

Abstract
Reported herein is a method that can be used to study the role of cholesterol in the microclustering of a ubiquitous class of membrane receptors, termed integrins. Integrin microclustering was measured using a fluorescence resonance energy transfer assay that does not require direct attachment of fluorescent donors or acceptors onto the integrins, and thus minimizes unwanted perturbations to integrin clustering. Membrane cholesterol levels were reduced using methyl-β-cyclodextrin (mβCD), as confirmed by Amplex Red assays of total cellular lipid or plasma membrane lipid extract. Subsequent changes in integrin microclustering were measured in cells expressing wild-type (WT) or mutant integrins. Although less integrin microclustering was measured after 27% membrane cholesterol depletion in a cell line expressing WT integrins, there was no statistically significant change for cells expressing α-cytoplasmic integrin mutants after a 45% reduction in plasma membrane cholesterol, and a significant increase in clustering for cells expressing ligand-binding domain integrin mutants after a 57% decrease in membrane cholesterol. These results are explained by differences in WT and mutant integrin partitioning into lipid nanodomains. Restoration of original cholesterol levels was used to confirm that the measured changes in membrane properties were cholesterol-dependent. No correlations between lipid diffusion and integrin microclustering were measured by means of fluorescence recovery after photobleaching using a fluorescent lipid mimetic. Similar lipid diffusion coefficients were measured after cholesterol depletion, irrespective of the integrins being expressed.
Introduction
The survival, growth, proliferation, differentiation, and proper functioning of cells depend largely on a dynamic flow of information being maintained between the external and internal environments of the cell (1). To this end, cells employ receptors to relay information inside and outside of the cell (2–5). One of the mechanisms for signal transduction involves clustering of receptors within the cell membrane. Receptor clustering is involved in many vital processes, including immunological synapse formation, actin cytoskeleton regulation, and leukocyte regulation (6–8). Integrins comprise a class of receptors that are fundamentally important for many critical cellular functions (1,9). They are heterodimeric proteins composed of one α-subunit noncovalently associated with one β-subunit (10). Both subunits contain a large extracellular domain and (by comparison) a short cytoplasmic domain. They mediate signaling through the cytoplasm by binding to intracellular proteins, and through the extracellular matrix by binding to ligand (9).
Fluorescence microscopy has been used to study the clustering of integrins within the cell membrane (11–13). Observing clusters in live cells that are smaller in size than the diffraction limit of light requires an imaging technique such as fluorescence resonance energy transfer (FRET) (14–16). In previous studies, integrin microclustering was measured using a FRET assay that did not require direct attachment of donor and acceptor FRET pairs to the integrins (17,18). Energy transfer was measured using transmembrane reporter peptides, which were generated by cloning the FRET donor or acceptor fluorescent protein to the transmembrane and cytoplasmic domain of the integrin β-subunit. Hereafter, these will be referred to as FRET reporters. The FRET reporters were coexpressed with integrins (17,18). When the integrins cluster in the membrane, so do the FRET reporters. This decreases the average separation distance between the donor and acceptor FRET reporters, and increases energy transfer. Conversely, when the separation distance between the integrins increases, less energy transfer is measured from the FRET reporters. The mechanism for the coclustering of integrins and FRET reporters is still under investigation. However, possible mechanisms can be deduced from evidence in the literature. The β-transmembrane and cytoplasmic domains contained in the FRET reporters were shown to be sufficient for clustering with integrins at muscle termini in vivo; additionally, it was reported that a chimera containing the transmembrane domain of an unrelated protein and the cytoplasmic domain of the β-subunit also localized to muscle termini, but the transmembrane and cytoplasmic domains of the unrelated protein did not localize to muscle attachments (19). Finally, a peptide containing the transmembrane and cytoplasmic domains of the vertebrate β3 subunit was shown to form homotrimers by polyacrylamide gel electrophoresis in the absence of the extracellular domain (20). Control experiments showed that FRET reporters did not alter key integrin properties, and no energy transfer was measured when FRET reporters, but no integrins, were expressed in the membrane (17,18).
Cholesterol intercalates between the fatty acyl chains of the lipid bilayer and is known to maintain membrane structure, regulate membrane fluidity, and interact directly with some membrane proteins (21–25). Cholesterol is an important constituent in membrane nanodomains (i.e., lipid rafts), which are areas with nonuniform compositions of lipids and proteins relative to the bulk membrane. Membrane nanodomains have been shown to play a role in many signal transduction events, such as immunoglobulin E (IgE) signaling during the allergic immune response, T-cell activation, glial-cell-derived neurotrophic factor signaling, and integrin leukocyte function-associated antigen (LFA)-1-mediated cell binding (26–30). Cholesterol levels can be modulated in the cell membrane with the use of cyclodextrins, which partition cholesterol from cell membranes into their interior pores. Cyclodextrins can also partition other membrane components, but preferentially extract cholesterol over other lipid components (31).
In this study, we measured the effect of cholesterol on the microclustering of αPS2CβPS integrins expressed in S2 cells using the above-mentioned FRET assay. We measured total cellular cholesterol and plasma membrane cholesterol by extraction and Amplex Red assays. In combination, we measured alterations in lipid diffusion in live cells using fluorescence recovery after photobleaching (FRAP) under native and reduced cholesterol levels. Mechanisms underlying the role of cholesterol in αPS2CβPS clustering are postulated.
Materials and Methods
S2 cell culture
All experiments were performed using transformed Drosophila S2 cells. Cells were cultured in Shields and Sang M3 insect medium (Sigma-Aldrich, St. Louis, MO) with 10% heat-inactivated fetal bovine serum (Irvine Scientific, Santa Ana, CA), 12.5 mM streptomycin, 36.5 mM penicillin, and 0.2 μM methotrexate (Fisher Scientific, Pittsburgh, PA) in a 22°C incubator. For the FRET assays, cells were cotransfected to express α- and β-integrin subunits and FRET reporters. The complete protein sequences for the FRET reporters containing mVenus and mCherry fluorescent proteins, and mutant integrin subunits can be found elsewhere (17,32–34). For the FRAP assays, cells were transfected to express integrin subunits (i.e., no FRET reporters). All of the exogenous proteins contained the heat-shock promoter.
Heat-shock treatment
Cells were transferred from the cell culture dish to a polypropylene tube and heat-shocked for 30 min at 36°C to induce the expression of integrins and/or FRET reporters. To achieve maximum protein expression, the cells were then placed in a 22°C incubator for 3–4 h, as specified in the sections below. The heat-shock treatment was found to increase total cellular cholesterol by 90%; therefore, heat-shock treatment was performed before all lipid extractions, FRET, and FRAP analyses were conducted.
Cholesterol depletion and restoration
For cholesterol depletion, cells that had been heat-shocked and incubated for 3 h were centrifuged at ∼600×g and the resulting pellet was resuspended at a concentration of 2×106 cells/mL in serum-free M3 medium containing 2 mM methyl-β-cyclodextrin (mβCD) (Sigma-Aldrich, St. Louis, MO) and incubated at 22°C for 30 min. Culturing cells in the absence of serum was tested as a second method for reducing cellular cholesterol concentration. Although total cholesterol levels decreased by ∼40% (day 1), ∼60% (day 2), and ∼75% (day 3) in the absence of serum, there was a concomitant 54–65% increase in cell death relative to control cells grown in serum containing medium. Because of the increased cell death, this method was not further pursued as a means of modulating cellular cholesterol levels.
For cholesterol restoration, cholesterol-depleted cells were washed with serum-free M3 medium and then resuspended in serum-free M3 medium containing 100 μM cholesterol-loaded cyclodextrin (chol-mβCD) complex (CTD Inc., High Springs, FL) for 1 h at 22°C. Before FRET, FRAP, or lipid extraction were performed, the cholesterol-depleted and cholesterol-restored cells were washed in serum-free medium to remove the cyclodextrin.
Extraction of total cellular lipids
After heat shock and 3 h incubation, the total cellular lipids were extracted using the Bligh-Dyer method (35). Briefly, 3.0 mL chloroform/methanol (2:1 v/v) was added to 1.0 mL medium containing 106 cells followed by vigorous vortexing for 15 min. Then, 1.0 mL of 1.0 M NaCl was added to the solution and the sample was vortexed for 1 min. The solution was allowed to sit for 10 min, and the chloroform phase was collected and filtered using Whatman filter paper No. 1. The chloroform was evaporated under nitrogen, and the cellular lipids were resuspended in phosphate buffer containing 0.1 M potassium phosphate, pH 7.4, 50 mM NaCl, 5 mM cholic acid, and 0.1% Triton X-100.
Extraction of plasma membrane lipids
After heat shock and 3 h incubation, plasma membrane lipid extraction was performed as previously described (36), with the following minor changes: Cells (3×106) in serum-free M3 media were plated on polylysine-coated glass petri dishes and allowed to spread for 2 h at 22°C. All rinse steps were performed with BES Tyrodes buffer. After membrane disruption occurred, the membrane lipid was extracted with 5 mL chloroform/methanol (2:1 v/v) with continuous rocking for 1 h. The solution was collected and added to a glass test tube. The subsequent procedure was similar to that described above for the extraction of total cellular lipids.
Quantitative measurements of cholesterol
Cholesterol levels were quantified using an Amplex Red cholesterol assay (Life Technologies, Carsbad, CA) without cholesterol esterase (37). This was done to ensure that cholesterol esters would not contribute to the measurement. Fluorescence was measured with a Synergy HT fluorescence microplate reader (BioTek Instruments, Winooski, VT) using an excitation filter of 530/25 nm and emission filter of 590/35 nm. Each microplate contained a series of five cholesterol standards in triplicate (i.e., 15 standards/plate) and a blank that were used to construct a calibration curve. All values were background-subtracted. Analysis of calibration data from 10 replicate microplates indicated that heteroscedasticity was present and weighted linear fitting was required (38). Weighted 1/[cholesterol]2 linear curve fitting produced the lowest summed relative errors and was used to construct the best fit line for each calibration set (see Table S1 in the Supporting Material). The unknown cholesterol concentrations were calculated using values from triplicate measurements of the same sample, and the calibration function was measured using standards on the same microplate. The uncertainty in measuring the cholesterol concentration from the calibration functions was determined as described in quantitative-analysis textbooks (39). The cholesterol limit of detection (three times the standard deviation (SD) in the signal from the blank divided by the slope of the weighted calibration curve) was 0.7 μM. All unknowns were at the limit of detection or higher. Each data point represents weighted averages from three replicate measurements.
FRET assay
After heat shock and 3 h of incubation, the cells were subjected to cholesterol depletion/restoration as described above. The cell densities were adjusted to 5×105 cells/mL in serum-free medium before the cells were placed on a ligand-coated substrate, prepared as described previously (18). The cells were allowed to spread onto the ligand-coated substrate for 1 h, followed by FRET data collection within the next hour as described previously (17,18).
The FRET data were analyzed using an in-house-developed Java plug-in for the software ImageJ. After subtracting the background value from each pixel, the plug-in calculates a FRET (Eapp) value on a pixel-by-pixel basis using the following equation (40):
| (1) |
where IDA, IAA, and IDD are intensities obtained from the images with the FRET, acceptor, and donor filters, respectively. The terms a, b, c, and d account for the bleedthrough in the filter sets, as previously described (17,18). The subtracted background value was calculated for each image by averaging several pixels that did not contain cells. The background relative SD across an image was 7% for the acceptor filter, 6% for the donor filter, and 5% for the FRET filter. The G term in Eq. 1 is instrument-specific and correlates the decrease in donor fluorescence with the increase in acceptor fluorescence due to energy transfer (41). The G-value for the instrument setup used in these studies is 1.4.
The energy transfer measured for each cell is an average of all pixel values between the cell edge and the perinuclear region, where intracellular FRET reporters contribute to the signal. Analysis of the pixel values from a representative cell provides an average value of 508 and an SD of 22 among 770 pixels. The average diameter of the analyzed spread cells was 28 ± 5 μm (αβ Reporters), 33 ± 7 μm (αanaβ Reporters), and 26 ± 6 μm (αβV409D Reporters). These measured diameters are indicative of the cells used for the FRET analysis, but not the entire cell population. A minimum cell diameter of 20 μm is set for the lower threshold of analysis, which is required to distinguish between the perinuclear region and the cell edge. The similarity in cell diameters indicates that similar areas are analyzed for each cell.
Every reported FRET value represents an average of the values obtained from 100 individual cells obtained over the course of three replicate experiments. All FRET data were statistically analyzed using the software JMP 7 (SAS Institute, Cary, NC) with statistical consulting from the Department of Statistics, Iowa State University. The raw FRET data, being not normally distributed, were log-transformed and the means were calculated (42,43). The means of the log-transformed data were compared between FRET data sets of treated and untreated cells. After unequal variance among the data sets was confirmed, the statistical significance between the data sets was assessed by means of Welch t-tests. The significance of the statistical test is indicated by p-values. A statistically significant p-value is one that is <5%, indicating that the means from the two data sets are not the same. A p-value > 5% indicates that there is not enough statistical evidence to show dissimilarity between the two data sets. The data are reported in the original data scale by converting the means of the transformed data as discussed in standard statistics textbooks (44).
FRAP assay
After heat shock and 3 h incubation, the cells were subjected to cholesterol depletion/restoration as mentioned above. The cell densities were adjusted to 5×105 cells/mL in serum-free medium. A carbocyanine DiD dye (Life Technologies, Carsbad, CA) was added at a final concentration of 11.9 μM. The cells were immediately plated onto the ligand-coated substrate and allowed to spread in the dark for 1 h. The medium was then replaced with 20 mM BES Tyrodes buffer before FRAP analysis was performed.
The microscope used for these studies contains two excitation light paths: one for a 635 nm diode laser (Newport Corp., Irvine, CA) and one for a mercury (Hg) lamp. The Hg lamp was used to image the cells before and after the membrane-incorporated DiD dye was bleached with a laser focused to a 3 μm2 spot. A laser shutter and a CCD (Princeton Instruments, Trenton, NJ) shutter were synchronized with an external trigger. The trigger timing was 0.40 s between image captures, 0.35 s for image capture, and 0.35 s for the photobleaching laser pulse. The recovery curves from 10 replicate measurements were normalized so that the prephotobleached fluorescence intensity would be one. The normalized curves were then averaged and divided by a similarly averaged curve generated from a nonphotobleached area of each analyzed cell. This latter step accounts for photobleaching from the Hg lamp during the recovery period. The resultant curve was fit to a double exponential using Igor Pro 6.1 (WaveMetrics, Portland, OR), which provided a better fit than a single exponential curve as determined by the χ2 values. The diffusion coefficients and percentage of the population with the indicated diffusion coefficient were determined from the double exponential fit parameters. The listed uncertainty for these parameters was generated from the SD of the corresponding coefficients in the double exponential fit. The immobile fraction was measured using a previously reported equation for each photobleached corrected recovery curve (i.e., before averaging the 10 replicate measurements) and an average value was calculated (45). One-way analysis of variance (ANOVA) revealed that, for a given cell line, the differences in the measured immobile fraction were not statistically significant.
Results and Discussion
A schematic of the assay used to measure changes in integrin microclustering due to altered cholesterol levels is shown in Fig. 1. When the separation distance between integrins decreases, the separation distance between the donor and acceptor FRET peptides decreases, as previously reported (17). This results in more energy transfer between the donor and acceptor fluorescent proteins. Similarly, less energy transfer is measured when the integrin separation increases, since the separation distance between the FRET reporters also increases. The total cellular and plasma membrane cholesterol concentrations, lipid diffusion coefficients, and integrin microclustering levels were measured in three cell populations: 1), untreated cells containing native levels of cholesterol; 2), cells with depleted levels of cholesterol; and 3), cells that were first depleted of cholesterol and then restored to native levels. A comparison of the measured parameters in the three populations provides evidence for the role of cholesterol in altering the organization of integrins in the cell membrane.
Figure 1.
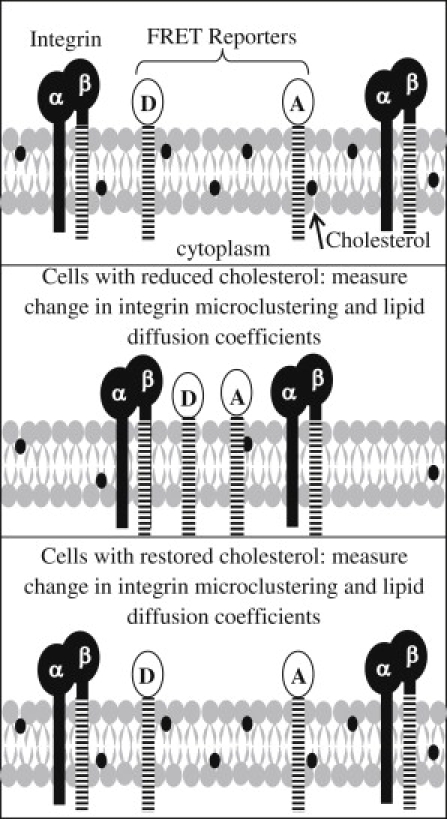
Schematic of the FRET assay used to measure integrin microclustering with donor and acceptor FRET reporters. Energy transfer is measured (top) before the plasma membrane cholesterol level is altered, (middle) after the concentration of cholesterol is reduced, and (bottom) after cholesterol levels are restored to approximately the starting concentration before treatment.
For these studies, we chose to use cell lines derived from Drosophila S2 cells transformed to express αPS2CβPS integrins because of the wealth of data available on the microclustering of this integrin (17,18). This enables a direct comparison with previous data obtained for the microclustering of this integrin upon alteration of cholesterol levels. There is substantial structural homology between vertebrate and invertebrate integrins, and many similarities between the integrin-signaling pathways (46). In many cases, the information gained regarding integrin microclustering in S2 cells can be used to advance our understanding of vertebrate integrins.
Unlike mammalian cells, Drosophila cells are sterol auxotrophs and derive sterols from their environment (47,48). In the case of cultured Drosophila cells, the source of the sterols is the fetal calf serum added to the growth medium. As confirmed by reverse phase high-performance liquid chromatography, the main sterol incorporated in the cell membrane of the cultured cells is cholesterol (Fig. S1). Fig. 2 A (black data bars) shows the weighted average total cellular cholesterol concentration per cell before cholesterol depletion. The cells utilized in these studies expressed wild-type (WT) or mutant integrins and FRET reporter peptides. Two well-characterized integrin mutants were used in this study. The mutant αanaβ integrin contains a two-point mutation in the α-subunit near a site where cytoplasmic proteins are known to bind, and is considered to mimic the signal transduction from inside to outside the cell (49). The mutant αβV409D integrin contains a single point mutation located in the extracellular ligand-binding domain of the β-subunit, and is considered to mimic signal transduction from outside to inside the cell (49). An increased affinity for ligand (compared to WT integrins) has been measured for both mutants (17).
Figure 2.
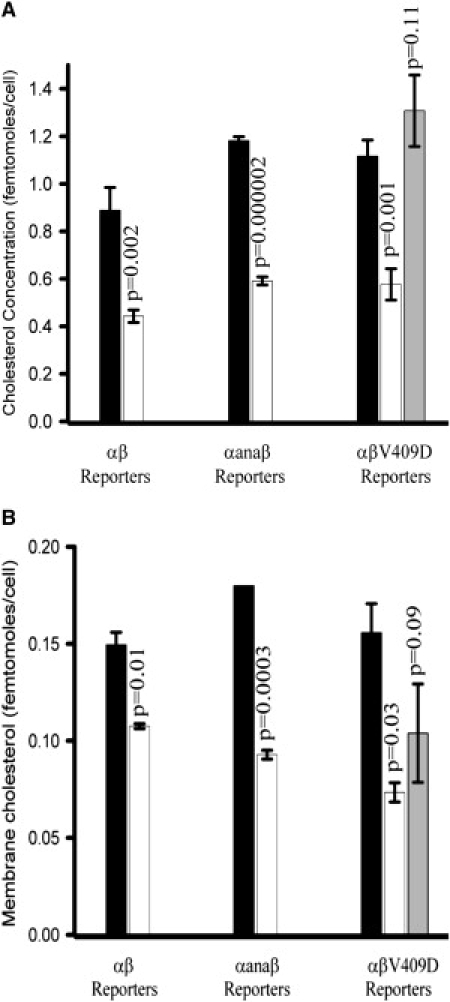
Graphs showing the weighted average of total cellular cholesterol per cell (A) and plasma membrane cholesterol (B) in three cell populations: (black) untreated cells before cholesterol reduction, (white) after reduction with mβCD to extract cholesterol, and (gray) after addition of chol-mβCD to the growth medium to restore cholesterol levels (αβV409D cell line only). Error bars represent weighted SDs from three replicate experiments; p-values indicate comparisons to untreated cells obtained using one-way ANOVA. Details of the cell lines are found in the text.
One-way ANOVA confirmed that there were statistically insignificant variations in cholesterol levels in untreated cells expressing WT and mutant integrins (Fig. 2 A, black data bars). The cholesterol concentration in all three cell lines is consistent with previous studies in which 10−14 to 10−16 moles cholesterol/cell were measured (50,51). For all three cell lines after cholesterol reduction with mβCD, there was an ∼50% reduction in total cellular cholesterol concentration (Fig. 2 A, white data bars) relative to untreated cells.
Plasma membrane cholesterol was measured in untreated and cholesterol-depleted cells (Fig. 2 B). The plasma membrane lipid extraction protocol isolates only a portion of the total cholesterol in the plasma membrane; however, it enables a comparison between cells expressing different integrins and cells that have undergone different treatments. Similar to the total cellular cholesterol concentration, the amount of cholesterol in the plasma membrane does not statistically vary for cells expressing WT or mutant integrins. However, after cholesterol depletion with mβCD, there is a statistically significant difference in plasma membrane cholesterol concentration. There is a 27% decrease in cells expressing WT integrins, a 45% decrease in cells expressing αanaβ integrins, and a 57% decrease in cells expressing αβV409D integrins. Since there is no statistically significant difference in cholesterol concentration in untreated cells or total cellular cholesterol concentrations after cholesterol depletion, and since the cells are not exposed to serum between cholesterol depletion and lipid extraction, there must be a decrease in intracellular cholesterol concentration upon cholesterol depletion with mβCD. Because only cholesterol in the plasma membrane is in direct contact with the mβCD solution, it is assumed that only plasma membrane cholesterol is available for partitioning into the mβCD. This strongly supports the notion that intracellular cholesterol partitions into the plasma membrane as cholesterol is extracted by the mβCD to different extents in the three cell lines.
Plasma membrane phospholipid content was measured as previously described in native and cholesterol-depleted cells (36). Although there was a small decrease in phospholipid content after cholesterol depletion compared to untreated cells, this difference was not determined to be significant by one-way ANOVA. The change in phospholipid concentration (ΔPhospholipid = Puntreated − Pcholesterol reduction) was 4.5 × 10−14 moles/cell (p = 0.3) αβ; 2.4 × 10−14 moles/cell (p = 0.8) αanaβ; and 1.9 × 10−13 moles/cell (p = 0.7) αβV409D.
Cholesterol extraction and lipid diffusion coefficients
Cholesterol has been shown to affect lipid diffusion in the cell membrane (52–54). To evaluate the role of cholesterol in lipid diffusion in WT and mutant integrin expressing S2 cells, we performed FRAP measurements to measure the diffusion coefficient of a fluorescent lipid analog: DiD. FRAP involves photobleaching of DiD in a defined area in the cell membrane, and then recording the time it takes to repopulate the photobleached species by the diffusion of the unbleached dye molecules. To avoid any spectral interference due to the fluorescence from the FRET reporters, we used cells expressing WT or mutant integrins with no FRET reporter for the FRAP studies. The average recovery curves from 10 replicate measurements in cells with native cholesterol levels, depleted cholesterol levels, and restored cholesterol levels (αβV409D cell line only) are provided in Fig. S2. The parameters calculated from the double exponential fit of the recovery curves are shown in Table 1.
Table 1.
Slow and fast lipid diffusion coefficients measured from the fluorescence recovery curves for untreated cells, cholesterol-depleted cells, and (αβV409D cell line only) cholesterol-restored cells
| Untreated cells∗ | Cholesterol-depleted cells∗ | Cholesterol-restored cells∗ | ||||
|---|---|---|---|---|---|---|
| Cell line | Diffusion coefficient (cm2/s) | Percent of population | Diffusion coefficient (cm2/s) | Percent of population | Diffusion coefficient (cm2/s) | Percent of population |
| αβ Reporters | 4.4 × 10−10 ± 0.7 × 10−10 | 50 ± 10 | 3.4 × 10−10 ± 0.5 × 10−10 | 46 ± 6 | - | - |
| 2.5 × 10−9 ± 0.3 × 10−9 | 50 ± 10 | 2.8 × 10−9 ± 0.3 × 10−9 | 57 ± 7 | - | - | |
| αanaβ Reporters | 5.0 × 10−10 ± 0.6 × 10−10 | 50 ± 20 | 6.1 × 10−10 ± 0.4 × 10−10 | 55 ± 4 | - | - |
| 2.0 × 10−9 ± 0.2 × 10−9 | 50 ± 10 | 4.0 × 10−9 ± 0.6 × 10−9 | 45 ± 1 | - | - | |
| αβV409D Reporters | 4.1 × 10−10 ± 0.5 × 10−10 | 50 ± 9 | 4.7 × 10−10 ± 0.9 × 10−10 | 40 ± 10 | 3.2 × 10−10 ± 0.6 × 10−10 | 44 ± 8 |
| 2.5 × 10−9 ± 0.3 × 10−9 | 50 ± 10 | 3.0 × 10−9 ± 0.4 × 10−9 | 60 ± 10 | 2.2 × 10−9 ± 0.2 × 10−9 | 56 ± 9 | |
Parameters calculated from double exponential fit to the average curve generated from 10 replicate measurements ± the SD calculated from the uncertainty of the corresponding coefficients obtained in the double exponential fit.
A slow and a fast diffusion coefficient are obtained from the double exponential fit parameters. The diffusion coefficients in Table 1 are consistent with the values obtained for other cell types, which range from 10−9 to 10−10 cm2/s (52–55). The slowly diffusing component represents ∼50% of the measured diffusing species, and the faster component represents the other 50% for all recovery curves analyzed. Analysis of the χ2 values indicates that additional exponential terms are not warranted in the fit; however, there may be additional species in the membrane that are not detectable by FRAP. The origin of the two diffusion coefficients can be understood from FRAP measurements using a similar fluorescent lipid mimetic in a single-component, solid-supported lipid bilayer (56). The recovery curve is fit to a double exponential fit to generate two diffusion coefficients that differ by approximately an order of magnitude. Membrane nanodomains would not exist in single-component bilayers, and the upper leaflet is exposed only to buffer. In this case, the two diffusion coefficients correspond to the leaflet exposed to the solid support, which hinders lipid diffusion, and the opposing leaflet. Although the two diffusion coefficients measured in the spread S2 cells are orders of magnitude slower than those measured for the lipid bilayer, they are most likely the result of one leaflet being in contact with the solid support (i.e., 50% of the measured diffusing species) and the opposing leaflet being exposed to the cytoplasm (i.e., the other 50% of the measured diffusing species).
Although there is generally an increase in the slow and fast lipid diffusion coefficient in all cell lines after cholesterol depletion (Table 1, Fig. S2), the magnitude of the change falls within the uncertainty of the measurement for all cell lines, except for the cells expressing the αanaβ integrins. For this cell line, the fast component increases by 50% and the slow component increases by ∼22% after cholesterol depletion, suggesting that cholesterol reduction increases lipid diffusion in the inner leaflet more than in the leaflet exposed to the substrate in this cell line. Overall, no correlation can be found between the amount of cholesterol extracted from the plasma membrane and the change in the lipid diffusion coefficients in this data set.
An immobile fraction can be measured from the fluorescence recovery curves. This parameter corresponds to species that do not diffuse out of the probe area, and produce <100% recovery of the fluorescence after photobleaching. No statistically significant differences are found among the immobile fractions measured for any of the cell lines with native or depleted cholesterol concentrations (Table 2).
Table 2.
Average percent immobile fraction measured from 10 replicate fluorescence recovery curves for untreated cells, cholesterol-depleted cells, and cholesterol-restored cells (αβV409D cell line only)
| Cell lines | Untreated cells∗ | Cholesterol-depleted cells∗ | Cholesterol-restored cells∗ |
|---|---|---|---|
| αβ Reporters | 13.1 | 12.9 (p = 0.95) | - |
| αanaβ Reporters | 11.9 | 16.1 (p = 0.18) | - |
| αβV409D Reporters | 12.2 | 9.2 (p = 0.3) | 13.9 (p = 0.5) |
Statistical significance was tested with one-way ANOVA, and the results are indicated by the p-values.
Cholesterol affects integrin microclustering
Cells were spread on a glass substrate coated with a ligand for the αPS2CβPS integrins at a ligand surface density of 3–5%. To ensure that integrin-ligand interactions were the only mechanism for cell spreading, nonspecific interactions with the glass substrate were inhibited by coating the remaining exposed glass with bovine serum albumin. Previous studies found that the properties of integrin microclustering are dependent on the ligand's surface density (17,18). Under these conditions, we attempted to elucidate the role of cholesterol in integrin microclustering with minimal binding to extracellular ligand.
Table 3 shows the average FRET values for the three cell lines before and after cholesterol depletion. At 3–5% ligand surface density, the mean FRET value for cells expressing WT or mutant integrins and FRET reporters indicates statistically similar levels of energy transfer for all three cell lines (untreated cells). These results indicate similar amounts of integrin microclustering within the assay detection limit in all cell lines before cholesterol depletion, and are consistent with previous studies (18).
Table 3.
Integrin microclustering levels measured by FRET for the untreated cells, cholesterol-depleted cells, and (αβV409D cell line only) cholesterol-restored cells
| Cell lines | Untreated cells∗ | Cholesterol-depleted cells∗ | Cholesterol-restored cells∗ |
|---|---|---|---|
| αβ Reporters | 0.010 | 0.005 (p = 0.02) | - |
| αanaβ Reporters | 0.011 | 0.014 (p = 0.36) | - |
| αβV409D Reporters | 0.008 | 0.026 (p = 0.0001) | 0.006 (p = 0.45) |
Statistical significance was tested with a Welch t-test, and the results are indicated by the p-values.
After the membrane cholesterol concentration is reduced, integrin microclustering is altered in two of the three cell lines studied (Table 3, Cholesterol-depleted cells). There is a statistically significant 50% decrease in energy transfer for cells expressing WT integrins after cholesterol reduction, indicating that there is less microclustering of αPS2CβPS integrins. For cells expressing αanaβ integrins, there is no statistically significant change in energy transfer after cholesterol depletion. There is a >3-fold increase in energy transfer for the cells expressing αβV409D mutant integrins after cholesterol depletion. The increase in energy transfer indicates that the αβV409D integrins reduce their separation distance by forming higher-order oligomers or new oligomers when there is less cholesterol in the membrane. No correlation can be made between the change in the measured energy transfer and changes in lipid diffusion after cholesterol depletion, since the only cell line that showed a statistically significant increase in lipid diffusion coefficients (αanaβ) did not show a statistically significant change in energy transfer.
The energy transfer measured using the FRET assay depicted in Fig. 1 is a result of both integrin-specific interactions with the FRET reporters and potentially nonintegrin-specific interactions from many possible sources. Assuming that the nonintegrin-specific contributions to energy transfer are similar for the three cells lines derived from the S2 parent cell line, the differences in energy transfer listed in Table 3 are primarily from integrin-specific interactions with the FRET reporters. This is supported by previous studies in which no energy transfer was measured within the instrument's detection limit in cells expressing FRET reporters and no integrins, suggesting that the energy transfer measured in cells expressing both FRET reporters and integrins is primarily integrin-dependent (17).
The changes in energy transfer measured after cholesterol depletion report on the amount of integrin microclustering that is cholesterol-dependent. There may be cholesterol-independent integrin microclusters present, which would not result in a change in energy transfer after cholesterol depletion. Although the amount of integrin microclustering in the cell membrane is similar for all three cell lines before cholesterol depletion, the dependence of these microclusters on cholesterol is not the same, as determined by different changes in energy transfer after cholesterol depletion. This may be due to differences in the integrins' ligand affinity: αβV409D has the highest ligand affinity, followed by αanaβ and then WT. Leitinger and Hogg (57) previously showed that lipid nanodomains are involved in the signaling events of many classes of integrins. They reported that a mutant LFA-1 integrin missing the I domain has characteristics that mimic integrins with high ligand affinity, and preferentially localizes into lipid nanodomains in T lymphocytes, whereas WT LFA-1 with a low affinity for ligand does not preferentially localize into lipid nanodomains under the conditions used in the study. After WT LFA-1 was exposed to Mn2+ or phorbol esters, which have been shown to increase ligand affinity for a number of integrin classes, the WT LFA-1 increased partitioning into lipid nanodomains. The protein and lipid composition of Drosophila membranes supports nanodomain formation, and lipid nanodomains enriched in sphingolipids, glycosylphosphatidylinositol-linked proteins, and sterols have been measured in Drosophila (58). If αβV409D integrins with a higher ligand affinity than WT αPS2CβPS integrins (49) exhibit greater partitioning into lipid nanodomains, this might explain the difference in cholesterol-dependent integrin microclustering measured in the three cells lines included in this study, as discussed below.
After cholesterol depletion, αβV409D shows the highest amount of integrin microclusters, followed by αanaβ and then WT. Of course, it may seem counterintuitive that the integrin with the highest ligand affinity (αβV409D) and possibly the greatest partitioning into lipid nanodomains would show the largest increase in integrin microclustering upon plasma membrane cholesterol reduction. A recent study in T cells showed that the amount of cholesterol extracted from the cell membrane affected the resulting change in membrane organization and cell signaling originating at the membrane (59). When cholesterol was depleted by <50% of its original value (the highest depletion included in the study), lipid nanodomains were found to aggregate. Given the amount of cholesterol depleted from the membrane in our studies (Fig. 2 B), it is reasonable to assume that the lipid nanodomains are aggregating. This would explain the increase in αβV409D microclustering upon cholesterol depletion if it preferentially partitions into nanodomains.
There are several possible explanations for the different amounts of integrin microclustering in cells expressing WT and αanaβ integrins. First, less cholesterol is extracted from the plasma membrane in these cell populations compared to the αβV409D cell line (Fig. 2 B). There may be different levels of lipid nanodomain aggregation when less cholesterol is extracted. Additionally, these integrins may partition into lipid nanodomains to different extents, or partition into a different population of lipid nanodomains. It has been shown that the extent to which certain proteins partition into lipid nanodomains depends on the amount of cholesterol in the membrane (59). The population of WT integrins in lipid nanodomains may decrease (by partitioning out or by nanodomain disruption) upon 27% cholesterol extraction from the plasma membrane. This would explain its decreased microclustering upon cholesterol depletion. Studies with additional integrin mutants could further test the hypothesized relationship between ligand affinity, partitioning into lipid nanodomains, and cholesterol-dependent microclustering.
Previous studies have shown that treatment with mβCD can extract membrane phospholipids along with cholesterol (31), and that restoration of membrane properties, such as diffusion coefficients, can be achieved by restoring the cholesterol levels to their original values (24). We confirmed the role of cholesterol in altering membrane properties by adding cholesterol back to the membrane of previously cholesterol-depleted cells and reevaluating those properties. The cholesterol-depleted, αβV409D-expressing cells were incubated with chol-mβCD, and the total cellular and plasma membrane cholesterol concentrations were measured. Fig. 2, A and B (gray data bars) show that the total cholesterol and plasma membrane cholesterol levels in cells expressing the αβV409D mutant integrins and FRET reporters can be restored to a level statistically similar to that of untreated cells at the 95% confidence level. After cholesterol restoration, the slow and fast lipid diffusion coefficients show a statistically significant decrease relative to the values obtained for the cholesterol-depleted cell line (Table 1). Similarly, the FRET results (Table 3) indicate that αβV409D integrin microclustering levels return to a value statistically similar to that of the original value obtained for the untreated cells. Although this provides evidence that cholesterol plays a role in the altered membrane properties measured in this study, it cannot be concluded that other membrane components are not playing a role. It is possible that mβCD may perturb another membrane component(s) that also affects integrin microclustering, and this change is convoluted with that obtained upon cholesterol depletion. If this is the case, the FRET assay utilized in this study will be an ideal analysis technique to identify other lipid or membrane components that affect integrin microclustering. Current research is being performed to identify additional membrane species with a role in altering integrin microclustering.
Conclusions
Integrins are ubiquitous membrane receptors that are important in nearly all cell-signaling cascades, including those that control basic cellular functions (60–62). Therefore, it is important to understand the molecular mechanism of integrin function. This includes not only the much-studied changes in ligand affinity and macroscale clustering, but also the less-studied changes in receptor microclustering. The data reported herein highlight a simple method that can be used to elucidate the role of cholesterol in integrin microclustering. Upon cholesterol depletion, the maximum increase in integrin microclustering was measured for cells expressing αβV409D integrins, which have the highest affinity for ligand of the three integrins included in this study. Partitioning into cholesterol-rich nanodomains may explain the difference in cholesterol-dependent integrin microclustering for WT and mutant integrins. Restoration of membrane cholesterol to native levels restored the levels of αβV409D integrin microclustering to values obtained for untreated cells. A similar methodology can be used to elucidate the role of cholesterol in the microclustering of integrins in other cell types, as well as the role of other membrane components in integrin microclustering and for other members of the integrin family.
Acknowledgments
The authors thank Roger Tsien (Howard Hughes Medical Institute, La Jolla, CA) for the original mCherry plasmid, Atsushi Miyawaki (Riken, Wako City, Saitama, Japan) for the original Venus plasmid, Christopher Gonwa-Reeves (Iowa State University, Ames, Iowa) for assisting with the statistical analysis, and Anthony Young and Andrew Pavel (Iowa State University) for their assistance with the experimental procedures.
Support for this work was provided by the National Science Foundation (CHE-0845236) and the Roy J. Carver Charitable Trust.
Supporting Material
References
- 1.Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 2.Tang C.K., Pietersz G.A. Intracellular detection and immune signaling pathways of DNA vaccines. Expert Rev. Vaccines. 2009;8:1161–1170. doi: 10.1586/erv.09.79. [DOI] [PubMed] [Google Scholar]
- 3.Su C.Y., Menuz K., Carlson J.R. Olfactory perception: receptors, cells, and circuits. Cell. 2009;139:45–59. doi: 10.1016/j.cell.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sorkin A., von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan W.N. B cell receptor and BAFF receptor signaling regulation of B cell homeostasis. J. Immunol. 2009;183:3561–3567. doi: 10.4049/jimmunol.0800933. [DOI] [PubMed] [Google Scholar]
- 6.van Kooyk Y., Figdor C.G. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 2000;12:542–547. doi: 10.1016/s0955-0674(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 7.Saito T., Yokosuka T. Immunological synapse and microclusters: the site for recognition and activation of T cells. Curr. Opin. Immunol. 2006;18:305–313. doi: 10.1016/j.coi.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 8.Vicente-Manzanares M., Sánchez-Madrid F. Role of the cytoskeleton during leukocyte responses. Nat. Rev. Immunol. 2004;4:110–122. doi: 10.1038/nri1268. [DOI] [PubMed] [Google Scholar]
- 9.Ginsberg M.H., Partridge A., Shattil S.J. Integrin regulation. Curr. Opin. Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Adair B.D., Yeager M. Electron microscopy of integrins. Methods Enzymol. 2007;426:337–373. doi: 10.1016/S0076-6879(07)26015-X. [DOI] [PubMed] [Google Scholar]
- 11.van Kooyk Y., van Vliet S.J., Figdor C.G. The actin cytoskeleton regulates LFA-1 ligand binding through avidity rather than affinity changes. J. Biol. Chem. 1999;274:26869–26877. doi: 10.1074/jbc.274.38.26869. [DOI] [PubMed] [Google Scholar]
- 12.Yauch R.L., Felsenfeld D.P., Hemler M.E. Mutational evidence for control of cell adhesion through integrin diffusion/clustering, independent of ligand binding. J. Exp. Med. 1997;186:1347–1355. doi: 10.1084/jem.186.8.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hato T., Pampori N., Shattil S.J. Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin αIIb β3. J. Cell Biol. 1998;141:1685–1695. doi: 10.1083/jcb.141.7.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selvin P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- 15.Buensuceso C., de Virgilio M., Shattil S.J. Detection of integrin αIIbβ 3 clustering in living cells. J. Biol. Chem. 2003;278:15217–15224. doi: 10.1074/jbc.M213234200. [DOI] [PubMed] [Google Scholar]
- 16.Kim M., Carman C.V., Springer T.A. The primacy of affinity over clustering in regulation of adhesiveness of the integrin αLβ2. J. Cell Biol. 2004;167:1241–1253. doi: 10.1083/jcb.200404160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith E.A., Bunch T.A., Brower D.L. General in vivo assay for the study of integrin cell membrane receptor microclustering. Anal. Chem. 2007;79:3142–3147. doi: 10.1021/ac062008i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dibya D., Sander S., Smith E.A. Identifying cytoplasmic proteins that affect receptor clustering using fluorescence resonance energy transfer and RNA interference. Anal. Bioanal. Chem. 2009;395:2303–2311. doi: 10.1007/s00216-009-3146-5. [DOI] [PubMed] [Google Scholar]
- 19.Martin-Bermudo M.D., Brown N.H. Intracellular signals direct integrin localization to sites of function in embryonic muscles. J. Cell Biol. 1996;134:217–226. doi: 10.1083/jcb.134.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li R., Babu C.R., DeGrado W.F. Oligomerization of the integrin αIIbβ3: roles of the transmembrane and cytoplasmic domains. Proc. Natl. Acad. Sci. USA. 2001;98:12462–12467. doi: 10.1073/pnas.221463098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klappauf E., Schubert D. Band 3-protein from human erythrocyte membranes strongly interacts with cholesterol. FEBS Lett. 1977;80:423–425. doi: 10.1016/0014-5793(77)80490-0. [DOI] [PubMed] [Google Scholar]
- 22.Mühlebach T., Cherry R.J. Influence of cholesterol on the rotation and self-association of band 3 in the human erythrocyte membrane. Biochemistry. 1982;21:4225–4228. doi: 10.1021/bi00261a006. [DOI] [PubMed] [Google Scholar]
- 23.McConnell H.M., Vrljic M. Liquid-liquid immiscibility in membranes. Annu. Rev. Biophys. Biomol. Struct. 2003;32:469–492. doi: 10.1146/annurev.biophys.32.110601.141704. [DOI] [PubMed] [Google Scholar]
- 24.Vrljic M., Nishimura S.Y., McConnell H.M. Cholesterol depletion suppresses the translational diffusion of class II major histocompatibility complex proteins in the plasma membrane. Biophys. J. 2005;88:334–347. doi: 10.1529/biophysj.104.045989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeagle P.L. Cholesterol and the cell membrane. Biochim. Biophys. Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 26.Baird B., Sheets E.D., Holowka D. How does the plasma membrane participate in cellular signaling by receptors for immunoglobulin E? Biophys. Chem. 1999;82:109–119. doi: 10.1016/s0301-4622(99)00110-6. [DOI] [PubMed] [Google Scholar]
- 27.Janes P.W., Ley S.C., Kabouridis P.S. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 2000;12:23–34. doi: 10.1006/smim.2000.0204. [DOI] [PubMed] [Google Scholar]
- 28.Krauss K., Altevogt P. Integrin leukocyte function-associated antigen-1-mediated cell binding can be activated by clustering of membrane rafts. J. Biol. Chem. 1999;274:36921–36927. doi: 10.1074/jbc.274.52.36921. [DOI] [PubMed] [Google Scholar]
- 29.Simons K., Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 30.Tansey M.G., Baloh R.H., Johnson E.M., Jr. GFRα-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 2000;25:611–623. doi: 10.1016/s0896-6273(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 31.Kilsdonk E.P., Yancey P.G., Rothblat G.H. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 1995;270:17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 32.Bunch T.A., Brower D.L. Drosophila PS2 integrin mediates RGD-dependent cell-matrix interactions. Development. 1992;116:239–247. doi: 10.1242/dev.116.1.239. [DOI] [PubMed] [Google Scholar]
- 33.Jannuzi A.L., Bunch T.A., Brower D.L. Disruption of C-terminal cytoplasmic domain of βPS integrin subunit has dominant negative properties in developing Drosophila. Mol. Biol. Cell. 2002;13:1352–1365. doi: 10.1091/mbc.01-08-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jannuzi A.L., Bunch T.A., Brower D.L. Identification of integrin β subunit mutations that alter heterodimer function in situ. Mol. Biol. Cell. 2004;15:3829–3840. doi: 10.1091/mbc.E04-02-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 36.Bezrukov L., Blank P.S., Zimmerberg J. An adhesion-based method for plasma membrane isolation: evaluating cholesterol extraction from cells and their membranes. Anal. Biochem. 2009;394:171–176. doi: 10.1016/j.ab.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amundson D.M., Zhou M. Fluorometric method for the enzymatic determination of cholesterol. J. Biochem. Biophys. Methods. 1999;38:43–52. doi: 10.1016/s0165-022x(98)00036-0. [DOI] [PubMed] [Google Scholar]
- 38.Almeida A.M., Castel-Branco M.M., Falcão A.C. Linear regression for calibration lines revisited: weighting schemes for bioanalytical methods. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002;774:215–222. doi: 10.1016/s1570-0232(02)00244-1. [DOI] [PubMed] [Google Scholar]
- 39.Harris D.C. 4th ed. W.H. Freeman and Co.; New York: 2009. Exploring Chemical Analysis. [Google Scholar]
- 40.Zal T., Gascoigne N.R. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 2004;86:3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon G.W., Berry G., Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bland J.M., Altman D.G. The use of transformation when comparing two means. BMJ. 1996;312:1153. doi: 10.1136/bmj.312.7039.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altman D.G., Gore S.M., Pocock S.J. Statistical guidelines for contributors to medical journals. Br. Med. J. (Clin. Res. Ed.) 1983;286:1489–1493. doi: 10.1136/bmj.286.6376.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zar J. Prentice Hall; Upper Saddle River, NJ: 1999. Biostatistical Analysis. [Google Scholar]
- 45.Axelrod D., Koppel D.E., Webb W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brower D.L. Platelets with wings: the maturation of Drosophila integrin biology. Curr. Opin. Cell Biol. 2003;15:607–613. doi: 10.1016/s0955-0674(03)00102-9. [DOI] [PubMed] [Google Scholar]
- 47.Clark A.J., Block K. The absence of sterol synthesis in insects. J. Biol. Chem. 1959;234:2578–2582. [PubMed] [Google Scholar]
- 48.Dobrosotskaya I.Y., Seegmiller A.C., Rawson R.B. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science. 2002;296:879–883. doi: 10.1126/science.1071124. [DOI] [PubMed] [Google Scholar]
- 49.Bunch T.A., Helsten T.L., Brower D.L. Amino acid changes in Drosophila αPS2βPS integrins that affect ligand affinity. J. Biol. Chem. 2006;281:5050–5057. doi: 10.1074/jbc.M508550200. [DOI] [PubMed] [Google Scholar]
- 50.Connor J., Bucana C., Schroit A.J. Differentiation-dependent expression of phosphatidylserine in mammalian plasma membranes: quantitative assessment of outer-leaflet lipid by prothrombinase complex formation. Proc. Natl. Acad. Sci. USA. 1989;86:3184–3188. doi: 10.1073/pnas.86.9.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hotta K., Bazartseren B., Yamada A. Effect of cellular cholesterol depletion on rabies virus infection. Virus Res. 2009;139:85–90. doi: 10.1016/j.virusres.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 52.Klein C., Pillot T., Drouet B. Determination of plasma membrane fluidity with a fluorescent analogue of sphingomyelin by FRAP measurement using a standard confocal microscope. Brain Res. Brain Res. Protoc. 2003;11:46–51. doi: 10.1016/s1385-299x(03)00016-3. [DOI] [PubMed] [Google Scholar]
- 53.Pucadyil T.J., Chattopadhyay A. Effect of cholesterol on lateral diffusion of fluorescent lipid probes in native hippocampal membranes. Chem. Phys. Lipids. 2006;143:11–21. doi: 10.1016/j.chemphyslip.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 54.Ramprasad O.G., Rangaraj N., Pande G. Differential regulation of the lateral mobility of plasma membrane phospholipids by the extracellular matrix and cholesterol. J. Cell. Physiol. 2008;215:550–561. doi: 10.1002/jcp.21339. [DOI] [PubMed] [Google Scholar]
- 55.Wolf D.E., Hagopian S.S., Ishijima S. Changes in sperm plasma membrane lipid diffusibility after hyperactivation during in vitro capacitation in the mouse. J. Cell Biol. 1986;102:1372–1377. doi: 10.1083/jcb.102.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith E.A., Coym J.W., Wirth M.J. Lipid bilayers on polyacrylamide brushes for inclusion of membrane proteins. Langmuir. 2005;21:9644–9650. doi: 10.1021/la051116h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leitinger B., Hogg N. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 2002;115:963–972. doi: 10.1242/jcs.115.5.963. [DOI] [PubMed] [Google Scholar]
- 58.Rietveld A., Neutz S., Eaton S. Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J. Biol. Chem. 1999;274:12049–12054. doi: 10.1074/jbc.274.17.12049. [DOI] [PubMed] [Google Scholar]
- 59.Mahammad S., Dinic J., Parmryd I. Limited cholesterol depletion causes aggregation of plasma membrane lipid rafts inducing T cell activation. Biochim. Biophys. Acta. 2010;1801:625–634. doi: 10.1016/j.bbalip.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 60.Harburger D.S., Calderwood D.A. Integrin signalling at a glance. J. Cell Sci. 2009;122:159–163. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huveneers S., Danen E.H. Adhesion signaling—crosstalk between integrins, Src and Rho. J. Cell Sci. 2009;122:1059–1069. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- 62.Legate K.R., Wickström S.A., Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23:397–418. doi: 10.1101/gad.1758709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.