

Abstract
Major histocompatibility complex class II (MHC-II) molecules sample peptides from the extracellular space, allowing the immune system to detect the presence of foreign microbes from this compartment. To be able to predict the immune response to given pathogens, a number of methods have been developed to predict peptide–MHC binding. However, few methods other than the pioneering TEPITOPE/ProPred method have been developed for MHC-II. Despite recent progress in method development, the predictive performance for MHC-II remains significantly lower than what can be obtained for MHC-I. One reason for this is that the MHC-II molecule is open at both ends allowing binding of peptides extending out of the groove. The binding core of MHC-II-bound peptides is therefore not known a priori and the binding motif is hence not readily discernible. Recent progress has been obtained by including the flanking residues in the predictions. All attempts to make ab initio predictions based on protein structure have failed to reach predictive performances similar to those that can be obtained by data-driven methods. Thousands of different MHC-II alleles exist in humans. Recently developed pan-specific methods have been able to make reasonably accurate predictions for alleles that were not included in the training data. These methods can be used to define supertypes (clusters) of MHC-II alleles where alleles within each supertype have similar binding specificities. Furthermore, the pan-specific methods have been used to make a graphical atlas such as the MHCMotifviewer, which allows for visual comparison of specificities of different alleles.
Keywords: epitope prediction, human leucocyte antigen, major histocompatibility complex class II, major histocompatibility complex peptide binding, T helper responses
Introduction
Major histocompatibility complex (MHC) molecules play an essential role in host–pathogen interactions determining the outcome of many host immune responses. Only a small fraction of the possible peptides that can be generated from proteins of pathogenic organisms actually generate an immune response. MHC class II molecules present peptides derived from proteins taken up from the extracellular environment. They stimulate cellular and humoral immunity against pathogenic micro-organisms through the actions of helper T lymphocytes. For a peptide to stimulate a helper T lymphocyte response, it must bind MHC class II in the endocytic organelles.1
Protein uptake by professional antigen-presenting cells through endocytosis or phagocytosis leads to formation of endosomes, which become increasingly acidic as they progress and eventually fuse with lysosomes containing MHC class II molecules (see Fig. 1). These vesicles contain aspartic and cysteine proteases, which are activated as the acidity increases and thereby degrade the protein into peptides. The protease activity can generate and destroy potential MHC class II epitopes. The peptides susceptible to destructive processing might survive if they can be loaded to MHC class II molecules early.2 The MHC class II molecules themselves are resistant to proteolysis2; therefore the core peptide is completely protected while the rest of the peptide can be trimmed by endopeptidases hydrolysing internal amide bonds and exoproteases hydrolysing one or two amino acids from either the N- or C-terminal.3,4 A type II membrane protein, called the invariant chain (Ii), is associated with newly synthesized MHC class II protein in the endoplasmic reticulum. The Ii stabilizes MHC molecules and directs transportation to early endosomes. Proteolytic cleavage of Ii is important for the correct peptide loading of MHC class II. Part of Ii, called CLIP (class II associated invariant peptide), occupies the peptide-binding groove of the MHC class II molecule. Interaction of MHC class II with an MHC class II-like molecule (called human leucocyte antigen HLA-DM in humans) catalyses the release of CLIP, allowing other peptides to bind before the MHC molecule’s migration to the plasma membrane.5
Figure 1.
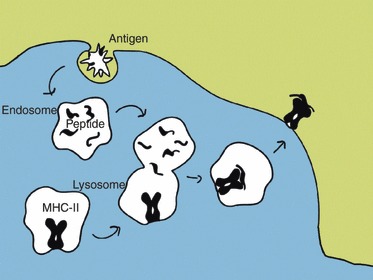
The major histocompatibility complex (MHC) class II antigen presentation pathway. Specialized antigen-presenting cells ingest exogenous antigens into endosomes by endocytosis or phagocytosis. The endosomes fuse with MHC class II containing lysosomes. The antigens (and MHC class II invariant chain) are degraded into peptides by proteases, and the release of the CLIP peptide allows the MHC class II molecule to bind antigen peptides before migration to the plasma membrane.
In the MHC class I antigen presentation pathway, the antigen degradation is predominantly caused by a single protease called the proteasome. Two distinctive forms of this protease exist in humans. The constitutive proteasome, which is expressed in all ‘healthy’ cells and the immuno-proteasome, which is expressed primarily in cells stimulated by interferon-γ. Both versions of the proteasome have a well-defined specificity, and predictive algorithms have been developed to characterize these. In contrast to this, the activities (and specificities) of the proteases in the MHC class II presentation pathway are poorly characterized.
The core binding motif of both MHC I and MHC II is approximately nine amino acids long.6 Whereas the peptide-binding groove in the MHC I molecule tends to be closed in both ends and MHC I rarely binds peptides much longer than nine amino acids, the ends of the MHC II binding groove are open (see Fig. 2). Consequently, MHC class II can accommodate much longer peptides – possibly even whole proteins.1,7 This difference has important implications for the development of algorithms predicting peptide binding to MHC class II. The specificity of an MHC I molecule can be derived by extracting the motif from a set of 9-mer peptides known to bind to a given allele. In contrast, a set of peptides binding MHC II will typically be of different lengths and therefore need to be correctly aligned before the nine amino acid core-binding motif can be identified.
Figure 2.
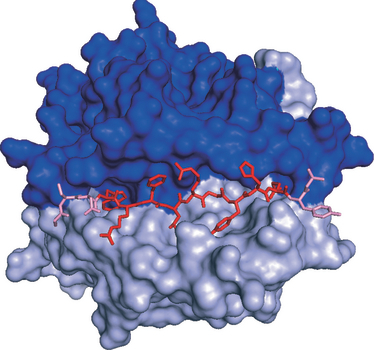
Protein crystal structure of major histocompatibility complex (MHC) class II molecule (HLA-DRB1*0101) in complex with peptide (PDB id: 1AQD). The MHC α-chain is shown in dark-blue, and the β-chain in light blue. The peptide (GSDWRFLRGYHQYA) is shown in red/pink, with the peptide-binding core (WRFLRGYHQ) in red, and the peptide-flanking amino acids in pink.
In humans, the MHC class II locus encodes for genes for an α- and a β-subunit of the MHC class II proteins HLA-DR, HLA-DQ and HLA-DP. The genes encoding the MHC class I and class II are highly polymorphic (i.e. many alleles exist at each gene locus). In the current release of the IMGT/HLA database, the number of registered DRα, DRβ, DQα, DQβ and DPα, DPβ proteins is 2, 637, 26, 77, 16 and 118, respectively.8 This can potentially generate more than 4000 combinations of HLA class II α and β subunits. This polymorphism complicates the task of deriving MHC prediction algorithms. These problems have been addressed by developing methods showing which MHC alleles have similar binding specificities, making it possible to find promiscuous peptides that bind to a series of MHC variants. Furthermore, pan-specific prediction algorithms have been developed that allow for binding predictions to be made even for alleles for which no binding data exist. Moreover, this polymorphism has large implications for vaccine design. The peptide-binding specificity of the allelic molecules (variants) is often very different. Every child inherits a set of α and β subunits for each of the three proteins from its parents, giving a potential set of 6–12 HLA class II molecules depending on the degree of heterozygosity. Different individuals will therefore typically react to different sets of peptides from a given pathogen. If a vaccine needs to contain a unique peptide for each of these molecules, it will need to comprise many different peptides.
MHC class II data, databases and data quality
Many databases exist hosting data describing the binding specificities for MHC molecules. The two main sources of data are the SYFPEITHI database of MHC ligands and T-cell epitopes9, and the immune epitope database (IEDB).10 As of November 2009, the SYFPEITHI database contained 1623 HLA class II ligands/epitopes covering 47 different HLA-DR, -DP and -DQ alleles. For HLA class I, the corresponding numbers are 3866 ligands/epitopes covering 119 HLA-A, -B and -C molecules. Also, in November 2009, the IEDB contained 15 556 HLA class II peptide-binding measurements covering 31 full-typed HLA-DR, and a small set of fully typed HLA-DQ alleles. There are other smaller databases hosting MHC epitope-related data such as MHCBN,11 and AntiJen.12 Data describing the binding specificity are therefore many folds scarcer for MHC class II molecules than for MHC class I.
Historically, MHC class II molecules were the first for which peptide binding could be demonstrated. In 1982, Werdelin demonstrated that chemically related antigens competed for antigen presentation and suggested that this was the result of competition for MHC;13 in 1983, Shimonkevitz et al.14demonstrated that peptides could replace protein antigens; and in 1984, Watts et al.15demonstrated that peptide and purified MHC molecules could replace antigen-presenting cells. However, it was not until 1985 that Babbitt et al.,16 using equilibrium dialysis, succeeded in generating biochemical evidence of a specific and saturable binding between a fluoresceinated hen egg lysozyme peptide, HEL46–61, and purified mouse MHC class II molecule, I-Ak. Noting the extreme stability of peptide–MHC class II complexes, Buus et al.17in 1986 used gel filtration chromatography, radiolabelled peptides and purified MHC class II molecules to generate an easy, robust and sensitive assay for peptide–MHC class II interaction. Since then, many different working principles for how to measure peptide–MHC class II interactions have been suggested (and it would be beyond the scope of this review to give a detailed account of all these alternative methods). Whereas the original Babbitt and Buus methods relied upon purified MHC class II molecules, many alternative methods have attempted to generate whole-cell (bound to cell surfaces cell lysates etc.) binding assays (for example ref. 18). The current IEDB release (December 2009) details almost 36 000 reported peptide–MHC class II interactions. About 90% of these data points emanate from studies using purified MHC class II molecules. The remaining 10% of the IEDB MHC class II data points have been derived from cell-based assays. Although this is only a minor part of the available data it is important to keep in mind as such whole-cell assays are at best semi-quantitative.19 One reason for this may be the frequent lack of proteolytic control, which may skew the peptide dose–response by orders of magnitude and conceal binding.20 Another important factor that may affect the reliability of the quantitative aspects of the IEDB data points is the historical era in which they were obtained. In the early days of measuring peptide–MHC class II interactions biochemically, a peptide binding with an affinity in the micromolar range was considered to be a strong binder, whereas today a peptide would have to bind in the nanomolar range to be considered a strong binder – and a peptide that binds with a micromolar affinity today would be considered borderline to being a non-binder. Clearly, the many improvements in MHC class II preparation, in peptide labelling, and in assay technology (e.g. Justesen et al.21) have increased the sensitivity of peptide–MHC class II assays and allowed the proper determination of nanomolar binding affinities. The IEDB incorporates all available data irrespective of the source of the data, and it is therefore bound to contain some data that may appear highly divergent. This should be kept in mind when using the IEDB data to develop predicting methods. Filtering the data to remove older and whole-cell-based data might yield more consistent data, and generate better predictors.
Predictive methods for MHC class II binding
Many different methods have been applied to predict peptide–MHC binding including simple binding motifs, quantitative matrices, hidden Markov models and artificial neural networks. For class I, these alignment-free methods can readily be applied because the binding motif is well characterized and most natural peptides that bind MHC class I are of close to equal length.9,22–25 However, the situation for MHC class II binding is quite different because of the great variability in the length of natural MHC-binding peptides. This length variability makes alignment a crucial and integrated part of estimating the MHC-binding motif and predicting peptide binding. Quantitative matrices estimated from experimentally derived position-specific binding profiles have given reasonable performance in prediction of MHC class II binding.26–29 However, such matrices are very costly to derive and more importantly they lack the flexibility of data-driven machine-learning methods to be refined in an iterative manner when more data become available. During the last decade, large efforts have therefore been invested in developing data-driven prediction methods for MHC class II. The early method by Brusic et al.30 applied a hybrid method for predicting peptide–MHC class II binding using an evolutionary algorithm to define the binding core and subsequently applied artificial neural networks to classify peptides as binding/non-binding. The work by Nielsen et al.31 extended the Gibbs sampler approach by Lawrence et al.32 to search for binding motifs in MHC class II ligand data. Later many other (often highly exotic) algorithms were proposed for MHC class II binding prediction, including ant colony33, hidden Markov models34, support vector machines35–37, and other motif search algorithms9–43 as well as consensus methods integrating the output from two or more, different prediction methods.44,45 However, most of these methods have been trained and evaluated on very limited data sets covering only a single or a few different MHC class II alleles. Further, the majority of the methods are trained on binary classified peptide data (binders versus non-binders). This type of qualitative prediction method is well suited to classify data, but does not allow a direct prediction of the peptide–MHC binding affinity.
A limited number of methods are publicly available for quantitative MHC class II prediction, namely the ARB38, SVRMHC35, MHCpred41 and NetMHCII.46,47 Other methods such as SVMHC48 and Propred49 are implementations of the TEPITOPE method,29 and provide prediction scores that are not in any direct way related to the peptide-binding affinity.
The binding of a peptide to a given MHC molecule is predominantly determined by the amino acids present in the peptide-binding core. However, peptide residues flanking the binding core (so-called peptide flanking residues, PFR) do also to some degree affect the binding affinity of a peptide.50,51 Several striking examples of the stabilizing effect of PFR can be found in the IEDB, for example the peptide RFYKTLRAEQASQ binds to the HLA-DRB1*0401 molecule with a 50% inhibition concentration (IC50) value of 5·67 nm whereas the truncated form YKTLRAEQA binds with an IC50 value of 33 100 nm. Most published methods for MHC class II binding prediction, however, focus on identifying the peptide-binding core only, ignoring the effects on the binding affinity of PFRs. The only methods that attempt for a direct incorporation of the effect of peptide flanking residues and peptide length are the algorithms developed by Chang et al.,40 Nielsen et al.46 and Nielsen and Lund.47 Of these, the NN-align algorithm47 is the only method that explicitly incorporates PFR and peptide length in the training, and in the work by Nielsen et al., it was demonstrated that the additional information provided by the PFR leads to significantly improved predictions.
All the methods described above are allele-specific and are therefore limited by whether sufficient experimental peptide-binding data are available for each allele in question. A minimum number of 200 peptides with characterized binding affinity are needed to derive an accurate description of the binding motif for MHC class II alleles (unpublished results). In the IEDB, only 14 HLA-DR, and two HLA-DQ alleles meet this criterion, leaving the vast majority of the more than 4000 different HLA class II molecules uncovered.
A seemingly promising approach that does not require binding data is to use three-dimensional (3D) structures of peptide–MHC complexes. Different MHC alleles have high sequence homology, and all solved MHC structures have a highly conserved fold, which opens the possibility to use homology modelling for those MHC alleles for which no 3D structure has been solved explicitly. Several approaches have been published that predict peptide binding to MHC molecules using known 3D structures.52–55 Threading-based approaches have been used to align peptides to known peptide–MHC structures and binders are selected using statistical pairwise potentials.56–58 Davies et al.59 used molecular dynamic and simulated annealing to sample the conformational space and predict binding of peptides to MHC class I molecules. Structure information has also been coupled with experimental data to predict peptide–MHC binding via quantitative structure–affinity relationship method.60 In a recent study,61 several structure-based approaches for predicting peptide binding to MHC class II molecules have been compared. Their prediction performance was evaluated on a large dataset of 3882 peptide-binding affinities to HLA-DRB1*0101. The implementation and evaluation of the different approaches led to overall comparable results: the different methods all made significantly better than random discriminations of binders from non-binders, but failed to compete with the accuracy of data-driven methods and, more importantly, failed to reach the prediction quality necessary for practical applications.
For MHC class I, several groups have proposed so-called pan-specific methods.62–67 All of these methods aim at integrating structural information with experimental peptide-binding data allowing for the generalization of binding predictions to MHC molecules characterized with few or even no peptide-binding data. The NetMHCpan method65,66 for instance is constructed in a way that takes both the peptide sequence and the MHC contact environment into account. The method leverages information from multiple MHC molecules in the prediction of the binding affinity of a peptide to a given query MHC molecule, and hence allows for both improved predictions for MHC molecules characterized by few binding data as well as for reasonably accurate predictions for previously uncharacterized MHC molecules. For MHC class II, the pan-specific methods are still in their infancy. Only two methods have been published claiming pan-specificity for MHC class II binding, the NetMHCIIpan method (Nielsen et al.68) and the Shift-invariant adaptive threading method by Zaitlen et al.69 However, the performance of both these method is hampered by the limited coverage of the HLA class II specificity space, and the applicability of the methods is in most cases limited to HLA-DR. Table 1 gives an overview of publicly available methods for prediction of peptide binding to MHC class II molecules.
Table 1.
Publicly available methods for prediction of peptide–major histocompatibility complex (MHC) class II binding
| Method | Link | Reference |
|---|---|---|
| SVMHC | http://www-bs.informatik.uni-tuebingen.de/Services/SVMHC | 48 |
| NetMHCII | http://www.cbs.dtu.dk/services/NetMHCII/ | 46, 47 |
| NetMHCIIpan | http://www.cbs.dtu.dk/services/NetMHCIIpan/ | 68 |
| Tepitope/Propred | http://www.imtech.res.in/raghava/propred/ | 29, 49 |
| SYFPEITHI | http://www.syfpeithi.de/ | 9 |
| IEDB_ARB | http://www.tools.immuneepitope.org/analyze/html/mhc_II_binding.html | 38 |
| IEDB_Comblib | http://www.tools.immuneepitope.org/analyze/html/mhc_II_binding.html | |
| IEDB_SMM-align | http://www.tools.immuneepitope.org/analyze/html/mhc_II_binding.html | 46 |
| IEDB_Cons | http://www.tools.immuneepitope.org/analyze/html/mhc_II_binding.html | 45 |
| Rankpep | http://www.bio.dfci.harvard.edu/Tools/rankpep.html | 98 |
| HLA-DR4pred | http://www.imtech.res.in/raghava/hladr4pred/index.html | 99 |
| EpiToolKit | http://www.epitoolkit.org/ | 100 |
MHC class II benchmark studies
Several studies have compared the predictive performance of MHC class II prediction methods. In the study by Wang et al.45 nine publicly available MHC class II prediction methods were compared using a large set of quantitative MHC class II binding data covering 14 HLA-DR and two mouse MHC alleles. The study by Lin et al.70 covered 21 prediction servers and was based on 103 peptides with measured binding to seven common HLA-DR molecules. In both benchmark studies, the pioneer MHC–peptide-binding prediction algorithm TEPITOPE27,29 came out as one of the best performing methods. Of the many data-driven methods, only the NetMHCII46 (Wang study), and NetMHCIIpan68 and Multipred (SVM)62 (Lin study) performed consistently better than TEPITOPE.
A major conclusion from these benchmark studies is that the state-of-the-art MHC class II prediction methods do not match the prediction capabilities of MHC class I predictors. In terms of AUC values (area under the receiver operator curve) state-of-the-art MHC class I prediction methods will often achieve predictive performance values in the range 0·85–0·95 depending on the training and evaluation data size and composition. For MHC class II, the corresponding performance values are significantly lower, often in the range 0·75–0·85 even for MHC molecules where many thousand peptide-binding data are available for training the prediction method.
Visualization of MHC binding motifs
A powerful way of visualizing the receptor-binding motif is by using so-called sequence logos. Sequence logos are a graphical representation of aligned multiple amino (or nucleic) acid sequences. Sequence logos were originally developed by Tom Schneider and Mike Stephens.71 For each position, the frequency of all 20 amino acids is displayed as a stack of letters. The total height of the stack represents the sequence conservation and the individual height of the symbols relates to the relative frequency of the corresponding amino acid at that position. The higher the stack at a given position, the more conserved the position is. The MHC Motif Viewer is a website which collects such sequence logo representations of the binding motif for HLA-DR molecules (as well as a large set of MHC class I molecules).72,73 The binding specificity for each HLA-DR molecule is predicted using the NetMHCIIpan peptide–MHC binding prediction methods, and is visualized in a format that allows for a comprehensive interpretation of binding motif anchor positions and amino acid preferences. In the logo plots used in the MHCMotifViewer website, the amino acids are coloured according to their physicochemical properties:
Acidic [DE]: red
Basic [HKR]: blue
Hydrophobic [ACFILMPVW]: black
Neutral [GNQSTY]: green
Two examples of HLA-DR binding motifs are shown in Fig. 3. The difference in binding specificity of the MHC molecules is apparent from these logo plots. Both molecules have a preference for hydrophobic amino acids at P1. The HLA-DR1*0301 molecule will bind peptides with acidic amino acids at P4, and basic amino acids at P6 and P9. The HLA-DRB1*1501 molecule, on the other hand, prefers hydrophobic and neutral amino acids at the P4 and P6 anchors. The large binding promiscuity of the HLA-DR molecule is clear from the logo representation. At each anchor position 1, 4, (6) and 9 multiple amino acids are observed with close to equal preference for binding. This is in contrast to MHC class I where most anchor positions are characterized by a preference for one or two different amino acids. For more details on sequence logos and how they are constructed see ref. 73.
Figure 3.
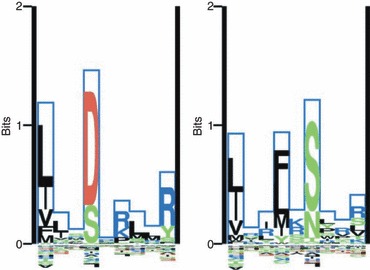
Sequence logo representation of the binding motif for two HLA-DR molecules. The Kullback–Leibler (KL) sequence logo is taken from the MHC Motif Viewer website.73 Left: HLA-DRB1*0301, Right: HLA-DRB1*1501. The KL information content is plotted along the nine-mer binding core (solid blue line). Amino acids with positive influence on the binding are plotted on the positive y-axis, and amino acids with a negative influence on binding are plotted on the negative y-axis. The height of each amino acid is given by their relative contribution to the binding specificity (for details see ref. 73). The primary anchor positions (P1, P4, P6 and P9) show clear and distinct amino acid preferences. At P1, both molecules prefer hydrophobic amino acids. HLA-DRB1*0301 has a preference for aspartic acid at P4 whereas at P6 and P9 basic amino acids are preferred. HLA-DRB1*1501, on the other hand, prefers hydrophobic and neutral amino acids at the P4 and P6 anchors.
MHC specificity clustering
As stated in the Introduction, each MHC molecule has a potentially unique binding specificity, and if a vaccine needs to contain a unique peptide for each of these molecules it will need to comprise several hundred peptides. Moreover, the task of deriving MHC prediction algorithms would be daunting. Fortunately, many MHC alleles have very similar binding specificities, and it is therefore often possible to find peptides that bind promiscuously to several MHC variants. This has important implications both for vaccine design and development of MHC binding prediction algorithms. As described above, this binding promiscuity has allowed for the development of pan-specific MHC binding prediction algorithms, and equally importantly, it limits the number of epitopes needed in order to have broad MHC allelic coverage in a vaccine design.
Most work on MHC specificity clustering has been performed on the MHC class I molecules.74–79 The general idea behind these clustering approaches is to identify groups (often called supertypes) so that all MHC molecules within one supertype will bind a similar set of peptides. For MHC class II, limited work has been made on specificity clustering because of the lack of data characterizing the specificity of the MHC class II molecules. Lund et al.76 used the experimentally derived specificity scoring matrices of the TEPITOPE method29 to define nine HLA-DR supertypes, and the work by Nielsen et al.68 refined this study using the pan-specific HLA-DR prediction method to define 12 HLA-DR supertypes. No study with broad allelic coverage has been made defining specificity clusters for HLA-DQ and HLA-DP.
Recent studies have challenged the MHC supertype concept, suggesting that supertypes often provide an oversimplification of the MHC specificity space80–83 These studies all demonstrate that the peptide-binding overlap between MHC molecules within a supertype is often far from 100%, and that peptides can bind promiscuously to MHC molecules belonging to different supertypes. However, and perhaps more importantly, the study by Perez et al.80 demonstrates that the observed promiscuity is predictable using advanced bioinformatical methods for pan-specific HLA-peptide binding. Another, and potentially more rational, approach to achieve broad allelic coverage in a vaccine design would therefore be to select a limited set of peptides restricted to as many alleles as possible. This should be within reach with pan-specific approaches that can make predictions for all alleles where the protein sequence is known.
Identification of CD4 epitopes and MHC class II ligands
An important question is how good the best available MHC class II prediction algorithms are in terms of predicting immunogenicity. The real test of MHC class II–peptide-binding prediction algorithms comes when trying to predict antigen presentation by MHC class II and CD4+ T-cell immunogenicity. It might well be that a method can be very accurate in predicting binding IC50 values, but poor when correlating prediction values to peptide immunogenicity.
Two types of studies are especially relevant in the context of antigen presentation and CD4+ immunogenicity. In the first type of study, antigen presentation is investigated. Peptides that are observed in complex with MHC on the cell surface (observed MHC ligands) must have passed through the antigen presentation pathway. One can ask how large a fraction of the peptide pool in the source protein needs to be tested to identify the observed MHC ligand. If a prediction method is perfect, the ligand should be ranked as the first, and therefore be identifiable with a false-positive rate of 0, and if a method is random, the false-positive rate would be 50%. Several papers have employed this type of benchmark approach correlating peptide-binding score to likelihood of antigen presentation.46,47,68,84–87 For MHC class II, the general observation in these studies is that the prediction methods perform significantly better than random, ranking the experimentally known ligands within top 10–15%. A typical protein contains 300 amino acids, and in terms of experimental work, these values translate into an effort of testing approximately 35 peptides to identify the ‘true’ ligand. In comparison, similar experiments for MHC class I prediction algorithms tend to rank the ligands within the top 1–2%, reducing the experimental effort to testing two to five peptides to identify the ‘true’ ligand.65,66,87,88
The second type of experimental validation of MHC class II binding prediction algorithms relates to CD4 T-cell immunogenicity. Even small pathogens will contain many thousands of peptides, and often large numbers of strains exist. This, combined with the large MHC specificity diversity in the human population, makes direct experimental immunology, where overlapping peptides covering the genome in question are experimentally investigated for their immunogenicity, an extremely costly process. Many studies have therefore applied in silico screening methods to the identification of T-cell epitopes.89–95 The majority of these studies, including even the recent ones, uses the TEPITOPE prediction algorithm for the in silico screening. This is surprising because many benchmark studies have shown that state-of-the-art data-driven methods significantly outperform the TEPITOPE method when it comes to identification of MHC class II ligands (see above). Nonetheless the general conclusion from these studies is that in silico screening is a useful tool for identification of CD4 T epitopes. In particular, the prediction algorithms have proven successful in identifying CD4 epitopes promiscuously restricted to multiple HLA-DR alleles. However, the relatively large false-positive rate in the studies further underlines the need for a continued search for improved prediction methods.
Discussion
During the last decade the accuracy and MHC coverage of data-driven peptide-binding methods have increased significantly. We have reviewed the quality of the state-of-the-art MHC class II peptide-binding prediction algorithms, and shown how such algorithms can provide a powerful tool to guide the rational search for CD4 T-cell epitopes. It is clear, however, that the quality of peptide-binding algorithms is significantly lower for MHC class II compared with MHC class I. One might speculate why this difference in accuracy persists? Many MHC class II alleles have been characterized by thousands of quantitative peptide-binding measurements, and stating that the difference in quality between class I and class II is caused by lack of data seems inadequate. In fact, we can show that limited improvement in prediction accuracy is observed once the number of peptide data surpasses 1000 (unpublished results). It is therefore natural to ask why we do not achieve a higher performance? It is clear that prediction of peptide binding to MHC class II is a much more complicated problem than peptide binding to MHC class I both because of the alignment problem imposed by the broad length distribution of MHC class II binders, and as a result of the vast peptide-binding promiscuity imposed by the relatively weak binding specificity of most MHC class II molecules. On the other hand, the peptide-binding event is highly reproducible. Repeated experiments measuring the binding affinity of a peptide to an MHC molecule under similar experimental conditions give similar results, indicating that the binding affinity should be predictable.
Several factors could explain this apparent discrepancy between the amount of data and the quality of the prediction methods. One important factor is the data quality. As mentioned earlier, data for MHC class II are generated using a diverse set of experimental assays by a large number of different groups. The data in the IEDB for MHC class I was primarily generated since 2006 by two experimental assays. More than 99% of the quantitative MHC class I binding data are generated by the two comparable assays developed in the laboratories of A. Sette and S. Buus. More than 95% of the class I data has been generated since 2005, and < 2% before 2001. For MHC class II the situation is different. Here, about 80% of the quantitative data are produced using one single assay type, whereas 20 groups using more than five different assay types produce the remaining 20%. Less than 80% of the data were produced after 2006, and more than 15% of the data were produced before 2001. Most binding data describing the specificity of MHC molecules are equilibrium binding affinity values. Binding affinity might not be the only relevant feature for the characterization of epitopes. Binding stability might be equally relevant because the avidity of the MHC peptide complex to bind T cells clearly depends both on the equilibrium binding constant and the stability of the complex,96 and complementing the MHC binding data with peptide stability measurements may lead to improved epitope predictions. As a result of the open ends of the MHC class II binding cleft, peptides might bind in multiple registers.97 Several conflicting studies have shown both positive and negative effects of including such multiple binding registers into the prediction of MHC class II binding, and no consensus has been reached in the field as to how big the effect of multiple binding registers would be for an accurate description of the binding specificity.40,70 Finally, for naturally processed MHC ligands and CD4 epitopes, factors other than peptide–MHC binding can influence the peptide immunogenicity, including susceptibility to proteolytic activity in the endosome/lysosome and peptide/antigen abundance in the antigen-presenting cell.
At this point, it is not clear to what extent improved data quality and development of accurate bioinformatical algorithms characterizing these other factors will improve the predictive performance for MHC class II epitope predictors. Recent large-scale epitope discovery projects have focused primarily on MHC class I epitopes, and large amounts of high-quality peptide-binding data have been generated since 2005 characterizing more than 60 different HLA-A and HLA-B alleles leading to great improvements in the prediction methods now available for MHC class I peptide binding. One can hope that MHC class II will gain more focus in future epitope discovery projects, hence allowing generation of high-quality MHC class II data and prediction methods.
Acknowledgments
The work has been supported by National Institutes of Health (contracts HHSN26620040006C and HHSN26620040006C) and EU 7th framework grant 222773 (PepChipOmics). The authors thank Tine Rugh Poulsen and Andreas Holm Mattsson for preparing the MHC class II pathway illustration.
Disclosures
The authors have no financial or conflicts of interests to disclose.
References
- 1.Castellino F, Zhong G, Germain RN. Antigen presentation by MHC class II molecules: invariant chain function, protein trafficking, and the molecular basis of diverse determinant capture. Hum Immunol. 1997;54:159–69. doi: 10.1016/s0198-8859(97)00078-5. [DOI] [PubMed] [Google Scholar]
- 2.Mouritsen S, Meldal M, Werdelin O, Hansen AS, Buus S. MHC molecules protect T cell epitopes against proteolytic destruction. J Immunol. 1992;149:1987–93. [PubMed] [Google Scholar]
- 3.Larsen SL, Pedersen LO, Buus S, Stryhn A. T cell responses affected by aminopeptidase N (CD13)-mediated trimming of major histocompatibility complex class II-bound peptides. J Exp Med. 1996;184:183–9. doi: 10.1084/jem.184.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman HA. Endosomal proteolysis and MHC class II function. Curr Opin Immunol. 1998;10:93–102. doi: 10.1016/s0952-7915(98)80038-1. [DOI] [PubMed] [Google Scholar]
- 5.Watts C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol. 2004;5:685–92. doi: 10.1038/ni1088. [DOI] [PubMed] [Google Scholar]
- 6.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 7.Sette A, Adorini L, Colon SM, Buus S, Grey HM. Capacity of intact proteins to bind to MHC class II molecules. J Immunol. 1989;143:1265–7. [PubMed] [Google Scholar]
- 8.Robinson J, Marsh SG. The IMGT/HLA database. Methods Mol Biol. 2007;409:43–60. doi: 10.1007/978-1-60327-118-9_3. (Clifton, NJ) [DOI] [PubMed] [Google Scholar]
- 9.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–9. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 10.Sette A, Fleri W, Peters B, Sathiamurthy M, Bui HH, Wilson S. A roadmap for the immunomics of category A–C pathogens. Immunity. 2005;22:155–61. doi: 10.1016/j.immuni.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Bhasin M, Singh H, Raghava GPS. MHCBN: a comprehensive database of MHC binding and non-binding peptides. Bioinformatics. 2003;19:665–6. doi: 10.1093/bioinformatics/btg055. [DOI] [PubMed] [Google Scholar]
- 12.Toseland CP, Clayton DJ, McSparron H, et al. AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immunome research. 2005;1:4. doi: 10.1186/1745-7580-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werdelin O. Chemically related antigens compete for presentation by accessory cells to T cells. J Immunol. 1982;129:1883–91. [PubMed] [Google Scholar]
- 14.Shimonkevitz R, Kappler J, Marrack P, Grey H. Antigen recognition by H-2-restricted T cells. I. Cell-free antigen processing. J Exp Med. 1983;158:303–16. doi: 10.1084/jem.158.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watts TH, Brian AA, Kappler JW, Marrack P, McConnell HM. Antigen presentation by supported planar membranes containing affinity-purified I-Ad. Proc Natl Acad Sci USA. 1984;81:7564–8. doi: 10.1073/pnas.81.23.7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babbitt BP, Allen PM, Matsueda G, Haber E, Unanue ER. Binding of immunogenic peptides to Ia histocompatibility molecules. Nature. 1985;317:359–61. doi: 10.1038/317359a0. [DOI] [PubMed] [Google Scholar]
- 17.Buus S, Sette A, Colon SM, Jenis DM, Grey HM. Isolation and characterization of antigen–Ia complexes involved in T cell recognition. Cell. 1986;47:1071–7. doi: 10.1016/0092-8674(86)90822-6. [DOI] [PubMed] [Google Scholar]
- 18.Rothbard JB, Busch R, Howland K, Bal V, Fenton C, Taylor WR, Lamb JR. Structural analysis of a peptide–HLA class II complex: identification of critical interactions for its formation and recognition by T cell receptor. Int Immunol. 1989;1:479–86. doi: 10.1093/intimm/1.5.479. [DOI] [PubMed] [Google Scholar]
- 19.Rothbard JB, Busch R. Binding of biotinylated peptides to MHC class II proteins on cell surfaces. Current protocols in immunology/edited by John E Coligan et al 2001;Chapter 18:Unit 18 1. [DOI] [PubMed]
- 20.Buus S, Werdelin O. Oligopeptide antigens of the angiotensin lineage compete for presentation by paraformaldehyde-treated accessory cells to T cells. J Immunol. 1986;136:459–65. [PubMed] [Google Scholar]
- 21.Justesen S, Harndahl M, Lamberth K, Nielsen LL, Buus S. Functional recombinant MHC class II molecules and high-throughput peptide-binding assays. Immunome research. 2009;5:2. doi: 10.1186/1745-7580-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–75. [PubMed] [Google Scholar]
- 23.Brusic V, Rudy G, Harrison LC. Prediction of MHC binding peptides using artificial neural networks. In: Stonier RJaYX., editor. Complex systems: mechanism of adaptation. Amsterdam: IOS Press; 1994. pp. 253–60. [Google Scholar]
- 24.Buus S, Lauemoller SL, Worning P, et al. Sensitive quantitative predictions of peptide–MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens. 2003;62:378–84. doi: 10.1034/j.1399-0039.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–17. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sette A, Buus S, Appella E, Smith JA, Chesnut R, Miles C, Colon SM, Grey HM. Prediction of major histocompatibility complex binding regions of protein antigens by sequence pattern analysis. Proc Natl Acad Sci USA. 1989;86:3296–2300. doi: 10.1073/pnas.86.9.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammer J, Bono E, Gallazzi F, Belunis C, Nagy Z, Sinigaglia F. Precise prediction of major histocompatibility complex class II–peptide interaction based on peptide side chain scanning. J Exp Med. 1994;180:2353–8. doi: 10.1084/jem.180.6.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall KW, Wilson KJ, Liang J, Woods A, Zaller D, Rothbard JB. Prediction of peptide affinity to HLA DRB1*0401. J Immunol. 1995;154:5927–33. [PubMed] [Google Scholar]
- 29.Sturniolo T, Bono E, Ding J, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17:555–61. doi: 10.1038/9858. [DOI] [PubMed] [Google Scholar]
- 30.Brusic V, Rudy G, Honeyman G, Hammer J, Harrison L. Prediction of MHC class II-binding peptides using an evolutionary algorithm and artificial neural network. Bioinformatics. 1998;14:121–30. doi: 10.1093/bioinformatics/14.2.121. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, Brunak S, Lund O. Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics. 2004;20:1388–97. doi: 10.1093/bioinformatics/bth100. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–14. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- 33.Karpenko O, Shi J, Dai Y. Prediction of MHC class II binders using the ant colony search strategy. Artif Intell Med. 2005;35:147–56. doi: 10.1016/j.artmed.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Noguchi H, Kato R, Hanai T, Matsubara Y, Honda H, Brusic V, Kobayashi T. Hidden Markov model-based prediction of antigenic peptides that interact with MHC class II molecules. J Biosci Bioeng. 2002;94:264–70. doi: 10.1263/jbb.94.264. [DOI] [PubMed] [Google Scholar]
- 35.Wan J, Liu W, Xu Q, Ren Y, Flower DR, Li T. SVRMHC prediction server for MHC-binding peptides. BMC Bioinformatics. 2006;7:463. doi: 10.1186/1471-2105-7-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salomon J, Flower DR. Predicting Class II MHC–peptide binding: a kernel based approach using similarity scores. BMC Bioinformatics. 2006;7:501. doi: 10.1186/1471-2105-7-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui J, Han LY, Lin HH, Zhang HL, Tang ZQ, Zheng CJ, Cao ZW, Chen YZ. Prediction of MHC-binding peptides of flexible lengths from sequence-derived structural and physicochemical properties. Mol Immunol. 2007;44:866–77. doi: 10.1016/j.molimm.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Bui HH, Sidney J, Peters B, et al. Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics. 2005;57:304–14. doi: 10.1007/s00251-005-0798-y. [DOI] [PubMed] [Google Scholar]
- 39.Murugan N, Dai Y. Prediction of MHC class II binding peptides based on an iterative learning model. Immunome research. 2005;1:6. doi: 10.1186/1745-7580-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang ST, Ghosh D, Kirschner DE, Linderman JJ. Peptide length-based prediction of peptide–MHC class II binding. Bioinformatics. 2006;22:2761–7. doi: 10.1093/bioinformatics/btl479. [DOI] [PubMed] [Google Scholar]
- 41.Doytchinova IA, Flower DR. Towards the in silico identification of class II restricted T-cell epitopes: a partial least squares iterative self-consistent algorithm for affinity prediction. Bioinformatics. 2003;19:2263–70. doi: 10.1093/bioinformatics/btg312. [DOI] [PubMed] [Google Scholar]
- 42.Rajapakse M, Schmidt B, Feng L, Brusic V. Predicting peptides binding to MHC class II molecules using multi-objective evolutionary algorithms. BMC Bioinformatics. 2007;8:459. doi: 10.1186/1471-2105-8-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hertz T, Yanover C. PepDist: a new framework for protein–peptide binding prediction based on learning peptide distance functions. BMC Bioinformatics. 2006;7(Suppl. 1):S3. doi: 10.1186/1471-2105-7-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karpenko O, Huang L, Dai Y. A probabilistic meta-predictor for the MHC class II binding peptides. Immunogenetics. 2008;60:25–36. doi: 10.1007/s00251-007-0266-y. [DOI] [PubMed] [Google Scholar]
- 45.Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4:e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10:296. doi: 10.1186/1471-2105-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donnes P, Elofsson A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics. 2002;3:25. doi: 10.1186/1471-2105-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–7. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 50.Godkin AJ, Smith KJ, Willis A, Tejada-Simon MV, Zhang J, Elliott T, Hill AV. Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide–MHC interactions. J Immunol. 2001;166:6720–7. doi: 10.4049/jimmunol.166.11.6720. [DOI] [PubMed] [Google Scholar]
- 51.Lovitch SB, Pu Z, Unanue ER. Amino-terminal flanking residues determine the conformation of a peptide–class II MHC complex. J Immunol. 2006;176:2958–68. doi: 10.4049/jimmunol.176.5.2958. [DOI] [PubMed] [Google Scholar]
- 52.Bordner AJ, Abagyan R. Ab initio prediction of peptide-MHC binding geometry for diverse class I MHC allotypes. Proteins. 2006;63:512–26. doi: 10.1002/prot.20831. [DOI] [PubMed] [Google Scholar]
- 53.Bui HH, Schiewe AJ, von Grafenstein H, Haworth IS. Structural prediction of peptides binding to MHC class I molecules. Proteins. 2006;63:43–52. doi: 10.1002/prot.20870. [DOI] [PubMed] [Google Scholar]
- 54.Schafroth HD, Floudas CA. Predicting peptide binding to MHC pockets via molecular modeling, implicit solvation, and global optimization. Proteins. 2004;54:534–56. doi: 10.1002/prot.10608. [DOI] [PubMed] [Google Scholar]
- 55.Fagerberg T, Cerottini JC, Michielin O. Structural prediction of peptides bound to MHC class I. J Mol Biol. 2006;356:521–46. doi: 10.1016/j.jmb.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 56.Altuvia Y, Margalit H. A structure-based approach for prediction of MHC-binding peptides. Methods. 2004;34:454–9. doi: 10.1016/j.ymeth.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Altuvia Y, Sette A, Sidney J, Southwood S, Margalit H. A structure-based algorithm to predict potential binding peptides to MHC molecules with hydrophobic binding pockets. Hum Immunol. 1997;58:1–11. doi: 10.1016/s0198-8859(97)00210-3. [DOI] [PubMed] [Google Scholar]
- 58.Singh SP, Mishra BN. Ranking of binding and nonbinding peptides to MHC class I molecules using inverse folding approach: implications for vaccine design. Bioinformation. 2008;3:72–82. doi: 10.6026/97320630003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davies MN, Sansom CE, Beazley C, Moss DS. A novel predictive technique for the MHC class II peptide-binding interaction. Mol Med. 2003;9:220–5. doi: 10.2119/2003-00032.sansom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doytchinova IA, Walshe VA, Jones NA, Gloster SE, Borrow P, Flower DR. Coupling in silico and in vitro analysis of peptide–MHC binding: a bioinformatic approach enabling prediction of superbinding peptides and anchorless epitopes. J Immunol. 2004;172:7495–502. doi: 10.4049/jimmunol.172.12.7495. [DOI] [PubMed] [Google Scholar]
- 61.Zhang H, Wang P, Papangelopoulos N, et al. Limitations of ab initio predictions of peptide binding to MHC class II molecules. Accepted for publication in PLoS One. 2010;5:e9272. doi: 10.1371/journal.pone.0009272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V. MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res. 2005;33:W172–9. doi: 10.1093/nar/gki452. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jacob L, Vert JP. Efficient peptide–MHC-I binding prediction for alleles with few known binders. Bioinformatics. 2008;24:358–66. doi: 10.1093/bioinformatics/btm611. [DOI] [PubMed] [Google Scholar]
- 64.Jojic N, Reyes-Gomez M, Heckerman D, Kadie C, Schueler-Furman O. Learning MHC I–peptide binding. Bioinformatics. 2006;22:e227–35. doi: 10.1093/bioinformatics/btl255. [DOI] [PubMed] [Google Scholar]
- 65.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nielsen M, Lundegaard C, Blicher T, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Lund O, Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC–peptide binding. Bioinformatics. 2009;25:1293–9. doi: 10.1093/bioinformatics/btp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, Buus S, Lund O. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol. 2008;4:e1000107. doi: 10.1371/journal.pcbi.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zaitlen N, Reyes-Gomez M, Heckerman D, Jojic N. Shift-invariant adaptive double threading: learning MHC II–peptide binding. J Comput Biol. 2008;15:927–42. doi: 10.1089/cmb.2007.0183. [DOI] [PubMed] [Google Scholar]
- 70.Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinformatics. 2008;9(Suppl. 12):S22. doi: 10.1186/1471-2105-9-S12-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rapin N, Hoof I, Lund O, Nielsen M. MHC motif viewer. Immunogenetics. 2008;60:759–65. doi: 10.1007/s00251-008-0330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rapin N, Hoof I, Lund O, Nielsen M. The MHC motif viewer: a visualization tool for MHC binding motifs. Curr Protoc Immunol. 2010 doi: 10.1002/0471142735.im1817s88. Chapter 18:Unit18.17. [DOI] [PubMed] [Google Scholar]
- 74.Hertz T, Yanover C. Identifying HLA supertypes by learning distance functions. Bioinformatics. 2007;23:e148–55. doi: 10.1093/Bioinformatics/btl324. [DOI] [PubMed] [Google Scholar]
- 75.Doytchinova IA, Guan P, Flower DR. Identifiying human MHC supertypes using bioinformatic methods. J Immunol. 2004;172:4314–23. doi: 10.4049/jimmunol.172.7.4314. [DOI] [PubMed] [Google Scholar]
- 76.Lund O, Nielsen M, Kesmir C, et al. Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics. 2004;55:797–810. doi: 10.1007/s00251-004-0647-4. [DOI] [PubMed] [Google Scholar]
- 77.Sette A, Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 1999;50:201–12. doi: 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- 78.Sette A, Sidney J. HLA supertypes and supermotifs: a functional perspective on HLA polymorphism. Curr Opin Immunol. 1998;10:478–82. doi: 10.1016/s0952-7915(98)80124-6. [DOI] [PubMed] [Google Scholar]
- 79.Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perez CL, Larsen MV, Gustafsson R, et al. Broadly immunogenic HLA class I supertype-restricted elite CTL epitopes recognized in a diverse population infected with different HIV-1 subtypes. J Immunol. 2008;180:5092–100. doi: 10.4049/jimmunol.180.7.5092. [DOI] [PubMed] [Google Scholar]
- 81.Frahm N, Yusim K, Suscovich TJ, et al. Extensive HLA class I allele promiscuity among viral CTL epitopes. Eur J Immunol. 2007;37:2419–33. doi: 10.1002/eji.200737365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hillen N, Mester G, Lemmel C, et al. Essential differences in ligand presentation and T cell epitope recognition among HLA molecules of the HLA-B44 supertype. Eur J Immunol. 2008;38:2993–3003. doi: 10.1002/eji.200838632. [DOI] [PubMed] [Google Scholar]
- 83.Lamberth K, Roder G, Harndahl M, Nielsen M, Lundegaard C, Schafer-Nielsen C, Lund O, Buus S. The peptide-binding specificity of HLA-A*3001 demonstrates membership of the HLA-A3 supertype. Immunogenetics. 2008;60:633–43. doi: 10.1007/s00251-008-0317-z. [DOI] [PubMed] [Google Scholar]
- 84.Peters B, Bulik S, Tampe R, Endert PMV, Holzhutter HG. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol. 2003;171:1741–9. doi: 10.4049/jimmunol.171.4.1741. [DOI] [PubMed] [Google Scholar]
- 85.Doytchinova IA, Guan P, Flower DR. EpiJen: a server for multistep T cell epitope prediction. BMC Bioinformatics. 2006;7:131. doi: 10.1186/1471-2105-7-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tenzer S, Peters B, Bulik S, et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci. 2005;62:1025–37. doi: 10.1007/s00018-005-4528-2. [DOI] [PubMed] [Google Scholar]
- 87.Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. 2005;35:2295–303. doi: 10.1002/eji.200425811. [DOI] [PubMed] [Google Scholar]
- 88.Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mustafa AS, Shaban FA. ProPred analysis and experimental evaluation of promiscuous T-cell epitopes of three major secreted antigens of Mycobacterium tuberculosis. Tuberculosis (Edinburgh, Scotland) 2006;86:115–24. doi: 10.1016/j.tube.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 90.Al-Attiyah R, Mustafa AS. Computer-assisted prediction of HLA-DR binding and experimental analysis for human promiscuous Th1-cell peptides in the 24 kDa secreted lipoprotein (LppX) of Mycobacterium tuberculosis. Scand J Immunol. 2004;59:16–24. doi: 10.1111/j.0300-9475.2004.01349.x. [DOI] [PubMed] [Google Scholar]
- 91.Nyarady Z, Czompoly T, Bosze S, et al. Validation of in silico prediction by in vitro immunoserological results of fine epitope mapping on citrate synthase specific autoantibodies. Mol Immunol. 2006;43:830–8. doi: 10.1016/j.molimm.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 92.Iwai LK, Yoshida M, Sidney J, et al. In silico prediction of peptides binding to multiple HLA-DR molecules accurately identifies immunodominant epitopes from gp43 of Paracoccidioides brasiliensis frequently recognized in primary peripheral blood mononuclear cell responses from sensitized individuals. Mol Med. 2003;9:209–19. [PMC free article] [PubMed] [Google Scholar]
- 93.Tongchusak S, Brusic V, Chaiyaroj SC. Promiscuous T cell epitope prediction of Candida albicans secretory aspartyl proteinase family of proteins. Infect Genet Evol. 2008;8:467–73. doi: 10.1016/j.meegid.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 94.Drouin EE, Glickstein L, Kwok WW, Nepom GT, Steere AC. Searching for borrelial T cell epitopes associated with antibiotic-refractory Lyme arthritis. Mol Immunol. 2008;45:2323–32. doi: 10.1016/j.molimm.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Calvo-Calle JM, Strug I, Nastke MD, Baker SP, Stern LJ. Human CD4+ T cell epitopes from vaccinia virus induced by vaccination or infection. PLoS pathogens. 2007;3:1511–29. doi: 10.1371/journal.ppat.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van der Burg SH, Visseren MJ, Brandt RM, Kast WM, Melief CJ. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC–peptide complex stability. J Immunol. 1996;156:3308–14. [PubMed] [Google Scholar]
- 97.Seamons A, Sutton J, Bai D, et al. Competition between two MHC binding registers in a single peptide processed from myelin basic protein influences tolerance and susceptibility to autoimmunity. J Exp Med. 2003;197:1391–7. doi: 10.1084/jem.20022226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Reche PA, Glutting JP, Reinherz EL. Prediction of MHC class I binding peptides using profile motifs. Hum Immunol. 2002;63:701–9. doi: 10.1016/s0198-8859(02)00432-9. [DOI] [PubMed] [Google Scholar]
- 99.Bhasin M, Raghava GP. SVM based method for predicting HLA-DRB1*0401 binding peptides in an antigen sequence. Bioinformatics. 2004;20:421–3. doi: 10.1093/bioinformatics/btg424. [DOI] [PubMed] [Google Scholar]
- 100.Feldhahn M, Thiel P, Schuler MM, Hillen N, Stevanovic S, Rammensee HG, Kohlbacher O. EpiToolKit – a web server for computational immunomics. Nucleic Acids Res. 2008;36:W519–22. doi: 10.1093/nar/gkn229. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]