

Abstract
Cytoskeleton is ubiquitous throughout the cell and is involved in important cellular processes such as cellular transport, signal transduction, gene transcription, cell-division, etc. Partial wave spectroscopic microscopy is a novel optical technique that measures the statistical properties of cell nanoscale organization in terms of the disorder strength. It has been found previously that the increase in the disorder strength of cell nanoarchitecture is one of the earliest events in carcinogenesis. In this study, we investigate the cellular components responsible for the differential disorder strength between two morphologically (and hence microscopically) similar but genetically altered human colon cancer cell lines, HT29 cells and Csk shRNA-transfected HT29 cells that exhibit different degrees of neoplastic aggressiveness. To understand the role of cytoskeleton in nanoarchitectural alterations, we performed selective drug treatment on the specific cytoskeletal components of these cell types and studied the effects of cytoskeletal organization on disorder strength differences. We report that altering the cell nanoarchitecture by disrupting cytoskeletal organization leads to the attenuation of the disorder strength differences between microscopically indistinguishable HT29 and CSK constructs. We therefore demonstrate that cytoskeleton plays a role in the control of cellular nanoscale disorder.
Introduction
Carcinogenesis is a complex multistep process that eventually results in the emergence of a malignant phenotype that often progresses into a fatal metastatic stage (1). Recent studies indicate that there are molecular changes (such as DNA methylation, genomic and proteomic alterations) in cells that occur before the microscopically detectable morphological alterations in early cancerization (2–5). Hence, it is intuitive to imagine that these molecular changes should impact various biochemical, biophysical, and transport processes within the cell to modify its morphology. However, there are several macromolecular compounds and organelles in a cell that work in synchronization to regulate these important physiological processes and maintain the homeostasis of a cell (6). Among all the functional components of a living cell, cytoskeleton is considered to be the backbone of a cell as it provides the cell its shape and structure (7). It is pivotal in controlling inter- and intracellular transportation and also plays an important role during cell division and differentiation (1). The cytoskeletal components participate in the signal transduction and also influence gene transcription in response to the extracellular factors (6,8,9). The cytoskeletal components in the nucleus are involved in DNA cross-linking, transcription control, and chromosome morphology (10–13). A few reports indicate that there are alterations in the expression and interactions among several cytoskeletal proteins during early carcinogenesis (14). Moreover, a number of genes have been identified to regulate the multimolecular focal complexes associated with cytoskeletal proteins and cell cycle progression (15). Some experiments show the relationship between basement membrane and type IV procollagen mRNA expression that support a direct interaction among extracellular matrix, cytoskeleton, and nuclear matrix (16). Hence, one could anticipate that these genetic/epigenetic alterations would result in active nanoarchitectural changes within a cell during early carcinogenesis. Therefore, it is imperative to understand the changes in cytoskeleton organization during early carcinogenesis. Researchers have been using several imaging techniques such as immunofluorescence, fluorescence correlation spectroscopy (17,18), and quantification by immunohistochemistry with nanoparticles to study the cytoskeleton organization (19). For example, Nadakavukaren et al. (20) visualized cytoskeletal elements such as actin, keratin, and vimentin in rat epithelial cells by indirect immunofluorescence. Similarly, Wang et al. (21) studied GFP mutant-enhanced cyan fluorescent protein and its tubulin fusion using fluorescence correlation spectroscopy. They also showed that analysis of the molecular brightness can provide a new avenue for studying the polymerization state of tubulin. Immunofluorescence microscopy was used to study microtubule cytoskeleton during conjugate division in the dikaryon Pleurotus ostreatus N001 (22). However, studying these nanoscale modifications in cellular organelles have been stymied due to the need for an extrinsic contrast agents or due to the diffraction limited resolution of traditional light microscopy that limits the smallest structure (∼300 nm) that can be visualized.
Light scattering, on the other end, offers the potential to probe cell and tissue architecture due to its ability to detect the structure dependent signatures in wavelength (23–27). However, light scattered by cells and tissues is typically recorded after its three-dimensional propagation within tissue and hence its sensitivity to subwavelength length scales decreases due to Fourier transform relationship between the scattering signal and scattering potential (28). Our group developed recently a spectroscopic microscopy technique called single cell partial wave spectroscopic (PWS) microscopy, which can quantify the statistical properties of nanoarchitecture of an isolated cell (29,30). This technique takes the advantage of both the diffraction-limited localized structural information provided by microscopy and the subdiffractional sensitivity of spectral signatures (29,31). PWS has proved its potential in capturing early carcinogenic events in the uninvolved mucosa from the field cancerization in the colon, the lung, and the pancreas (32). This technique is based on the mesoscopic light transport theory according to which a signal in one dimension arising due to multiple interferences of light waves within a sample of weak refractive index is sensitive to any length scale of refractive index fluctuations. Because the optical refractive index is linearly related to the local mass density of macromolecules, (that is, refractive index n = no + αρ where no is the refractive index of the medium, ρ is the portion of intracellular solids by volume that is mass density, and α is a proportionality coefficient with a constant value of ∼0.186 for most of the intracellular compounds (6,33,34)), the spectrum of the one-dimensional scattering signal contains information about particles whose length scales are well below the wavelength of visible light (35–37). Due to this unique sensitivity to the structures of submicron size, PWS was capable of measuring nanoscale refractive index fluctuations within genetically modified human colon cancer cell lines, HT29 cells and its genetic knock-downs, Csk shRNA construct (more aggressive than HT29 cells), and EGFR shRNA construct (less aggressive than HT29 cells) by quantifying their nanoscale disorder (29). PWS was able to differentiate among these three cell-types based on the disorder strength of intracellular nanoarchitecture. The disorder strength (Ld) is defined as the product of the variance 〈Δn2〉 and the correlation length lc of the refractive index fluctuations: Ld = 〈Δn2〉 lc (29,30). It is to be noted that 〈Δn2〉 is linearly proportional to the variance of the spatial fluctuations of local macromolecular concentration within a cell (that is, Ld = α2 〈Δρ2〉 lc); whereas lc corresponds to the size of these macromolecular compounds.
In this study, we attempt to understand the biological origin of the increase in the nanoscale disorder strength that was observed in human colon adenocarcinoma cell line, HT29 control cells and Csk shRNA-transfected HT29 cells. We first study the contribution of the nucleus and the cytoplasm to the intracellular disorder strength. Our results indicate that both the nucleus and the cytoplasm participate in determining the disorder strength differences between the two cell types indicating that the differences are not confined to a specific region but are distributed throughout the cell. Because the cytoskeleton is ubiquitous throughout the cell, we next investigate the role of the cytoskeleton in determining the differences in the disorder strength between the control HT29 and the Csk shRNA-transfected HT29 cells.
In most eukaryotic cells, cytoskeleton is made up of three major components: the actin microfilaments (∼6 nm in diameter) that are important for maintaining shape of the cell, inter- and intracellular transport, etc.; the microtubules (∼23–25 nm in diameter) that contain tubulin protein and are important for intracellular transport and during cell division by forming spindle fibers; and the intermediate filaments (∼10 nm in diameter) that are made of family of proteins (such as keratin, desmin, peripherin, etc.), which play role in maintaining cell shape and internal organization of the cell (6,38).
To investigate if cytoskeleton is responsible for the control of the disorder strength, we achieved pharmacological disruption of cytoskeleton using cytochalasin D (to rupture actin microfilaments) and colchicine (to perturb tubulin polymerization and hence microtubule organization) and demonstrated the attenuation of difference between disorder strengths in HT29 and Csk shRNA-transfected HT29 cells. Although there have been some notable efforts in investigating the cytoskeleton dynamics in HT29 cell line (39,40) to understand apoptotic processes and cell differentiation, to our knowledge, this is the first study that explores the underlying mechanism that gives rise to the disorder strength differences in genetically altered cell lines exhibiting differential neoplastic behavior—HT29 and Csk shRNA constructs.
Materials and Methods
Measurements using PWS
The schematic of the first generation PWS system is reported in Subramanian et al. (29,30). In the earlier instrumentation, the image of a single isolated cell on a cytology slide was obtained by spatially scanning the slit of a spectrograph and imaging the slit onto a charge-coupled device (CCD) camera. However, the spatial scanning of the spectrograph with a scanning stage leads to slow data collection (∼4–5 min for a single cell). To mitigate this issue, we have designed the second generation instrument that replaces the spectrometer-scanning stage combination with a liquid crystal tunable filter (LCTF; CRi, Woburn, MA) resulting in a faster acquisition rate (∼4–5 s for a single cell). The schematic of this newer version of the PWS system is shown in Fig. 1. A nearly plane wave of the white light from the Xenon lamp (150 W; Oriel, Stratford, CT) is collimated by a 4f-lens relay system and focused onto a sample by a low numerical aperture (NA) objective (NA of objective = 0.4, NA of illumination = 0.2, NA of collection = 0.4; objective lens from Edmund Optics, Barrington, NJ). The objective lens contains a correction collar to compensate for any aberrations. The backscattered image is then projected through the LCTF (spectral resolution = 7 nm) on to a CCD camera. The LCTF collects the backscattering image of a cell at a particular wavelength between 400 nm and 700 nm and projects it onto the CCD camera thereby avoiding the need for spatially scanning each pixel in the cell as was done previously (29,30). Hence this generates a three-dimensional data cube for the reflected intensity I(λ; x,y) where (x,y) refers to a specific pixel in the object plane and λ is the wavelength. After normalizing each pixel by the corresponding incident light profile (using a mirror), we generate R(λ; x,y) that is referred to as the fluctuating part of the reflection coefficient. The spectral fluctuations between wavelength range of 500–700 nm (to avoid the lower intensity regions at the lower wavelengths and to get rid of the noise from the camera) are further analyzed by means of 1D mesoscopic light transport theory (32,35,36,41) to obtain Ld:
| (1) |
where 〈R〉 is calculated by taking the root mean-square value of the spectrum R(k) whereas C(Δk) is the autocorrelation function of the spectrum R(k).
| (2) |
Figure 1.
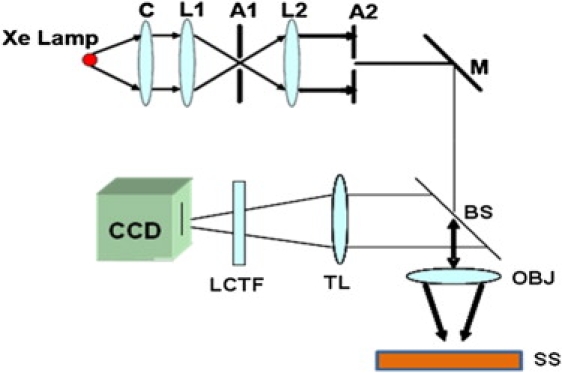
Schematic of the second generation PWS system. A1, A2, apertures [D(A1) = 2 mm, D(A2) = 4 mm]; BS, beam splitter; C, condenser; L1, L2, lenses (f = 150 mm); F, flipper mirror; LCTF, liquid crystal tunable filter; M, mirror; OBJ, objective lens; SS, sample stage; TL, tube lens (f = 450 mm).
Furthermore, is obtained by fitting a linear slope to −ln(CΔk)) versus (Δk)2. Thus a map of disorder strength Ld (x,y) is obtained from each pixel (x,y). From the two-dimensional map Ld (x,y) of each cell the mean intracellular disorder strength (the average Ld (x,y) over x and y) and the standard deviation (SD) of intracellular disorder strength values, σ(c) was obtained. The averages of over a group of cells (∼50–70 cells for each set of cell preparation), are termed as the group mean of the disorder strength, whereas its SD is defined as σ(g). This average disorder strength and the standard error calculated from its SD, σ(g) are depicted in all the bar-plots in this study.
Statistical methods
All p values were calculated using standard Student's t-tests. The effect-size between two groups was calculated from the average disorder strength, and its SD, σ(g). It is defined as, where μ1 and σ1 can be thought as the and σ(g) for the HT29 cell population. Similarly, μ2 and σ2 can be assumed as the and σ(g) for the CSK constructs. It has been used to take into account the statistical significance of the average disorder strength difference, i.e., ΔLd, for untreated and the treated HT29 control cells and CSK constructs.
Sample preparation
Cell culture
HT29 cells were grown in McCoy's medium mixed with 10% fetal bovine serum + 50 μg/mL penicillin/streptomycin under 5% CO2 environment at 37°C. CSK (C-terminus Src kinase) shRNA-transfected HT29 cells were grown under similar conditions in the same medium with addition of hygromycin (600 μg/mL). For the experiments, ∼25,000 cells were plated in each chamber of the sterile glass chamber slides and incubated for 6 h under standard conditions. All the experiments were carried out for cells between passages 9–15. The variability between different passages was negligible.
Cytochalasin D and colchicine treatment
Plated cells were treated with 0.15 μM cytochalasin D (Sigma-Aldrich, St. Louis, MO) for 15 min and 10 μM colchicine (Sigma-Aldrich) for 30 min separately. We carried out dose-response studies using microscopic abnormalities as an endpoint and picked this specific minimal dose that reliably would not alter microscopic appearance of the cells to avoid confounding effects such as toxicity to drug. The aim of the concentration-duration versus growth curve experiments was to arrive at the minimum concentration and time duration required to induce nanoarchitectural alterations in the cells to achieve morphologically comparable populations of HT29 cells and CSK constructs without any obvious microscopic differences after treatment. The chamber slides were washed with cold phosphate buffered saline and cells were fixed under chilled 70% ethanol. Chamber slides, thus prepared, were stored at 4°C keeping cells submerged in 70% ethanol.
For PWS analysis, we randomly selected 50–70 cells for each cell type from two to three different set of slides for both the untreated as well as the cytochalasin D and colchicine treated cases. The SD of the average intracellular disorder strength for different set of slides within each group was negligible. The slide-to-slide variability for different preparation turned out to be statistically nonsignificant (Student's t-test, p > 0.25).
Confocal microscopy
Approximately 50,000 cells were grown overnight on glass coverslips. Cells were then fixed in 4% paraformaldehyde for 20 min on ice and washed twice with Dulbecco's phosphate buffered saline (American Type Culture Collection, Manassas, VA) for 5 min each. Cells were then permeabilized using 0.1% Triton X-100 in PBS for 20 min at room temperature. Blocking was carried out using 1% bovine serum albumin in PBS for 20 min. Cells were then either incubated for 2 h with AlexaFluor 555 conjugated to anti-β-tubulin (Cell Signaling, Beverly, MA) or for 30 min with AlexaFluor 488 conjugated to phalloidin (Invitrogen, Carlsbad, CA). Chromatin staining was then carried out using TOPRO3 (Invitrogen) for 15 min. Finally, the coverslips were mounted in ProLong Gold antifade reagent (Invitrogen) onto glass slides. Specimens were analyzed using an inverted confocal microscope (Leica SP5 with Leica Systems software). The optimum z resolution was used for acquiring z-stack files of individual cells.
We then carried out a three-dimensional reconstruction of the Confocal images for all the cell types using a specialized imaging software, Volocity (PerkinElmer, Wellesley, MA). MATLAB (The MathWorks, Natick, MA) was used to estimate the percentage of cytoplasm layer on top and bottom of a cell nucleus and to determine its contribution toward the nuclear disorder strength. As indicated in Fig. 2, b and d, there appears to be a total of 10–15% of cytoplasm on top and bottom of the nucleus and the disorder strength calculations suggest that this is a negligible amount compared to the ΔLd between HT29 and Csk shRNA-transfected HT29 cells.
Figure 2.
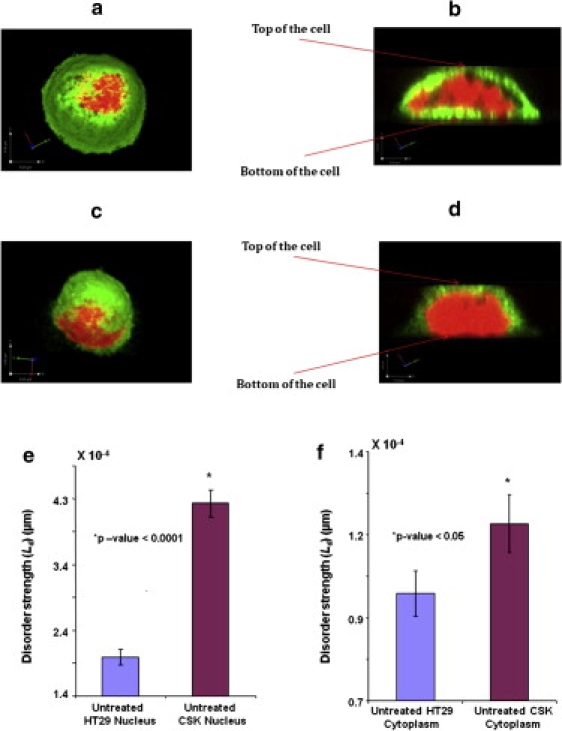
Representative confocal images of HT29 and CSK cells. (a and c) Three-dimensional top view of the HT29 cells and CSK constructs respectively. These cells appear microscopically and morphologically similar. The red region represents nucleus being stained with TOPRO3 (nuclear chromatin stain). The green region represents cytoplasm stained with anti-β-tubulin. (b and d) Slice cut through the cell in the x-z plane. There appears to be negligible cytoplasm (∼10–15%) on top and bottom of the nucleus. (e and f) Results of PWS analysis carried out independently on nuclear and cytoplasmic region of both the cell constructs respectively. There appears statistically highly significant difference (p < 0.00001) in the average disorder strength, calculated from the nuclear region of HT29 cells and CSK constructs. Also, there is a significant (p < 0.05) difference, ΔLd observed in cytoplasmic region of these cell constructs.
Results and Discussion
We first carried out PWS analysis on the entire cellular region of the colon cancer cell line, HT29 cells and its genetic variant, Csk shRNA-transfected HT29 cells, which showed statistically significant (effect-size = 100%, p < 0.00001) increase in the disorder strength for the more aggressive CSK constructs compared to HT29 cells. These results indicate that the increase in disorder strength parallels the genetic events within the cells that lead to rapid proliferation of the Csk shRNA constructs compared to HT29 cells (29).
Next, we studied the contributions of nucleus and cytoplasm independently toward disorder strength of the cells. To identify the nuclear and cytoplasmic region accurately, we imaged the cells using confocal microscope according to the standard protocol (detailed in Materials and Methods). Fig. 2, a and c, shows a typical three-dimensional confocal microscopy image of an HT29 and a Csk shRNA-transfected HT29 cell that has been stained using TOPRO3 (nuclear chromatin stain) and anti-β-tubulin (cytoplasmic tubulin stain). Aided with this information, the disorder strength was calculated separately for nucleus and cytoplasm. It is to be noted that although the nuclear and cytoplasmic stains were used to obtain confocal images, independent PWS measurements were obtained from cells that were not stained with any type of staining reagents.
The PWS quantification on the nuclear region (depicted in Fig. 2) indicated a highly statistically significant increase (effect-size = 117%, p < 0.00001) in the average disorder strength for the nuclei of untreated Csk shRNA-transfected HT29 cells compared to the control HT29 cells (Fig. 2 e). A similar trend of statistically significant increase in the average disorder strength (effect-size = 25.3%, p < 0.05) was also observed in the cytoplasmic region of these cell-types (Fig. 2 f). These results suggest that both nucleus as well as the cytoplasm contribute to the increase in disorder of the highly proliferative Csk shRNA constructs as compared to HT29 cells. It seems that disorder is more pronounced in the nuclear region (effect-size of 117%) compared to the cytoplasmic region (effect-size of 25.3%) that can be attributed to modifications in the nuclear organization of the more aggressive CSK constructs compared to the control HT29 cells.
To establish the biological origin of disorder strength, we hypothesized cytoskeleton to be the major contributor toward the disorder strength, due mainly to its presence everywhere in the cell. The correlation between disorder strength and cytoskeleton was obtained by specifically targeting microfilaments and microtubular components of cytoskeletal organization using specific pharmacological agents.
Treatment with cytochalasin D
Cytochalasin D, a fungal metabolite known to inhibit actin polymerization, was used to disrupt microfilament organization in HT29 and Csk shRNA-transfected HT29 cells. The purpose of targeting actin filaments was its widespread presence throughout the cell. Therefore, disrupting the actin microfilaments will change the cytoskeletal organization in both the nucleus and the cytoplasm. We treated cells with minimum concentration and duration (0.15 μM for 15 min) to avoid any confounding and to achieve microscopically comparable populations of HT29 and CSK constructs with minimal changes in their morphology.
We first measured the average disorder strength obtained from the entire cell of HT29 and Csk shRNA-transfected HT29 cells. A significant decrease in the average disorder strength was observed in the both cell types compared to their untreated counterparts. This indicates that the cytoskeleton disruption induced by cytochalasin D treatment has translated into decreased average disorder strength. A more important question, however, is the biological mechanism underlying the difference in disorder strength, ΔLd between the cell types. On analyzing the nuclear region of the treated cells separately, we observed a minimal difference in the disorder strength between HT29 and CSK constructs (Fig. 3 a). That is, the significant ΔLd, that was otherwise observed between the nuclei of untreated HT29 and CSK cells was nullified (p value = 0.54) and the effect-size reduced from 117% in untreated cells to 8.1% in cytochalasin D-treated cells. This critical result implied that cytoskeleton indeed plays an important role in determining the difference in disorder strength observed in the HT29 cells and Csk shRNA constructs. There are conflicting reports about the presence and nature of actin in the nucleus. However, there is convincing evidence of the active role that nuclear actin has in gene transcription and signal transduction (8,13,42,43), DNA cross-linking (10), and in determining higher-order chromatin structure and chromosome morphology (12). Hence, we hypothesize that actin is an integral part of nuclear architecture and disrupting its network with cytochalasin D leads to attenuation of the difference in the nuclear disorder strengths of HT29 and Csk shRNA-transfected HT29 cells.
Figure 3.
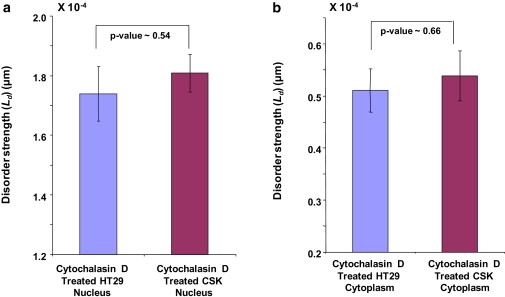
(a) Disorder strength (Ld) quantification for the nucleus of cytochalasin D-treated HT29 and CSK constructs show almost equal average disorder strength values. The ΔLd is statistically not significant (p = 0.54). This can be attributed to the effect of cytochalasin D on nuclear actin. (b) The ΔLd for cytoplasmic region of HT29 and CSK constructs is also minimal and is statistically nonsignificant (p = 0.66) that may be attributed to the rupture of filamentous actin in the cytoplasm.
We then carried out PWS analysis on the cytoplasmic region of the same cells. As indicated by Fig. 3 b, the ΔLd reduced substantially (effect-size = 5%, p value = 0.66). Comparing the differences in the cytoplasm for the untreated cells, this was a significant reduction of disorder strength difference. Attenuation of ΔLd in the cytoplasmic region of HT29 cells and Csk shRNA-transfected HT29 cells is the direct result of disruption of actin microfilament organization (44) in the cytoplasm of HT29 cells and Csk shRNA-transfected HT29 cells.
Treatment with colchicine
We next selected treatment with colchicine to selectively inhibit the microtubule polymerization due to its specific binding with tubulin (39). This treatment was carried out to evaluate the ΔLd between the two cell types if we were to induce cytoskeletal changes only in the cytoplasm without disturbing the cytoskeletal elements of the nucleus as microtubules are only present in the cytoplasm. We therefore hypothesized that treatment with colchicine should render significant ΔLd in nuclear region while attenuating it in the cytoplasmic region.
We treated both the HT29 cells and CSK constructs with a minimum dosage of colchicine (10 μM for 30 min) to induce microscopically comparable morphological changes. On carrying out PWS analysis on the nucleus of both the cell types, a statistically significant ΔLd was observed between the HT29 and CSK constructs (effect-size = 42.2%, p value = 0.002), a similar trend to the nuclear region of untreated cell types (Fig. 4 a). This significant ΔLd indicates that the colchicine treatment had little effect on the nucleus. However, as shown in Fig. 4 b, the difference in the cytoplasmic disorder strength between HT29 and Csk shRNA-transfected HT29 cells was reduced significantly (effect-size = 2.1%, p value = 0.87) due to the treatment with colchicine. These results confirmed the hypothesis that ΔLd of the cytoplasm would diminish while leaving the nuclear ΔLd intact. Also, this selective rupture of cytoplasmic cytoskeleton strengthened our original hypothesis regarding the role of cytoskeleton in determining the difference in disorder strength between the two cell types.
Figure 4.
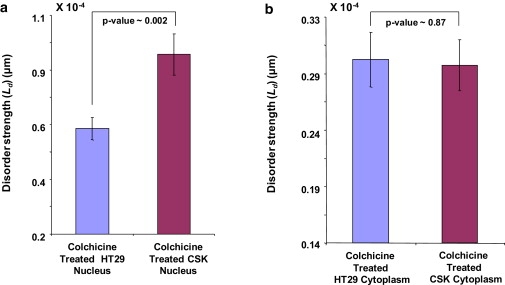
(a) Treatment with colchicine renders statistically significant (p = 0.002) ΔLd for the nuclear region of HT29 cells and CSK constructs as there are no microtubules present in the nucleus of these undividing cells. (b) The ΔLd reduces significantly (p = 0.87) in the cytoplasm of the colchicine-treated HT29 and CSK constructs that could be attributed to the rupture of microtubules present only in the cytoplasm.
In summary, the goal of this study was to identify the biological mechanism/s responsible for the disorder strength differences in the morphologically comparable cells with different aggressiveness; using human colon cancer cell line, HT29 cells, and Csk shRNA-transfected HT29 cells as a model system. By carrying out cytochalasin D and colchicine treatments selectively on these cell-types, we arrive at two major conclusions: 1), the significant difference in disorder strength between the untreated HT29 cells and CSK constructs is observed in both the nucleus and the cytoplasm instead of being limited to a specific region (albeit the effect-size was greater in the nucleus); and 2), one of the underlying biological origins of this difference in disorder strength is due to the cytoskeleton organization in these cellular compartments. By disrupting the actin microfilaments using cytochalasin D, the difference in disorder strength between these genetically modified cell lines is nullified in both the nucleus and cytoplasm of the cells. This falls in line with our hypothesis that because actin filaments are ubiquitously present throughout the cell, the disruption of their network with cytochalasin D would lead to the attenuation of nuclear as well as cytoplasmic disorder strength differences between HT29 cells and CSK constructs. Treating the HT29 and CSK constructs with colchicine negates the differences in disorder strength in the cytoplasm, while leaving the differences in nuclear disorder strength intact. This can be explained on the basis of strictly cytoplasmic localization of microtubular network in undividing cells. Overall, these results indicate that cytoskeleton is one of the major determinants of the disorder strength. In addition, they indicate that cells with different neoplastic aggressiveness could be expected to have different cytoskeletal architecture and hence different disorder strengths despite having comparable microscopic appearances. Moreover, this study implies that PWS instrument could be a potential tool for assessing alterations in cytoskeletal organization even in microscopically similar-appearing cells.
Herein, for the untreated HT29 and CSK constructs, it seems that there is a statistically significant gradient increase in the disorder strength that parallels higher neoplastic potential. The increase in disorder strength can be attributed to the increase in the intracellular mass-density (refractive-index) fluctuations and/or increase in the size of the macromolecular complexes (lc). As reported previously (29), the increase in lc can be correlated to the increase in fractal dimension of subcellular structures (45–47) and to the increase in the average and SD of the log-normal size distribution of particles (48,49).
It is well known that cell morphology, from nano- to microscale, is linked strongly to various biochemical, biomechanical, biophysical, and transport processes within the cell. Moreover, cytoskeleton shares a complex relationship with cellular processes (6). Cytoskeleton organization has been found to be defective in the cultured fibroblasts of patients with inherited adenocarcinoma in colon and rectum (50). There are reports that quantify altered amounts of different types of actin in nucleus and cytoplasm during tumorigenesis and during transformation and differentiation in bladder cells (1,51). The difference in disorder between HT29 cells and Csk shRNA constructs can be thought of as an indication of an early stage when these nanoscale molecular changes originate and get translated to nanoarchitectural modifications.
Although our results show one of the molecular mechanisms that can be responsible for elucidating intracellular disorder strength differences between HT29 and CSK constructs, there may be several other independent or correlated mechanisms that could play a critical role in determining the disorder differences between HT29 and Csk shRNA-transfected cells. Future studies will be required to elucidate these potential effects.
Acknowledgments
This work was supported by the National Institutes of Health (U54CA143869, R01 EB003682) and the National Science Foundation (CBET-0937987).
References
- 1.Rao J.Y., Bonner R.B., Hemstreet G.P., 3rd Quantitative changes in cytoskeletal and nuclear actins during cellular transformation. Int. J. Cancer. 1997;70:423–429. doi: 10.1002/(sici)1097-0215(19970207)70:4<423::aid-ijc9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 2.Ushijima T. Epigenetic field for cancerization. J. Biochem. Mol. Biol. 2007;40:142–150. doi: 10.5483/bmbrep.2007.40.2.142. [DOI] [PubMed] [Google Scholar]
- 3.Alberts D.S., Einspahr J.G., Bartels P.H. Karyometry of the colonic mucosa. Cancer Epidemiol. Biomarkers Prev. 2007;16:2704–2716. doi: 10.1158/1055-9965.EPI-07-0595. [DOI] [PubMed] [Google Scholar]
- 4.Chen L.C., Hao C.Y., Lee N.M. Alteration of gene expression in normal-appearing colon mucosa of APC(min) mice and human cancer patients. Cancer Res. 2004;64:3694–3700. doi: 10.1158/0008-5472.CAN-03-3264. [DOI] [PubMed] [Google Scholar]
- 5.Hao C.Y., Moore D.H., Lee N.M. Altered gene expression in normal colonic mucosa of individuals with polyps of the colon. Dis. Colon Rectum. 2005;48:2329–2335. doi: 10.1007/s10350-005-0153-2. [DOI] [PubMed] [Google Scholar]
- 6.Adam Wax V.B. McGraw-Hill; Burr Ridge, IL: 2009. Biomedical Applications of Light Scattering. [Google Scholar]
- 7.Zheng Y.X., Oegema K. Cell structure and dynamics. Curr. Opin. Cell Biol. 2008;20:1–3. [Google Scholar]
- 8.McDonald D., Carrero G., Hendzel M.J. Nucleoplasmic beta-actin exists in a dynamic equilibrium between low-mobility polymeric species and rapidly diffusing populations. J. Cell Biol. 2006;172:541–552. doi: 10.1083/jcb.200507101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollard T.D. Genomics, the cytoskeleton and motility. Nature. 2001;409:842–843. doi: 10.1038/35057029. [DOI] [PubMed] [Google Scholar]
- 10.Miller C.A., 3rd, Cohen M.D., Costa M. Complexing of actin and other nuclear proteins to DNA by cis-diamminedichloroplatinum(II) and chromium compounds. Carcinogenesis. 1991;12:269–276. doi: 10.1093/carcin/12.2.269. [DOI] [PubMed] [Google Scholar]
- 11.Kolber M.A., Broschat K.O., Landa-Gonzalez B. Cytochalasin B induces cellular DNA fragmentation. FASEB J. 1990;4:3021–3027. doi: 10.1096/fasebj.4.12.2394319. [DOI] [PubMed] [Google Scholar]
- 12.Lachapelle M., Aldrich H.C. Phalloidin-gold complexes: a new tool for ultrastructural localization of F-actin. J. Histochem. Cytochem. 1988;36:1197–1202. doi: 10.1177/36.9.3403970. [DOI] [PubMed] [Google Scholar]
- 13.Ankenbauer T., Kleinschmidt J.A., Franke W.W. Identification of a widespread nuclear actin binding protein. Nature. 1989;342:822–825. doi: 10.1038/342822a0. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein H., Payne C.M., Garewal H. A proteomic study of resistance to deoxycholate-induced apoptosis. Carcinogenesis. 2004;25:681–692. doi: 10.1093/carcin/bgh072. [DOI] [PubMed] [Google Scholar]
- 15.Nobes C.D., Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 16.Schlussel R.N., Droller M.J., Liu B.C.S. Type IV procollagen mRNA regulation: evidence for extracellular matrix/cytoskeleton/nuclear matrix interactions in human urothelium. J. Urol. 1991;146:428–432. doi: 10.1016/s0022-5347(17)37818-7. [DOI] [PubMed] [Google Scholar]
- 17.Hess S.T., Huang S., Webb W.W. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 2002;41:697–705. doi: 10.1021/bi0118512. [DOI] [PubMed] [Google Scholar]
- 18.Webb W.W. Fluorescence correlation spectroscopy: inception, biophysical experimentations, and prospectus. Appl. Opt. 2001;40:3969–3983. doi: 10.1364/ao.40.003969. [DOI] [PubMed] [Google Scholar]
- 19.Lipski A.M., Pino C.J., Shastri V.P. The effect of silica nanoparticle-modified surfaces on cell morphology, cytoskeletal organization and function. Biomaterials. 2008;29:3836–3846. doi: 10.1016/j.biomaterials.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadakavukaren K.K., Chen L.B., Griffith O.H. Photoelectron microscopy and immunofluorescence microscopy of cytoskeletal elements in the same cells. Proc. Natl. Acad. Sci. USA. 1983;80:4012–4016. doi: 10.1073/pnas.80.13.4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z.F., Shah J.V., Berns M.W. Fluorescence correlation spectroscopy investigation of a GFP mutant-enhanced cyan fluorescent protein and its tubulin fusion in living cells with two-photon excitation. J. Biomed. Opt. 2004;9:395–403. doi: 10.1117/1.1646416. [DOI] [PubMed] [Google Scholar]
- 22.Torralba S., Pisabarro A.G., Ramirez L. Immunofluorescence microscopy of the microtubule cytoskeleton during conjugate division in the dikaryon Pleurotus ostreatus N001. Mycologia. 2004;96:41–51. [PubMed] [Google Scholar]
- 23.Backman V., Gurjar R., Feld M.S. Imaging and measurement of cell structure and organization with submicron accuracy using light scattering spectroscopy. Optical Biopsy IV. 2002;4613:101–110. [Google Scholar]
- 24.Backman V., Wallace M.B., McGillican T. Detection of preinvasive cancer cells. Nature. 2000;406:35–36. doi: 10.1038/35017638. [DOI] [PubMed] [Google Scholar]
- 25.Gurjar R.S., Backman V., Feld M.S. Imaging human epithelial properties with polarized light-scattering spectroscopy. Nat. Med. 2001;7:1245–1248. doi: 10.1038/nm1101-1245. [DOI] [PubMed] [Google Scholar]
- 26.Perelman L.T., Backman V., Feld M.S. Observation of periodic fine structure in reflectance from biological tissue: A new technique for measuring nuclear size distribution. Phys. Rev. Lett. 1998;80:627–630. [Google Scholar]
- 27.Itzkan I., Qiu L., Perelman L.T. Confocal light absorption and scattering spectroscopic microscopy monitors organelles in live cells with no exogenous labels. Proc. Natl. Acad. Sci. USA. 2007;104:17255–17260. doi: 10.1073/pnas.0708669104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Born M., Wolf E. Cambridge University Press; New York: 1999. Principles of Optics: Electromagnetic Theory of Propagation, Interference and Diffraction of Light. [Google Scholar]
- 29.Subramanian H., Pradhan P., Backman V. Optical methodology for detecting histologically unapparent nanoscale consequences of genetic alterations in biological cells. Proc. Natl. Acad. Sci. USA. 2008;105:20118–20123. doi: 10.1073/pnas.0804723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian H., Pradhan P., Backman V. Partial-wave microscopic spectroscopy detects subwavelength refractive index fluctuations: an application to cancer diagnosis. Opt. Lett. 2009;34:518–520. doi: 10.1364/ol.34.000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y., Li X., Backman V. Elastic backscattering spectroscopic microscopy. Opt. Lett. 2005;30:2445–2447. doi: 10.1364/ol.30.002445. [DOI] [PubMed] [Google Scholar]
- 32.Subramanian H., Roy H.K., Backman V. Nanoscale cellular changes in field carcinogenesis detected by partial wave spectroscopy. Cancer Res. 2009;69:5357–5363. doi: 10.1158/0008-5472.CAN-08-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barer R., Ross K.F., Tkaczyk S. Refractometry of living cells. Nature. 1953;171:720–724. doi: 10.1038/171720a0. [DOI] [PubMed] [Google Scholar]
- 34.Davies H.G., Wilkins M.H. Interference microscopy and mass determination. Nature. 1952;169:541. doi: 10.1038/169541a0. [DOI] [PubMed] [Google Scholar]
- 35.Haley S.B., Erdös P. Wave propagation in one-dimensional disordered structures. Phys. Rev. B Condens. Matter. 1992;45:8572–8584. doi: 10.1103/physrevb.45.8572. [DOI] [PubMed] [Google Scholar]
- 36.Pradhan P., Kumar N. Localization of light in coherently amplifying random media. Phys. Rev. B Condens. Matter. 1994;50:9644–9647. doi: 10.1103/physrevb.50.9644. [DOI] [PubMed] [Google Scholar]
- 37.Rammal R., Doucot B. Invariant imbedding approach to localization. 1. General framework and basic equations. J. Phys. (Paris) 1987;48:509–526. [Google Scholar]
- 38.Frixione E. Recurring views on the structure and function of the cytoskeleton: a 300-year epic. Cell Motil. Cytoskeleton. 2000;46:73–94. doi: 10.1002/1097-0169(200006)46:2<73::AID-CM1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 39.Cohen E., Ophir I., Shaul Y.B. Induced differentiation in HT29, a human colon adenocarcinoma cell line. J. Cell Sci. 1999;112:2657–2666. doi: 10.1242/jcs.112.16.2657. [DOI] [PubMed] [Google Scholar]
- 40.Fantini J., Rognoni J.B., Marvaldi J. Suramin-treated HT29-D4 cells grown in the presence of glucose in permeable culture chambers form electrically active epithelial monolayers. A comparative study with HT29-D4 cells grown in the absence of glucose. Eur. J. Cell Biol. 1990;51:110–119. [PubMed] [Google Scholar]
- 41.Anderson P.W., Thouless D.J., Fisher D.S. New method for a scaling theory of localization. Phys. Rev. B. 1980;22:3519–3526. [Google Scholar]
- 42.Percipalle P., Visa N. Molecular functions of nuclear actin in transcription. J. Cell Biol. 2006;172:967–971. doi: 10.1083/jcb.200512083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parfenov V.N., Davis D.S., Murti K.G. Nuclear actin filaments and their topological changes in frog oocytes. Exp. Cell Res. 1995;217:385–394. doi: 10.1006/excr.1995.1101. [DOI] [PubMed] [Google Scholar]
- 44.Cooper J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wax A., Yang C., Feld M.S. Cellular organization and substructure measured using angle-resolved low-coherence interferometry. Biophys. J. 2002;82:2256–2264. doi: 10.1016/S0006-3495(02)75571-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wax A., Pyhtila J.W., Stoner G.D. Prospective grading of neoplastic change in rat esophagus epithelium using angle-resolved low-coherence interferometry. J. Biomed. Opt. 2005;10:051604. doi: 10.1117/1.2102767. [DOI] [PubMed] [Google Scholar]
- 47.Chalut K.J., Ostrander J.H., Wax A. Light scattering measurements of subcellular structure provide noninvasive early detection of chemotherapy-induced apoptosis. Cancer Res. 2009;69:1199–1204. doi: 10.1158/0008-5472.CAN-08-3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson J.D., Foster T.H. Mie theory interpretations of light scattering from intact cells. Opt. Lett. 2005;30:2442–2444. doi: 10.1364/ol.30.002442. [DOI] [PubMed] [Google Scholar]
- 49.Mourant J.R., Johnson T.M., Freyer J.P. Polarized angular dependent spectroscopy of epithelial cells and epithelial cell nuclei to determine the size scale of scattering structures. J. Biomed. Opt. 2002;7:378–387. doi: 10.1117/1.1483317. [DOI] [PubMed] [Google Scholar]
- 50.Kopelovich L., Conlon S., Pollack R. Defective organization of actin in cultured skin fibroblasts from patients with inherited adenocarcinoma. Proc. Natl. Acad. Sci. USA. 1977;74:3019–3022. doi: 10.1073/pnas.74.7.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rao J.Y., Hemstreet G.P., 3rd, Jones P.L. Cellular F-actin levels as a marker for cellular transformation: correlation with bladder cancer risk. Cancer Res. 1991;51:2762–2767. [PubMed] [Google Scholar]