, 14). This involves PARP1 having a role in DNA single-strand break (SSB) repair (15), which results in suppression of HR (16). PARP inhibitors may increase the amount of SSBs, which collapse into DNA double-strand breaks (DSB) at replication forks, requiring HR for repair (17). In the absence of HR, these DSBs are not repaired, resulting in HR defective cells such as BRCA1- and BRCA2-mutated cancers, which are therefore hypersensitive to PARP inhibitors (13). In the clinic, PARP inhibitors efficiently killed BRCA1- and BRCA2-defective tumours in a phase I-II clinical trial (18). However, acquired resistance to PARP inhibitor is a problem and may involve either additional mutations in the BRCA1 or BRCA2 genes that result in restoration of the C-terminal part of the proteins (19-21) or up-regulation of Abcb1a/b genes encoding P-glycoprotein efflux pumps (22). Although the extent of resistance that can be acquired by these mechanisms is unclear in humans, it suggests that the discovery of agents that may overcome such resistance mechanisms merit further investigation.
MATERIALS AND METHODS
Chemicals
All chemicals were obtained from Sigma unless stated otherwise, AG014699 was provided Pfizer GRD, La Jolla, and KU0058948 and olaparib by KuDOS Pharmaceuticals, (Cambridge, UK). AG014699 and KU0058948 compounds were dissolved at 10 mM in 100% dry DMSO and stored at −20°C, it was diluted in culture medium to give the final desired drug concentration in 1% DMSO with control cultures exposed to 1% DMSO alone. The NCI diversity and mechanistic set were obtained from NCI (Bethesda, MA) and stocks were maintained in 96-well plates in DMSO (stock concentration 1 mM (mechanistic set) and 10 mM (diversity set).
Cell culture and isolation of PARP inhibitor resistant cells
The HCT116 and HCT116+Chr3 were obtained from Dr Bert Vogelstein, U2OS cell line was obtained from ATCC, Capan-1 and resistant Capan-1 clones from Dr Toshiyasu Taniguchi, AA8, irs1SF and CXR3 cells from Dr Larry Thompson and V-C8 and V-C8B2 cells previously isolated (4). All cells were grown in Dulbecco's modified Eagle's Medium (DMEM) with 10% foetal bovine serum and penicillin/ streptomycin at 37°C under an atmosphere containing 5% CO2.
shRNA depletion of BRCA2
Depletion of BRCA2 expression in U2OS or HCT116 cells was obtained from the stably integrated regulatable expression of BRCA2 shRNA using the BLOCK-iT™ Inducible H1 RNAi Entry Vector Kit from Invitrogen, according to the manufacturers protocol (Invitrogen, Sweden). The target sequence introduced was AAC AAC AAU UAC GAA CCA AAC UU (23).
Western Blot
Proteins from cell lysates were separated and detected using Western blotting as previously described (24). The primary antibody was an anti-rabbit BRCA2 antibody (Santa Cruz, H299) diluted 1:500 in blocking solution.
Screening procedure
U2OS BRCA2 shRNA cells, were grown in the presence or absence of 2 μg/ml of dox for 2 days, plated in 96 wells plates (2000 cells per well) in the presence or absence of doxycyclin, and the next day treated with test compounds (at a concentration 10 μM) from the NCI library. After 72 hours WST-1 cell proliferation reagent was used in order to determine the cells viability as described earlier (25). Compounds from the library that selectively suppressed the growth of BRCA2 defective cells, but had only modest effects on BRCA2 proficient cells were selected.
Colony formation assay
Cells were plated into 6 well plates at a density of 200 cells/well. The next day the cells were treated with selected compounds at a range of concentrations, for different time periods (from 3 h up to 6 days). After 1 week (for VC-8 and VC-8+B2 cells and PIR clones) or 2 weeks (for U2OS cells), when colonies could be observed, the colonies were fixed and stained with methylene blue in methanol (4 g/l). Colonies consisting of more than 30 cells were subsequently counted. In addition, exponentially growing VC-8, VC-8+B2 and PIR clones 1C and 2B were exposed to varying concentrations of AG014699 for 24 hr or 6TG for 48 hr prior to seeding in 90 mm dishes in drug-free medium for colony formation. Colonies were fixed and stained with crystal violet 10-14 days later and counted on an automatic colony counter (Oxford Optronix, Oxford UK)
Pulsed-field gel electrophoresis
Cells were treated with 6TG, collected by trypsinisation, resuspended in 1% InCert-agarose (in 37°C PBS) to a final concentration of 1.5 million cells/100μl and agarose plugs were separated by Pulsed-field gel electrophoresis as previously described (26).
Immunofluorescence
Cells were grown on coverslips, treated, fixed, immunostained and analysed as previously described (24). The primary antibodies used were: mouse monoclonal anti-gammaH2AX at a dilution of 1:1000 and rabbit polyclonal anti-Rad51 (H-92, Santa Cruz) at a dilution of 1:1000.
PI staining and FACS analysis
1×106 cells were treated (or untreated) with compounds (such as B9 or 6-TG), collected by trypsinisation, and fixed in ice-cold 70% ethanol overnight at −20°C. The cells were then rehydrated in PBS and stained with 50 μg/ml PI (propidium iodide) and 100 μg/ml RNAse A in PBS for 30 min RT. Samples were further analyzed on a BD Biosciences FACScan. The data was analyzed by the WinMDI software version 2.8.
In vivo experiments
1 × 107 exponentially growing VC8 or VC8-B2 cells were injected intramuscularly into the thigh of each CD1 nude (Charles River) mouse in 50 μl of PBS and handled and analysed as previously described (13). Tumour growth was assessed using the ratio of the diameter of the right (tumour bearing) to the left (normal) thighs. When thigh ratio reached between 1.3 and 1.5 mice were divided into groups for the following treatments: AG014699 10 mg/kg (made up on day of use at 1 mg/ml in water) or 6TG 1.5 mg/kg (made up on day of use at 0.15 mg/ml in PBS) or saline (control) administered daily × 10 i.p.
Generation of mammary tumors. Brca1Δ5-13/Δ5-13;p53Δ2-10/Δ2-10 mammary tumors were generated in K14cre;Brca1F5-13/F5-13;p53F2-10/F2-10 mice and genotyped as described (27). Orthotopic transplantations of tumor fragments into syngeneic animals and caliper measurements of mammary tumors were reported previously (22)
RESULTS
BRCA2 defective cells are hypersensitive to 6-thioguanine
Individuals with inherited mutations in either BRCA1 or BRCA2 alleles have a high risk of developing breast cancer (1, 28). Here, we wanted to identify novel compounds to selectively kill BRCA2-defective cells. We developed an shRNA system to deplete the BRCA2 protein upon removal of doxycycline in U2OS sarcoma cells and we assayed the mechanistic and diversity set compound libraries from the NCI for compounds that selectively killed BRCA2-depleted cells. Mercury-(2-amino-1, 9-dihydro-6H-purine-6-thionato-N7,S6)hexyl (B9) was identified in the screen as the most efficient compound to selectively kill BRCA2-depleted U2OS cells (Supplementary Figure 1). We also found that this compound selectively killed BRCA2 defective V-C8 cells as compared to V-C8+B2 the isogenic cell line expressing BRCA2WT protein (V-C8+B2) (4) (Supplementary Figure 1). The reason for selective killing of BRCA2-defective cells was likely to be explained by the role of BRCA2 in HR since cells defective in the RAD51 paralog XRCC3 were similarly sensitive to the B9 compound (Supplementary Figure 1).
Structural analysis of the B9 compound revealed a strong resemblance to 6-thioguanine (6TG), which is a well established chemotherapy used to treat haematological malignancies in children and adults (29). We therefore decided to test the sensitivity of BRCA2-deficient cells to 6TG and found that survival was significantly lower than that of BRCA2-expressing cells (Figure 1A). Furthermore, we found that the BRCA2 protein is required to prevent apoptosis induced by 6TG, measured by subG1 population (Figure 1B) and the TUNEL assay (Figure 1C).
Figure 1. homologous recombination defective cells are hypersensitive to chemical 6TG.
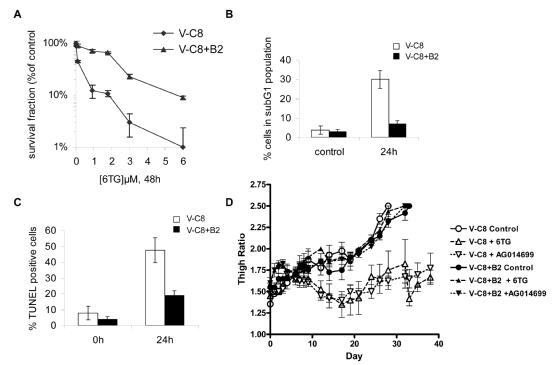
(a) 6TG selectively kill BRCA2 defective VC-8 cells in a colony formation assay. 6TG-induces apoptosis in HR defective VC-8 cells as measured by FACS analysis of the (b) subG1 population and (c) TUNEL staining. The average and standard deviation from at least three experiments is shown. (d) Tumour outgrowth in xenograft mice (5 per group) following injection of V-C8 and BRCA2 complemented V-C8+B2 upon i.p. treatment with 6TG and PARP inhibitor.
6-thioguanine selectively kills BRCA2 defective tumours
To test the hypothesis that 6TG is useful for selective treatment of BRCA2-deficient tumours, we treated mice bearing xenografts derived from BRCA2-deficient V-C8 and wt BRCA2-complemented V-C8+B2 cells. Consistent with the hypothesis, we found that neither the PARP inhibitor, AG014699, nor 6TG had any effect on the outgrowth of the BRCA2-proficient tumours (Figure 1D); in contrast, all mice with BRCA2-defective tumours responded to both 6TG and the PARP inhibitor equally, with significant growth delay and 3/5 complete tumour regressions in both groups. These results suggest that 6TG is as effective as the PARP inhibitor, AG014699, in selectively killing BRCA2-defective tumours. However, it should be noted that 6TG at 1.5 mg/kg caused greater loss of body weight than AG014699 (Supplementary Table I) and was not tolerated at a higher dose of 3 mg/kg (data not shown).
6-thioguanine induced DNA double-strand breaks are repaired by homologous recombination
6TG is an anti-metabolite of purine metabolism and is incorporated into the DNA of mammalian cells in place of guanine during DNA replication (29). The incorporated 6TG (about 1 in 104 bases) is then methylated in situ to 6-meTG by endogenous S-adenosylmethionine and becomes a substrate for mismatch repair (MMR) in the second replication round to mediate its toxicity (30-33). As a measure of formation of DSBs in DNA, we looked at the nuclear levels of γH2AX in cells treated with 6TG and we found that γH2AX foci formed with similar frequency in both BRCA2-deficient and -proficient cells (Figure 2a,b). This lack of discrepancy suggests that the hypersensitivity of BRCA2-deficient cells to 6TG is due to an inability to repair DNA damage rather than a difference in the amount of damage introduced. We also found that γH2AX foci co-localised with RAD51 foci after 6TG treatment in BRCA2-proficient cells (Figure 2c), suggesting that the RAD51 protein is recruited to DSBs to repair the lesion by HR. In contrast, RAD51 foci did not form at 6TG-induced γH2AX foci in BRCA2-defective V-C8 cells, suggesting that DSB repair is deficient in these cells (2c,d). Using pulsed-field gel electrophoresis, we found that BRCA2 defective V-C8 cells were unable to repair 6TG-induced DSBs compared to BRCA2-expressing cells (Supplementary Figure 2). To further investigate the role of HR in the 6TG induced DNA damage, we looked at the survival of cells defective in the RAD51 paralog, XRCC3 (irs1SF), and we found that these cells were considerably more sensitive to 6TG than AA8 control cells (Figure 2e). Altogether, these findings suggest that the sensitivity of BRCA2-defective cells to 6TG is due to their inability to perform HR repair. HR is important in repairing DNA damage caused by a wide range of mono- or bi-functional alkylating anti-cancer agents, for example the commonly used drugs, cisplatin and mitomycin C (34). To our knowledge, these results with 6TG represent the first time HR has been implicated in repair of thiopurine anti-metabolite drugs. Interestingly, it has previously been shown that HR-defective cells are more sensitive to O6-methyl guanine lesions than cell lines with defects in other repair pathways (35, 36), a finding which corroborates the importance of HR in repairing lesions on the O6 position of guanine.
Figure 2. homologous recombination is needed to repair the damage induced by 6TG.
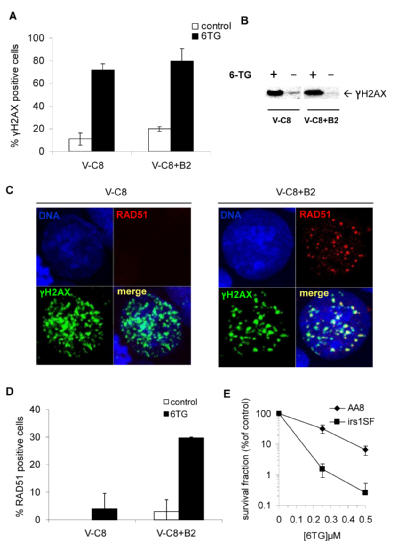
Equal amount of DNA damage in V-C8 and V-C8+B2 cells following 6-thioguanine treatment, as determined by (a) γH2AX foci (Cells containing more than 10 foci were scored as positive) or (b) by Western blot. (c) RAD51 foci do not form in V-C8 cells upon 6TG induced DNA damage. RAD51 foci do form in V-C8+B2 cells and co-localise with γH2AX foci. (d) Quantification of RAD51 foci formed in V-C8 and V-C8+B2 cells after 6TG treatment. (e) 6TG selectively kills XRCC3 defective irs1SF cells in a colony formation assay as compared to wild type control (AA8).
BRCA1-defective tumours that gained resistance to PARP inhibitors through P-glycoprotein expression remain 6-thioguanine sensitive
Increased expression from the Abcb1a and Abcb1b genes encoding the mouse drug efflux transporter P-glycoprotein (P-gp) explains resistance to the PARP inhibitor olaparib in BRCA1;p53-defective mammary tumours (22). Here, we wanted to determine whether 6TG can target such PARP inhibitor resistant tumours. To test this, Brca1Δ5-13/Δ5-13;p53Δ2-10/Δ2-10 mammary tumours derived from K14cre;Brca1F5-13/F5-13;p53F2-10/F2-10 mice were grown out and treated with olaparib as previously described (22). Small tumour fragments of an olaparib resistant tumour (T6-28) with 80-fold increased expression of the Abcb1b gene were transplanted orthotopically into syngeneic wild-type female mice. The animals were then treated with 6TG when the tumour volume reached ~200 mm3. Interestingly, we found that tumours responded to 6TG (Figure 3). This shows that spontaneous BRCA1;p53-defective mammary tumours are sensitive to 6TG, and importantly, that PARP inhibitor resistant tumours in which resistance is caused by increased P-glycoprotein-mediated drug efflux remain sensitive to 6TG. After the 10 day treatment with 6TG, tumours eventually grew back. However, such tumours were still responding to a second line treatment with 6TG, indicating that the tumours did not easily obtain resistance to 6TG.
Figure 3. Response of the PARP inhibitor resistant Brca1Δ5-13/Δ5-13;p53Δ2-10/Δ2-10 tumour T6-28 to 6TG.
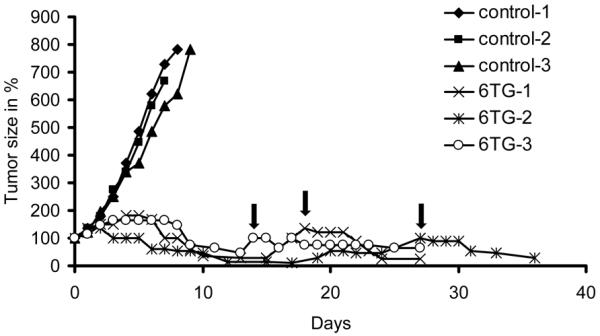
Animals carrying orthotopically transplanted tumours were treated with 1.5mg 6TG per kg i.p. daily on days 0-9 or 50mg olaparib per kg i.p. daily when the tumours reached a volume of 150-250mm3 (100% day0). When tumours relapsed back to 100% (arrows), a second treatment of 1.5mg 6TG per kg daily for 4 consecutive days was tolerated. rs.
BRCA2-defective cells and tumours that gain resistance to PARP inhibitors through genetic reversion respond to 6-thioguanine treatment
Mechanisms of acquired resistance to PARP inhibitors can also evolve through genetic reversion in BRCA2-defective cancer cells (20, 21). In such cases, a mutation in the BRCA2 gene results in that the C-terminal part of the protein is retained and the protein is overall functional in HR, despite missing a ssDNA binding domain (20, 21). In order to investigate this mechanism of resistance further, we made use of BRCA2-defective V-C8 clones selected for resistance to a PARP inhibitor (Gottipati et al 2010, submitted manuscript). All PARP inhibitor resistant (PIR) V-C8 clones harbour the same mutation, restoring the correct reading frame for BRCA2 and at the same time introducing a mutation within a highly conserved region in exon 15. This mutation affects a highly conserved arginine that was also identified in a family with breast and ovarian cancers (37) and was described in mitomycin C (MMC) resistant V-C8 cells (38). Thus, the reverted BRCA2 still has a defective ssDNA domain in the C-terminal part of the protein, as described earlier (20, 21), which restored HR as indicated by ability to form RAD51 foci in response to PARP inhibitor treatment (Supplementary Figure 3). We tested the sensitivity of PIR resistant clones 1C and 2B to the PARP inhibitor AG014699 and found that both clones had lost their sensitivity to PARP inhibitors (Figure 4a).
Figure 4. PARP-inhibitor and cisplatin- resistant BRCA2 defective cells and tumours respond to 6-thioguanine.
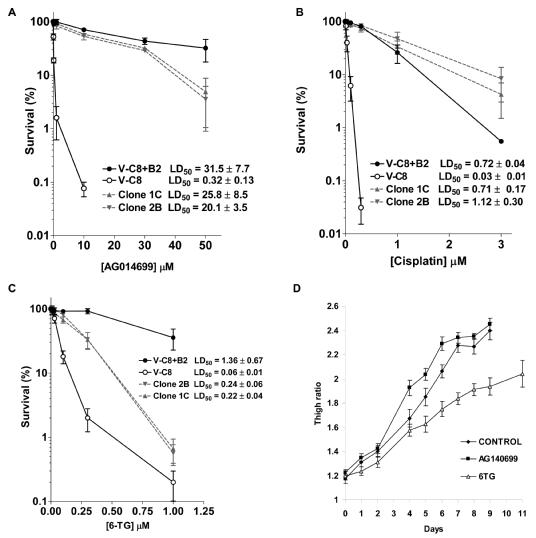
Clonogenic survival in BRCA2 defective V-C8, BRCA2 complemented V-C8+B2 and V-C8 PARP inhibitor resistant clones following treatment (a) PARP inhibitor AG014699; (b) cisplatin; (c) 6TG. The average and standard deviation of at least three experiments is shown. (d) Tumour outgrowth in xenograft mice following injection of PIR V-C8 clone 2B upon i.p. treatment with 6TG and PARP inhibitor. 6TG retards PIR V-C8 clone 2B tumour outgrowth (statistically significant in Mann Whitney test p<0.01). The average and standard error from ten mice in each group is shown.
We then tested the sensitivity of PIR V-C8 cells for cross-resistance to cisplatin and 6TG. Parental V-C8 cells are highly sensitive to both cisplatin and 6TG, compared to the BRCA2-expressing V-C8+B2 cells (Figure 4b,c). As expected from previous studies (20, 21), PIR V-C8 clones exhibited resistance to cisplatin (Figure 4b). Surprisingly, we found that PIR V-C8 cells had not fully reverted to resistance to 6TG (Figure 4c), suggesting that 6TG may still kill PIR BRCA2 defective tumours that gained resistance through genetic reversion.
We confirmed that BRCA2 revertant cells, that have acquired resistance to cisplatin, retain sensitivity to 6TG by using BRCA2 defective human pancreatic cancer cell line, Capan1, and 4 different independent cisplatin resistant Capan1 clones (20). Clones 6 and 12 acquired resistance through an additional mutation in the BRCA2 gene that corrected the frameshift caused by the 6174delT mutation in Capan1 cells, while clones 10 and 11 do not have an additional BRCA2 mutation, lack BRCA2 protein expression and are likely to have reverted to cisplatin resistance through other unknown pathways (20). Interestingly, all 4 cisplatin resistant clones showed equal sensitivity to 6TG as parental Capan1 cells (Supplementary Figure 4), providing additional evidence that PIR and cisplatin-resistant BRCA2-defective cancer cells are sensitive to treatment with 6TG.
Next we wanted to test whether 6TG can also retard outgrowth of PIR BRCA2 defective tumours that have gained resistance through genetic reversion. To test this we treated mice bearing xenografts derived from PIR clone 2B, with a 10 day treatment with AG014699 or 6TG and found that PIR clone 2B tumours only responded to the 6TG treatment and not to the PARP inhibitor (Figure 4d; statistically significant in Mann Whitney test p<0.01).
Differential sensitivity to anti-cancer drugs in genetically reverted BRCA2-defective cells
To gain further insights into the function of the restored BRCA2 protein carrying a mutation in the ssDNA binding domain, we further investigated the sensitivity of the PIR clones to a range of cytotoxic agents (Supplementary Figure 5a,b,c,d,e,f). The PIR clones exhibited similar levels of resistance to temozolomide, camptothecin and ionising radiation as the V-C8+B2 cells compared to the more sensitive V-C8 cells. Interestingly, the PIR clones were slightly more resistant to doxorubicin than the V-C8+B2, which were in turn more resistant than the V-C8 cells (Supplementary Figure 5d). Surprisingly, the V-C8 and the PIR clones were less sensitive to gemcitabine and paclitaxel than the V-C8+B2 cells (Supplementary Figure 5e,f). These data suggest that recombination defective BRCA1 and BRCA2 tumours will respond poorly to gemcitabine and paclitaxel treatments. The retained sensitivity to 6TG and resistance to gemcitabine and paclitaxel in the PIR clones is likely to be explained by the reverted BRCA2 gene which did not revert back to wild type, but retained a mutation in the ssDNA domain, which may impair HR induced by these agents. This ssDNA domain may be required for the BRCA2 response to 6TG and may prevent efficient repair of gemcitabine and paclitaxel induced lesions.
6-thioguanine induces both mismatch repair dependent and independent lesions that require HR repair
Next, we wanted to understand the mechanism for PIR cells maintaining their sensitivity to 6TG. We analysed RAD51 foci and found that PIR V-C8 cells induced RAD51 foci as efficiently as V-C8+B2 cells in response to treatment with a PARP inhibitor, but not in response to treatment with 6TG, suggesting that the BRCA2 reverted protein is not fully proficient for 6TG-induced HR (Figure 5a). We also investigated the translocation of the RAD51 protein into the chromatin fraction, as this may be associated with the efficiency of RAD51 loading on to DNA and subsequent HR (39). We found that the RAD51 protein is more efficiently recruited to DNA after 6TG in V-C8+B2 cells than in the 1C clone and conversely that the RAD51 protein is more efficiently recruited to DNA in the 1C clone than V-C8+B2 following treatment with the PARP inhibitor, KU0058948 (Supplementary Figure 6). Next, we investigated the repair of 6TG induced DNA lesions by γH2AX foci formation and find that, BRCA2 is required for efficient repair (Figure 5b). Interestingly, neither the 1C or 2B clones fully repaired the 6TG-induced DNA damage, suggesting their lack of efficient HR repair is the reason for their 6TG sensitivity.
Figure 5. PARP-inhibitor resistant BRCA2 defective cells partially activates HR in response to 6TG.
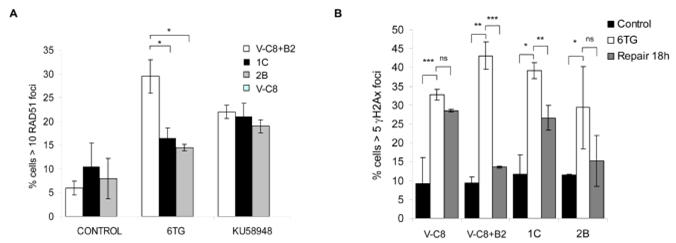
(a) Quantification of 6TG and PARP inhibitor induced RAD51 foci. BRCA2 complemented V-C8+B2 cells showed a significant increase in RAD51 foci compared to PIR V-C8 cells upon 6TG treatment (24 hours after an 18 hour treatment with 1μM of 6-TG) (b) Quantification of repair of 6TG-induced γH2AX foci. HR defective V-C8 cells fail to repair 6TG-induced γH2AX foci, while V-C8+B2 cells repairs a majority of 6TG-induced γH2AX foci. PIR V-C8 clones show an intermediate repair of 6TG-induced γH2AX foci. The average and standard error of three independent experiments is depicted. Values marked with asterisks are statistically significant in T-test (* p<0.05; ** p<0.01; *** p<0.001).
The reason for the differential response to 6TG and PARP inhibitors may be related to the production of different recombinogenic lesions in DNA. To test this hypothesis, we treated mismatch repair-defective HCT116 colorectal cancer cells and the same cells with restored mismatch repair function (HCT116+Ch3) (40) with 6TG, the PARP inhibitor 4-amino-1,8-napthalimide (ANI) and cisplatin. We found that only the cytotoxicity of 6TG was dependent upon a functional mismatch repair pathway (Figure 6a; Supplementary Figure 7). Furthermore, the level of recombinogenic DSBs, measured by γH2AX foci, were dependent on mismatch repair after 6TG treatment, whereas generation of these lesions was unaffected by PARP inhibitor and cisplatin treatment in these cell lines (Figure 6b). Our data are in line with evidence showing that toxic DSBs induced by 6TG are mismatch repair dependent (30), and it has previously been shown that HR induced by O6-guanine methylating agents depend on mismatch repair (41). This is also in line with our previous observation that 6TG-induced DSBs require HR for repair (Figure 5B; Supplementary Figure 2).
Figure 6. BRCA2 suppresses 6TG toxicity in mismatch repair deficient cells.

(a) Survival following continuous treatment with PARP inhibitor ANI, 6TG and cisplatin in HCT116 and HCT116+Ch3 cells. (b) γH2AX foci formation in hMLH1 defective HCT116 and hMLH1 complemented HCT116+Ch3 cells after a 24 hour treatment with 6TG and cisplatin or a 4 hour treatment with PARP inhibitor ANI. (c) Clonogenic survival in HCT116 and BRCA2 depleted HCT116 to increasing doses of 6TG. The average and standard deviation of three independent experiments is depicted. Values marked with asterisks are statistically significant in T-test (*** p<0.001).
There is a possibility that 6TG may produce another, mismatch repair independent HR substrate, given that the PIR V-C8 clones show an intermediate HR response to 6TG (Figure 5). To test this we shRNA depleted BRCA2 in HCT116 cells to test if the absence of 6TG-induced DSBs also abolish the requirements for HR for survival. Surprisingly, we find that mismatch repair defective HCT116 still requires BRCA2 for survival to 6TG, showing that 6TG also produces a recombinogenic lesion that is independent of mismatch repair (6c).
DISCUSSION
Here, we report that cells and/or tumours defective in the HR genes BRCA1, BRCA2 or XRCC3 are hypersensitive to 6TG and that in the case of BRCA2 this can be reversed by introduction of a vector expressing the BRCA2 protein. We show that 6TG induced RAD51 foci at 6TG-induced DSBs and that the DSBs are less efficiently repaired in BRCA2 defective cells, which correlates with increased toxicity in HR defective cells. This is to our knowledge the first time HR has been implicated in the repair of 6TG-induced DSBs.
Interestingly, the opposite result was previously reported: that expression of BRCA1WT in BRCA1 mutated HCC1937 breast cancer cells increases sensitivity to 6TG (42). However, this is unrelated to any role of BRCA1 in HR (42) and the BRCA1 mutation in HCC1937 cells is unlikely to affect HR, as RAD51 foci are efficiently induced by IR in these cells (43).
Although PARP inhibitors have been shown to efficiently kill both BRCA1 and BRCA2 defective tumours, resistance to therapy may develop within 18 to 77 weeks (18). Although, the exact mechanisms for PIR in patients remain unknown, they may involve expression of P-glycoprotein efflux pumps as in mammary mice tumours (22) or through genetic reversion of either BRCA1 or BRCA2 (19-21). Here, we show that 6TG efficiently kills PIR BRCA1 defective mammary tumours (Figure 3), which is likely explained by 6TG not being a substrate for P-glycoprotein (44). Furthermore, we show that genetically reverted BRCA2 defective cells and tumours respond to 6TG. Altogether, these findings suggest that 6TG may be efficient in also killing advanced and drug resistant BRCA1 or BRCA2 defective tumours.
Here, we find that PIR V-C8 clones do not completely revert back to a functional HR phenotype in response to 6TG (Figure 5), which likely explains their 6TG sensitivity. This suggests that there may be several lesions formed following 6TG treatment that trigger HR. For instance, we recently showed that HR is involved in restart at stalled replication forks, which does not involve DSB repair (45, 46). Thus, there is a possibility that 6TG may cause replication lesions other than mismatch repair dependent DSBs that trigger HR. In support for this notion, we find that mismatch repair defective HCT116 cells are sensitised to 6TG by depletion of BRCA2 in spite of already being defective in mismatch repair. This shows that both mismatch repair dependent and independent HR lesions are formed by 6TG. Also, this would explain the intermediate response in PIR V-C8 clones to 6TG.
In conclusion, we show that 6TG can be efficiently used to selectively kill BRCA2 defective tumours and that 6TG may be used as treatment for BRCA1 or BRCA2 mutant tumour which are resistant to cisplatin chemotherapy and/or PARP inhibitor therapy by various mechanisms.
Supplementary Material
Acknowledgements
We thank the NCI, Drs Toshiyasu Taniguchi, Bert Vogelstein, Larry Thompson, Zdenek Hostomsky and Mark O'Connor for materials and discussions. This study was supported by The Swedish Cancer Society, the Swedish Children's Cancer Foundation the Swedish Research Council, the Swedish Pain Relief Foundation, Cancer Research UK, the NIHR Biomedical Research Centre, Oxford, and the Medical Research Council. The authors declare that they have no competing financial interest.
REFERENCES
- 1.Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–92. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 2.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 3.Xia F, Taghian DG, DeFrank JS, et al. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc Natl Acad Sci U S A. 2001;98:8644–9. doi: 10.1073/pnas.151253498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraakman-van der Zwet M, Overkamp WJ, van Lange RE, et al. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol. 2002;22:669–79. doi: 10.1128/MCB.22.2.669-679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraakman-van der Zwet M, Wiegant WW, Zdzienicka MZ. Brca2 (XRCC11) deficiency results in enhanced mutagenesis. Mutagenesis. 2003;18:521–5. doi: 10.1093/mutage/geg032. [DOI] [PubMed] [Google Scholar]
- 6.Meijers-Heijboer H, van den Ouweland A, Klijn J, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–9. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 7.Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 8.Rahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid S, Schindler D, Hanenberg H, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–4. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 10.Xia B, Dorsman JC, Ameziane N, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–61. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 11.Seal S, Thompson D, Renwick A, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–41. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 12.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–90. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose)polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 14.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 15.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–8. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 16.Schultz N, Lopez E, Saleh-Gohari N, Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003;31:4959–64. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–45. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 18.Fong PC, Boss DS, Yap TA, et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 19.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581–6. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 22.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruun D, Folias A, Akkari Y, Cox Y, Olson S, Moses R. siRNA depletion of BRCA1, but not BRCA2, causes increased genome instability in Fanconi anemia cells. DNA Repair. 2003;2:1007–13. doi: 10.1016/s1568-7864(03)00112-5. [DOI] [PubMed] [Google Scholar]
- 24.Bryant HE, Petermann E, Schultz N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28:2601–15. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–8. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 26.Lundin C, Erixon K, Arnaudeau C, et al. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol. 2002;22:5869–78. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Holstege H, van der Gulden H, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A. 2007;104:12111–6. doi: 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 29.Karran P, Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer. 2008;8:24–36. doi: 10.1038/nrc2292. [DOI] [PubMed] [Google Scholar]
- 30.Swann PF, Waters TR, Moulton DC, et al. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273:1109–11. doi: 10.1126/science.273.5278.1109. [DOI] [PubMed] [Google Scholar]
- 31.Griffin S, Branch P, Xu YZ, Karran P. DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells: implications for tolerance to DNA methylation damage. Biochemistry. 1994;33:4787–93. doi: 10.1021/bi00182a006. [DOI] [PubMed] [Google Scholar]
- 32.Waters TR, Swann PF. Cytotoxic mechanism of 6-thioguanine: hMutSalpha, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry. 1997;36:2501–6. doi: 10.1021/bi9621573. [DOI] [PubMed] [Google Scholar]
- 33.Yan T, Berry SE, Desai AB, Kinsella TJ. DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin Cancer Res. 2003;9:2327–34. [PubMed] [Google Scholar]
- 34.Caldecott K, Jeggo P. Cross-sensitivity of gamma-ray-sensitive hamster mutants to cross-linking agents. Mutat Res. 1991;255:111–21. doi: 10.1016/0921-8777(91)90046-r. [DOI] [PubMed] [Google Scholar]
- 35.Lundin C, North M, Erixon K, et al. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roos WP, Nikolova T, Quiros S, et al. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair (Amst) 2009;8:72–86. doi: 10.1016/j.dnarep.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Santarosa M, Dolcetti R, Magri MD, et al. BRCA1 and BRCA2 genes: role in hereditary breast and ovarian cancer in Italy. Int J Cancer. 1999;83:5–9. doi: 10.1002/(sici)1097-0215(19990924)83:1<5::aid-ijc2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 38.Wiegant WW, Overmeer RM, Godthelp BC, van Buul PP, Zdzienicka MZ. Chinese hamster cell mutant, V-C8, a model for analysis of Brca2 function. Mutat Res. 2006;600:79–88. doi: 10.1016/j.mrfmmm.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Sorensen CS, Hansen LT, Dziegielewski J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 40.Koi M, Umar A, Chauhan DP, et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–12. [PubMed] [Google Scholar]
- 41.Zhang H, Marra G, Jiricny J, Maher VM, McCormick JJ. Mismatch repair is required for O(6)-methylguanine-induced homologous recombination in human fibroblasts. Carcinogenesis. 2000;21:1639–46. doi: 10.1093/carcin/21.9.1639. [DOI] [PubMed] [Google Scholar]
- 42.Yamane K, Schupp JE, Kinsella TJ. BRCA1 activates a G2-M cell cycle checkpoint following 6-thioguanine-induced DNA mismatch damage. Cancer Res. 2007;67:6286–92. doi: 10.1158/0008-5472.CAN-06-2205. [DOI] [PubMed] [Google Scholar]
- 43.Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59:3547–51. [PubMed] [Google Scholar]
- 44.Pieters R, Hongo T, Loonen AH, et al. Different types of non-P-glycoprotein mediated multiple drug resistance in children with relapsed acute lymphoblastic leukaemia. Br J Cancer. 1992;65:691–7. doi: 10.1038/bjc.1992.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010 doi: 10.1093/carcin/bgq064. doi: bgq064 [pii] 10.1093/carcin/bgq064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.