

Abstract
Patients with systemic lupus erythematosus (SLE) produce antibodies to many different self-antigens. Here, we investigated antibodies in SLE sera using an antigen microarray containing many hundreds of antigens, mostly self-antigens. The aim was to detect sets of antibody reactivities characteristic of SLE patients in each of various clinical states – SLE patients with acute lupus nephritis, SLE patients in renal remission, and SLE patients who had never had renal involvement. The analysis produced two novel findings: (i) an SLE antibody profile persists independently of disease activity and despite long-term clinical remission, and (ii) this SLE antibody profile includes increases in four specific immunoglobulin G (IgG) reactivities to double-stranded DNA (dsDNA), single-stranded DNA (ssDNA), Epstein–Barr virus (EBV) and hyaluronic acid; the profile also includes decreases in specific IgM reactivities to myeloperoxidase (MPO), CD99, collagen III, insulin-like growth factor binding protein 1 (IGFBP1) and cardiolipin. The reactivities together showed high sensitivity (> 93%) and high specificity for SLE (> 88%). A healthy control subject who had the SLE antibody profile was later found to develop clinical SLE. The present study did not detect antibody reactivities that differentiated among the various subgroups of SLE subjects with statistical significance. Thus, SLE is characterized by an enduring antibody profile irrespective of clinical state. The association of SLE with decreased IgM natural autoantibodies suggests that these autoantibodies might enhance resistance to SLE.
Keywords: autoantibodies, autoimmune diseases, informatics, microarray, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) can affect many of the body’s organ systems, including the kidneys, skin, joints, nervous system, serous membranes, blood cells and blood vessels. SLE is thought to be an autoimmune disease: over 100 different self-molecules have been found to bind autoantibodies in different patients;1 indeed, anti-nuclear antibodies and autoantibodies to double-stranded DNA (dsDNA), phospholipids and Sm proteins are among the 11 criteria used for diagnosing SLE.2 However, many patients diagnosed with SLE lack these autoantibodies, especially when they are in clinical remission.
The aim of the present study was to investigate antibody reactivity profiles in SLE patients using an antigen microarray device we developed for measuring patterns of antibody binding that is at least 10-fold to 100-fold more sensitive than standard enzyme-linked immunosorbent assays (ELISAs) or fluorescence assays.3 This sensitivity and the range of antigen-chip laser activation along with informatic analysis made it possible to obtain informative results on antibody binding at a low serum dilution (1 : 10) and without fixed thresholds for determining a positive result. We could thus study the antibody profiles of SLE subjects without ignoring the natural autoantibodies present in healthy persons – the immunological homunculus.4,5 This approach made it possible to detect decreased immunoglobulin M (IgM) antibody reactivities as well as increased IgG reactivities in SLE subjects and the persistence of an SLE profile independent of disease activity and despite long-term clinical remission.
Materials and methods
Human subjects
The study was approved by the institutional review board of each participating clinical unit; informed consent was obtained from all participants. Three groups of SLE patients and a control group were studied: 15 patients in renal remission; 14 patients with active lupus nephritis; 11 patients without renal involvement; and 16 healthy controls matched with the lupus subjects for age and sex. Blood samples and clinical data were collected from SLE patients arriving at the Rheumatology Unit and Hematology Department of the Sheba Medical Center, Israel; the Rheumatology Unit at the Hadassah Medical Center, Ein Kerem, Jerusalem, Israel; and the Cellular Biology and Immunogenetics Unit at the Corporacion para Investigaciones Biologicas, Medellín, Colombia. All patients fulfilled the American Collage of Rheumatology criteria for SLE.2 SLE patients with active lupus nephritis were defined by an systemic lupus erythematosus disease activity index (SLEDAI) of ≥ 8 and one of the following: new onset proteinuria of ≥ 1 g/24 hr; an increase in the urinary protein:creatinine ratio of ≥ 2; or an increase of ≥ 50% in serum creatinine from baseline. SLE patients in renal remission were individuals who were once diagnosed as having active lupus nephritis as defined above, but now had an SLEDAI ≤ 4 and one of the following: a return to baseline serum creatinine with a decrease in proteinuria to within 25% of the baseline level, or a return to baseline proteinuria and a return of serum creatinine to within 25% of the baseline level. All patients in remission remained stable for at least 6 months; the mean time in remission was 8 years; the range was 3 months to 30 years. Patients without known renal involvement were known not to have had kidney involvement in the past and during a follow-up period of at least 1 year. The mean time from diagnosis was 7 years; the range was from 0·5 to 27 years. Additional patient data are shown in Table 1.
Table 1.
The clinical characteristics of the patients with systemic lupus erythematosus (SLE)1
| Characteristic | Renal involvement | ||
|---|---|---|---|
| None (n = 11) | Remission (n = 15) | Active (n = 14) | |
| Sex (% female) | 100% | 87% | 86% |
| Age (years) | 40·6 ± 4·4 | 36·1 ± 3·2 | 32·4 ± 2·5 |
| SLEDAI | 1·8 ± 1 | 1·1 ± 0·4 | 14·6 ± 2·2 |
| Anti DNA Ab (% positive) | 18% | 40% | 86% |
| Complement (% decreased) | 9% | 7% | 83% |
| Creatinine (mg/dl) | 0·75 ± 0·03 | 0·97 ± 0·08 | 1·2 ± 0·26 |
| Duration of disease (months) | 84 ± 29 | 150 ± 23 | 118 ± 25 |
Values are presented as mean ± standard error or per cent.
Ab, antibody; SLEDAI, systemic lupus erythematosus disease activity index.
Antigen microarrays and serum testing
Antigen microarray chips were prepared as described elsewhere.4,6,7 Briefly, 694 antigens, each at its optimal concentration (mostly at 1 mg/ml), were spotted in triplicate on epoxy-activated glass substrates using a 48-pin robot (Microgrid 600; Genomics Solutions, Ann Arbor, MI). These antigens included proteins, synthetic peptides from the sequences of selected proteins, nucleotides, phospholipids, and other self and non-self molecules. The microarrays were then blocked for 1 hr at 37° with 1% bovine serum albumin. Test serum in blocking buffer (1 : 10 dilution) was incubated under a coverslip for 1 hr at 37°. The arrays were then washed and incubated for 1 hr at 37° with a 1 : 500 dilution of two detection antibodies, mixed together: a goat anti-human IgG Cy3-conjugated antibody, and a goat anti-human IgM Cy5-conjugated antibody (both purchased from Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Image acquisition was performed by laser (Agilent Technologies, Santa Clara, CA) and the results were analysed using Quantarray software (Packard BioChip Technologies, Billerica, MA) and software developed by us. The quantitative range of signal intensity of binding to each antigen spot was 0–65 000; this range of detection made it possible to obtain reliable data at the 1 : 10 dilution of test samples.3 Anti-DNA antibodies were also measured separately using ELISA (QUANTA-Lite; Inova, San Diego, CA) and the Farr assay.8
Image analysis and data processing
Technically faulty spots were manually excluded by visual inspection of each slide. The foreground and background intensities of multiple spots of each antigen were then averaged, and a log-base-10 value of the difference between the foreground and the background was calculated; differences < 500 were clamped to 500 and then log transformed. To control for differences between different slides, the average laser intensity value of each slide (in the corresponding IgM or IgG channel) was then subtracted. The value of each antigen was then shifted such that its minimal value over the entire data set equalled zero. The resulting value was taken as the antigen reactivity of the antibodies binding to that spotted antigen. Antigens that showed zero reactivity in more than 80% of the slides were excluded, as were antigens whose coefficient of variation across slides was lower than 20%. In this way, the 694 IgM and the 694 IgG antigen reactivities were reduced to about 930 reactivities, almost evenly split between IgM and IgG channels.
Statistical analysis
Statistical analysis was carried out to identify antigen reactivities and groups of antigen reactivities that could distinguish between the different groups of subjects. The different comparisons were performed in the same manner, designated generically here as groups A and B, each consisting of nA and nB subjects, respectively (each subject being a separate microarray slide). To estimate the quality of the differentiation between groups A and B, we applied a leave-one-out (LOO) procedure.9 One subject from nA + nB was left out, and the remainder of the nA + nB − 1 subjects were used to select candidate antigens for separating groups A and B. We applied a t-test between groups A and B (excluding the one subject), and selected the d antigens that passed a false discovery rate (FDR) threshold of 5%.10 These antigens were then used to classify the left-out subject using the K-nearest neighbours algorithm,9 with K = 3; the left-out subject seeks its three nearest subject data points in the d-dimensional space containing the other nA + nB − 1 subjects, and is classified according to the majority class of these three subjects. This procedure was repeated for all nA + nB subjects; the performance, which appears in Table 2, was the number of misclassifications, quantified as specificity (1 – false positive rate) and sensitivity (1 – false negative rate) measures.
Table 2.
The antigen microarray differentiates between subjects with systemic lupus erythematosus (SLE) and healthy controls
| Healthy controls compared with | Sensitivity (%) | Specificity (%) |
|---|---|---|
| All SLE subjects | 90 | 81 |
| SLE subjects in renal remission | 93 | 100 |
| SLE subjects with active lupus nephritis | 86 | 100 |
The composition of the list of antigen reactivities that separate the groups can vary depending on the specific subject that is left out in the particular LOO cross-validation run. To further identify antigen reactivities that play an important role in separating groups A and B, we selected the antigen reactivities that appeared in the candidate lists in at least 90% of the LOO tests. We repeated the LOO procedure with each of these selected antigens independently, by classifying the left-out subject using a simple threshold criterion: we found, for the reactivity values of the nA + nB − 1 training subjects, the threshold value that maximized the specificity, and then classified the left-out point using this threshold. The average LOO specificity and sensitivity of this classification over the nA + nB subjects appear for each of the selected antigens in Tables 3 and 4. To test the performance of combinations of antigen reactivities, we projected the particular combination onto a one-dimensional space using principal component analysis (PCA)9 and then treated the combination as we did above for single reactivities.
Table 4.
Antibody reactivities that distinguish the healthy control subjects from the patients with systemic lupus erythematosus (SLE) in renal remission
| Antigen | Immunoglobulin isotype | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| SLE up-regulated | |||
| ssDNA | IgG | 87 | 94 |
| dsDNA | IgG | 47 | 87 |
| Hyaluronic acid | IgG | 93 | 86 |
| EBV | IgG | 73 | 88 |
| SLE down-regulated | |||
| MPO | IgM | 87 | 94 |
| Collagen III | IgM | 73 | 83 |
| CD99 | IgM | 77 | 93 |
| All seven antigens | IgM+IgG | 100 | 94 |
| All four IgG antigens | IgG | 93 | 94 |
| All three IgM antigens | IgM | 93 | 94 |
dsDNA, double-stranded DNA; ssDNA, single-stranded DNA; EBV, Epstein–Barr virus; MPO, myeloperoxidase.
In addition to the LOO test, we took the subjects in groups A and B, together with their list of differentiating antigen reactivities, and used them to classify a test set C via three nearest neighbours. For example, we took the entire list of frequent antigen reactivities that separated healthy controls from SLE subjects in renal remission and used that set of reactivities to classify the other two SLE groups – those in renal relapse and those without renal involvement. Figure 2 shows a projection of these antigen reactivities onto a three-dimensional space using a PCA representation.
Figure 2.
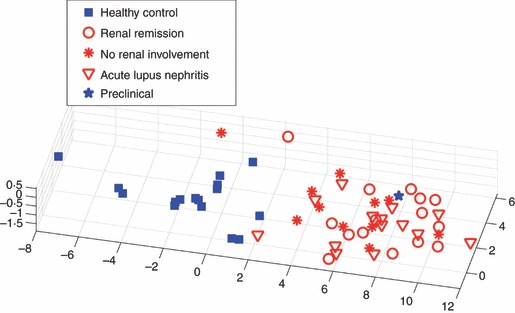
The antigen profile of subjects with systemic lupus erythematosus (SLE) in renal remission also characterizes subjects with SLE in acute renal relapse and those without renal involvement. A three-dimensional principal component analysis (PCA) based on the seven antigen reactivities in Table 4 is shown. Classification of the 25 subjects in acute renal relapse and those without renal involvement yields two errors – a specificity of 92% (see text). The blue star represents the profile of a subject who was healthy at the time of serum collection but who later developed SLE.
Results
Antigen microarray reactivities differentiate SLE subjects from healthy controls
Table 2 shows a global analysis of the 930 total antibody reactivities of the healthy control subjects compared with those of three SLE groups based on the LOO test: all SLE subjects; SLE subjects in renal remission; and SLE subjects with active lupus nephritis. The analysis showed that the microarray reactivities clearly separated the three groups of SLE subjects from the healthy controls. A comparison between healthy controls and SLE subjects without renal involvement does not appear in the table because no antigen exceeded an FDR level of 5% in some of the LOO tests. Moreover, the various subgroups of SLE subjects, with the limited numbers available for testing here, also could not be separated from one another because none of the antigen reactivities exceeded an FDR level of 5% in any of the LOO tests between the groups.
SLE subjects show both up-regulation and down-regulation of individual reactivities
Table 3 lists the particular antibody reactivities that distinguished all the SLE patients as a group from the healthy control subjects. Figure 1 shows the relative amounts of antibody reactivities to these antigens in each serum. The differences between the two groups for each of these antigens exceeded an FDR level of 5% (P < 0·0007). Four IgG antibody reactivities were up-regulated in the SLE group: classic reactivities to dsDNA and ssDNA, reactivity to Epstein–Barr virus (EBV), which has previously been found to be strongly associated with SLE,11,12 and a novel reactivity to hyaluronic acid.
Table 3.
Antibody reactivities that distinguish the healthy control subjects from all patients with systemic lupus erythematosus (SLE)
| Antigen | Immunoglobulin isotype | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| SLE up-regulated | |||
| dsDNA | IgG | 58 | 87 |
| ssDNA | IgG | 75 | 94 |
| Hyaluronic acid | IgG | 86 | 86 |
| EBV | IgG | 70 | 88 |
| SLE down-regulated | |||
| MPO | IgM | 78 | 88 |
| IGFBP1 | IgM | 59 | 82 |
| CD99 | IgM | 61 | 93 |
| Cardiolipin | IgM | 60 | 81 |
| All eight antigens | IgM+IgG | 93 | 88 |
| All four IgG antigens | IgG | 90 | 88 |
| All four IgM antigens | IgM | 68 | 88 |
The performance is based on a leave-one-out (LOO) procedure, as explained in the text.
dsDNA, double-stranded DNA; ssDNA, single-stranded DNA; EBV, Epstein–Barr virus; MPO, myeloperoxidase; IGFBP1, insulin-like growth factor binding protein 1.
Figure 1.
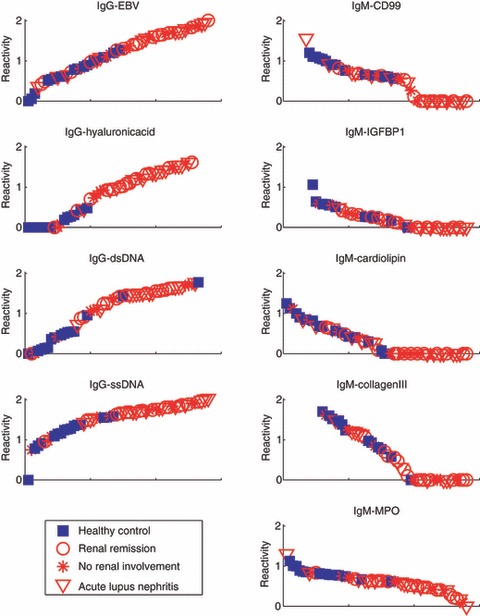
Antibody reactivities of individual subjects to the antigen reactivities that characterize patients with systemic lupus erythematosus (SLE). Sera from healthy controls (blue closed squares) and from SLE subjects in renal remission (red open circles), with acute lupus nephritis (red open triangles), or without renal involvement (red asterisks) were tested for antibody reactivities to the designated antigen. The relative amount of antibody reactivity is shown on the y-axis. The x-axis orders the subjects according to their relative reactivity.
Four novel antigen reactivities, all IgM, were found to be down-regulated in SLE: insulin-like growth factor binding protein 1 (IGFBP1), CD99, cardiolipin and myeloperoxidase (MPO). The IgM antibody reactivities of SLE subjects to these antigens tended to be low or undetectable compared with the healthy controls (Fig. 1). In contrast to the decreased IgM reactivities to MPO and to cardiolipin, increased IgG antibodies to these antigens have been associated with SLE and other vasculitis-related diseases.13–16
Table 3 shows that, except for IgG reactivity to hyaluronic acid, the other individual reactivities showed sensitivity for SLE of < 80%. However, the specificities of each reactivity, whether increased IgG or decreased IgM, were > 80%. The combination of all eight reactivities increased the sensitivity to > 90%; the combination of the four IgG increased reactivities was more sensitive than the combination of the four IgM decreased reactivities: 90% compared with 68%, respectively. The specificities of each of the combined sets were equal at 88%.
SLE subjects in remission maintain an SLE profile
An important question is whether clinical renal remission is associated with a return of the SLE antibody pattern to a healthy state. Table 4 shows that SLE patients in clinical remission still maintained an SLE profile. These patients showed significantly up-regulated IgG reactivities to the same four antigens that characterized the general set of SLE subjects: dsDNA, ssDNA, hyaluronic acid and EBV. Moreover, those in remission showed down-regulation of three IgM reactivities, of which two were characteristic of the SLE group as a whole: decreased IgM reactivities to CD99 and to MPO were present in both groups, but those in remission showed decreased IgM reactivity to collagen III rather than to cardiolipin and to IGFBP1 (Fig. 1).
Table 4 also shows that combining the four increased IgG and the three decreased IgM reactivities led to 100% sensitivity and 94% specificity. Thus, a combination of reactivities may provide a higher degree of accuracy than any of the component reactions alone. Note too that the set of combined decreased IgM reactivities performed as well as did the set of combined increased IgG reactivities. Thus, a loss of specific IgM reactivities appears to be a characteristic of SLE.
The SLE remission profile also characterizes other SLE groups
To determine whether the set of antigen reactivities characteristic of SLE in remission might be applicable to SLE generally, we used the seven antigens that separated subjects in remission from healthy controls (Table 4) to classify the 14 SLE patients with active lupus nephritis and the 11 SLE patients without renal involvement. These 25 SLE patients were classified via a three nearest neighbours algorithm, based on 15 SLE patients in remission and 16 healthy controls. Twenty-three of these 25 SLE subjects were correctly classified, generating a sensitivity of 92%.
Figure 2 displays a three-dimensional PCA representation (projected from the space spanned by the seven separating antigens) of healthy control subjects and those with various subgroups of SLE. The healthy controls were clearly separated by the seven antigen reactivities from the SLE subjects in remission. Moreover, the individuals in long-term remission, in acute lupus nephritis, and without renal involvement were completely overlapping. In other words, the remission list of antigen reactivities in Table 4 constitutes an SLE antibody profile that includes SLE subjects with active lupus nephritis and those without renal involvement.
Note the subject marked with a blue star, who was projected into the SLE domain of Fig. 2; serum from this subject was obtained when she was in a healthy state and was negative for standard anti-DNA antibodies, but 8 months later she began showing nonspecific symptoms and was eventually found, after seven more months, to fulfill the criteria for a diagnosis of SLE. Thus the antigen microarray profile might also detect pre-clinical SLE.
Discussion
In this study, we deployed an antigen microarray device to survey the IgG and IgM antibody reactivities that distinguish SLE patients from healthy controls. We measured a total of 930 antibody reactivities, mostly to self-antigens. The results based on these combined reactivities showed that SLE patients, including those in remission, can be separated from healthy individuals by their antibody repertoires with a high degree of sensitivity and specificity (Table 2). This suggests that the antibody repertoires of SLE subjects, and hence their adaptive immune systems, may be fundamentally different from those of healthy people. This notion, however, needs to be tested in more extensive studies; the antigens we spotted on the antigen chip probably constitute only a fraction of a subject’s antibody reactivities.
We extended our informatic analysis and detected a small group of IgG and IgM antibody reactivities that significantly distinguished between SLE and control subjects, both as individual reactivities and as sets of combined reactivities. Four IgG reactivities were elevated in SLE subjects (Fig. 1, Tables 3 and 4): ssDNA, dsDNA, hyaluronic acid and EBV. Reactivities to DNA are classically associated with SLE; however, the antigen chip appeared to be more sensitive for detecting anti-DNA than were the standard assays performed in the same subjects: anti-DNA reactivity detected either by the standard Farr assay or ELISA was positive in only 18% of the SLE subjects without renal involvement and in 40% of the subjects in renal remission (Table 1). In the antigen microarray assay used here, we studied dilutions of serum of only 1 : 10; this low dilution may have made it possible to detect anti-DNA with a relatively higher degree of sensitivity, even in remission (Table 4). In addition, the antigen chip was found to be intrinsically more sensitive than an ELISA of reactivity in subjects with multiple sclerosis.3
Reactivity to EBV has been known for many years to be associated with SLE, and chronic EBV infection has been proposed as a possible inducing event in susceptible persons.11,12
The present study highlights reactivity to hyaluronic acid as a relatively sensitive and specific marker for SLE (Fig. 1 and Tables 3 and 4). Hyaluronic acid has several immunologically interesting properties that could connect anti-hyaluronic acid antibodies to the pathogenesis of SLE and in particular to lupus nephritis: hyaluronic acid is a component of the kidney glomerulus17 and of the extracellular matrix.18 Moreover, hyaluronic acid is cross-reactive with DNA.19,20 Indeed, anti-DNA antibodies, measured in the traditional ways, are most prevalent in SLE patients with renal involvement and are thought to play a part in the pathogenesis of lupus nephritis.21 In addition to being a self-antigen for humans,20 hyaluronic acid is a component of the capsule of group A streptococcus and a virulence factor for the bacterium.22,23 Indeed, immunization of rabbits with encapsulated streptococci induced antibodies reactive with both mammalian and streptococcal hyaluronate.24 However, infection with group A Streptococcus pyogenes is associated with acute rheumatic fever and other autoimmune sequelae,25 but very rarely with SLE.26
In addition to the four elevated IgG reactivities, we detected a set of three or four significantly decreased IgM reactivities in SLE compared with the natural IgM antibodies present in the sera of healthy persons.4 The specific decreases in IgM included reactivity to MPO, to IGPBP1, to CD99, to collagen III, and to cardiolipin. Other microarray studies too have detected reduced IgM reactivity in SLE,27,28 but these other studies did not identify the specific antigens as determined here. In any case, the association of SLE with reduced IgM autoantibodies suggests that these autoantibodies might actually mark resistance to SLE.29 We have recently shown that IgM autoreactivities to DNA and other disease-associated autoantigens are prevalent in healthy human cord blood.4 Indeed, IgM and IgG autoantibodies are present in all healthy immune repertoires.30–32 Thus the development of an autoimmune disease cannot be explained by the mere presence of autoimmune reactivity; emergence of clinical disease involves the transition from benign autoreactivity to pathogenic autoreactivity – a subject for further research.30
Our antigen microarray and informatic views of SLE differ considerably from the standard ways of characterizing antibodies in lupus and from the ways in which others have deployed antigen microarrays to study SLE. Nevertheless, the data appear to be meaningful: the signal generated by the microarray was prominent and highly significant statistically. Indeed, our positive results did not depend on one particular informatic analysis; we obtained the same discriminations using various other analytical methods. We chose to present here only one of these methods because of its relative simplicity. Moreover, the subjects were recruited from three different centres on two continents, and the core reactivity profile was robust in being able to detect subjects in remission for as long as 30 years, as well as one subject who was diagnosed with clinical SLE more than 1 year after her serum sample was collected and tested. Microarray technology and informatic analysis thus provide a promising entry into immunomics – a global view of a subject’s immune state.6,7,32
Acknowledgments
This study was supported by grant number 712007- from the Israel Ministry of Health and by grants from the Rosario University and the Colombian Society of Rheumatology to J-M.A. ImmunArray Ltd, Israel, donated the antigen microarray chips.
Disclosures
None of the authors has a financial interest or any conflict of interest in the study or in its results.
References
- 1.Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. 2004;34:501–37. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 2. Criteria published by EM Tan et al: Arthritis Rheum 1982; 25:1271, updated by MC Hochberg, Arthritis Rheum 1997; 40:1725.
- 3.Quintana FJ, Farez MF, Viglietta V, et al. Antigen microarrays identify unique serum autoantibody signatures associated with different clinical forms and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci USA. 2008;105:18889–94. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merbl Y, Zucker-Toledano M, Quintana FJ, Cohen IR. Newborn humans manifest autoantibodies to defined self molecules detected by antigen microarray informatics. J Clin Invest. 2007;117:712–18. doi: 10.1172/JCI29943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen IR. The cognitive paradigm and the immunological homunculus. Immunol Today. 1992;13:490–4. doi: 10.1016/0167-5699(92)90024-2. [DOI] [PubMed] [Google Scholar]
- 6.Quintana FJ, Merbl Y, Sahar E, Domany E, Cohen IR. Antigen-chip technology for accessing global information about the state of the body. Lupus. 2006;15:428–30. doi: 10.1191/0961203306lu2328oa. [DOI] [PubMed] [Google Scholar]
- 7.Quintana FJ, Hagedorn PH, Elizur G, Merbl Y, Domany E, Cohen IR. Functional immunomics: microarray analysis of IgG autoantibody repertoires predicts the future response of mice to induced diabetes. Proc Natl Acad Sci USA. 2004;101(Suppl. 2):14615–21. doi: 10.1073/pnas.0404848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wold RT, Young FE, Tan EM, Farr RS. Deoxyribonucleic acid antibody: a method to detect its primary interaction with deoxyribonucleic acid. Science. 1968;161:806. doi: 10.1126/science.161.3843.806. [DOI] [PubMed] [Google Scholar]
- 9.Duda RO, Hart PE, Stork DG. Pattern Classification. New York: John Wiley and Sons, Inc; 2001. [Google Scholar]
- 10.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 11.James JA, Neas BR, Moser KL, Hall T, Bruner GR, Sestak AL, Harley JB. Systemic lupus erythematosus in adults is associated with previous Epstein–Barr virus exposure. Arthritis Rheum. 2001;44:1122–6. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 12.Barzilai O, Sherer Y, Ram M, Izhaky D, Anaya JM, Shoenfeld Y. Epstein–Barr virus and cytomegalovirus in autoimmune diseases: are they truly notorious? A preliminary report. Ann N Y Acad Sci. 2007;1108:567–77. doi: 10.1196/annals.1422.059. [DOI] [PubMed] [Google Scholar]
- 13.Manolova I, Dancheva M, Halacheva K. Predominance of IgG1 and IgG3 subclasses of autoantibodies to neutrophil cytoplasmic antigens in patients with systemic lupus erythematosus. Rheumatol Int. 2002;21:227–33. doi: 10.1007/s00296-002-0174-2. [DOI] [PubMed] [Google Scholar]
- 14.Sen D, Isenberg DA. Antineutrophil cytoplasmic autoantibodies in systemic lupus erythematosus. Lupus. 2003;12:651–8. doi: 10.1191/0961203303lu456rr. [DOI] [PubMed] [Google Scholar]
- 15.Love PE, Santoro SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med. 1990;112:682–98. doi: 10.7326/0003-4819-112-9-682. [DOI] [PubMed] [Google Scholar]
- 16.Moreland LW, Gay RE, Gay S. Collagen autoantibodies in patients with vasculitis and systemic lupus erythematosus. Clin Immunol Immunopathol. 1991;60:412–18. doi: 10.1016/0090-1229(91)90097-t. [DOI] [PubMed] [Google Scholar]
- 17.Yaoita E, Oguri K, Okayama E, Kawasaki K, Kobayashi S, Kihara I, Okayama M. Isolation and characterization of proteoglycans synthesized by cultured mesangial cells. J Biol Chem. 1990;5:522–31. [PubMed] [Google Scholar]
- 18.Hay ED. Extracellular matrix. J Cell Biol. 1981;91:205s–23s. doi: 10.1083/jcb.91.3.205s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faaber P, Capel PJ, Rijke GP, Vierwinden G, van de Putte LB, Koene RA. Cross-reactivity of anti-DNA antibodies with proteoglycans. Clin Exp Immunol. 1984;55:502–8. [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen C, Otto E, Kuhlemann K, Förster G, Kahaly GJ. Glycosaminoglycans in autoimmunity. Clin Exp Rheumatol. 1996;14(Suppl. 15):S59–67. [PubMed] [Google Scholar]
- 21.Yung S, Chan TM. Anti-DNA antibodies in the pathogenesis of lupus nephritis-the emerging mechanisms. Autoimmun Rev. 2008;7:317–21. doi: 10.1016/j.autrev.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Efstratiou A. Group A streptococci in the 1990s. J Antimicrob Chemother. 2000;45(Suppl):3–12. doi: 10.1093/jac/45.suppl_1.3. [DOI] [PubMed] [Google Scholar]
- 23.Stollerman GH, Dale JB. The importance of the group a streptococcus capsule in the pathogenesis of human infections: a historical perspective. Clin Infect Dis. 2008;46:1038–45. doi: 10.1086/529194. [DOI] [PubMed] [Google Scholar]
- 24.Fillit HM, McCarty M, Blake M. Induction of antibodies to hyaluronic acid by immunization of rabbits with encapsulated streptococci. J Exp Med. 1996;164:762–76. doi: 10.1084/jem.164.3.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Girschick HJ, Guilherme L, Inman RD, et al. Bacterial triggers and autoimmune rheumatic diseases. Clin Exp Rheumatol. 2008;1(Suppl. 48):S12–17. [PubMed] [Google Scholar]
- 26.Horwitz M, Shapiro N, Daya H. Streptococcal pharyngitis and systemic lupus erythematosus. S Afr Med J. 1966;1240:1008–10. [PubMed] [Google Scholar]
- 27.Li QZ, Zhou J, Wandstrat AE, et al. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol. 2007;147:60–70. doi: 10.1111/j.1365-2249.2006.03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, Mohan C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–39. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen IR, Cooke A. Natural autoantibodies might prevent autoimmune disease. Immunol Today. 1986;7:363–4. doi: 10.1016/0167-5699(86)90026-5. [DOI] [PubMed] [Google Scholar]
- 30.Cohen IR. Tending Adam’s Garden: Evolving the Cognitive Immune Self. UK: Academic Press, London; 2000. [Google Scholar]
- 31.Cohen IR, Young DB. Autoimmunity, microbial immunity and the immunological homunculus. Immunol Today. 1991;12:105–10. doi: 10.1016/0167-5699(91)90093-9. [DOI] [PubMed] [Google Scholar]
- 32.Cohen IR. Real and artificial immune systems: computing the state of the body. Nat Rev Immunol. 2007;7:569–74. doi: 10.1038/nri2102. [DOI] [PubMed] [Google Scholar]