

Abstract
The expression of major histocompatibility complex class II (MHC II) molecules is post-translationally regulated by endocytic protein turnover. Here, we identified the serine protease cathepsin G (CatG) as an MHC II-degrading protease by in vitro screening and examined its role in MHC II turnover in vivo. CatG, uniquely among endocytic proteases tested, initiated cleavage of detergent-solubilized native and recombinant soluble MHC II molecules. CatG cleaved human leukocyte antigen (HLA)-DR isolated from both HLA-DM-expressing and DM-null cells. Even following CatG cleavage, peptide binding was retained by pre-loaded, soluble recombinant HLA-DR. MHC II cleavage occurred on the loop between fx1 and fx2 of the membrane-proximal β2 domain. All allelic variants of HLA-DR tested and murine I-Ag7 class II molecules were susceptible, whereas murine I-Ek and HLA-DM were not, consistent with their altered sequence at the P1’ position of the CatG cleavage site. CatG effects were reduced on HLA-DR molecules with DRB mutations in the region implicated in interaction with HLA-DM. In contrast, addition of CatG to intact B-lymphoblastoid cell lines (B-LCLs) did not cause degradation of membrane-bound MHC II. Moreover, inhibition or genetic ablation of CatG in primary antigen-presenting cells did not cause accumulation of MHC II molecules. Thus, in vivo, the CatG cleavage site is sterically inaccessible or masked by associated molecules. A combination of intrinsic and context-dependent proteolytic resistance may allow peptide capture by MHC II molecules in harshly proteolytic endocytic compartments, as well as persistent antigen presentation in acute inflammatory settings with extracellular proteolysis.
Keywords: antigen presentation, APC, cathepsin G, MHC class II, proteolysis
Introduction
Major histocompatibility complex class II (MHC II) molecules bind self and non-self peptides in endocytic compartments of antigen-presenting cells (APCs) and present them on the cell surface for inspection by CD4+ T cells via T-cell antigen receptors (TCRs).1 MHC II expression is tightly controlled at several levels. Transcriptional regulation confines constitutive MHC II expression to professional APCs and thymic epithelial cells and allows up-regulation on other cell types after exposure to inflammatory cytokines.2
Post-translational events also regulate cellular localization of MHC II, thereby influencing MHC II half-life. In immature dendritic cells (DCs), MHC II molecules are efficiently targeted to lysosomes by the clathrin adaptor protein complex 2 (AP-2) and/or by the E3 ubiquitin ligase, membrane-associated RING-CH protein 1 (MARCH-I) and are degraded within a few hours; surface expression remains relatively low. DC activation stimulates a transient burst of MHC II synthesis, turn-off of MARCH-I and deposition of peptide/MHC II complexes at the plasma membrane, where they are long-lived (> 100 hr). Data from B-cell lines, melanoma lines and human monocytes implicate similar pathways in control of MHC II levels in these cell types.3–6
Expression levels of MHC II are also influenced by interaction with accessory molecules that regulate MHC II peptide loading: MHC II-associated invariant chain (Ii) and HLA-DM (DM). Nascent MHC II molecules assemble in the endoplasmic reticulum with Ii; in cells from animals lacking Ii, surface levels of most MHC II alleles are substantially reduced because of inefficient assembly and egress.7–9 After assembly, MHC II/Ii complexes travel to endocytic compartments, directed by sequences in the Ii cytoplasmic tail; there, Ii is sequentially degraded by cathepsins.10 Groove-bound Ii remnants, the class II-associated Ii peptides (CLIPs), are exchanged for antigenic peptides with the assistance of the peptide exchange factor DM.11 Chaperoning effects of DM provide further regulation of MHC II preservation/degradation1,2 (C. Rinderknecht and S. Roh, unpublished data). DM editing of peptides in favour of strong binders is also a factor, as the quality of peptide cargo is thought to influence MHC II half-life.12–14
Despite active regulation of expression at the level of proteolysis, MHC II molecules must be relatively resistant to proteolytic attack. MHC II molecules traverse acidic, proteolytic endosomal compartments, where peptide loading occurs, for several hours en route to the plasma membrane.15–17 Moreover, in inflammatory settings, myeloid and stromal cells may release proteases into the extracellular fluid, yet MHC II molecules are abundantly expressed in such settings and must remain functional to allow local antigen presentation. The paradox of regulated turnover in the face of inherent proteolytic resistance is only beginning to be addressed. Only limited information exists regarding the proteases involved in constitutive or regulated MHC II turnover, or the factors that render MHC II molecules at least partially resistant to proteolytic attack.
In the present study, we screened a panel of cathepsins, including cysteine, aspartyl and serine proteases, for their ability to degrade MHC II molecules. The serine protease CatG uniquely was able to cleave MHC II molecules in vitro. CatG is abundant in storage granules of neutrophils; it is released in inflammatory sites and contributes to innate protection from bacterial infection. Non-immune roles for CatG are suggested by subtle developmental defects in CatG-deficient mice.18 Notably, CatG is expressed in primary human APCs, such as B cells, monocytes, and myeloid and plasmacytoid DCs,19,20 where it has been shown to contribute to proteolytic antigen processing.21 Here, we characterized the specificity of CatG cleavage of MHC II molecules in vitro, and examined whether CatG contributes to MHC II turnover in vivo.
Materials and methods
Cells
The HLA-DM-deficient human B-LCLs 9.5.3 and 5.2.4, their parent line 8.1.6 and the 5.2.4-DR3 transfectant have been described previously.22–24 Transduced B-LCL 5.2.4 expressing the mutant HLA-DR3 molecules DRB R74Q, DRB D152N, DRB S197N and DRB E187K have been described.24,25 Schneider-2 Drosophila melanogaster (S2) cells expressing recombinant soluble HLA-DR molecules have been described previously.26,27 Mammalian cells were cultured in complete RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (HyClone Laboratories, Logan, UT) and 2 mm l-glutamine (Life Technologies, Carlsbad, CA). S2 cells were cultured as described previously.28 Human peripheral blood mononuclear cells (PBMC) were isolated from buffy coats of healthy donor blood. B cells and myeloid type 1 dendritic cells (mDC1s) were positively selected using immunomagnetic beads specific for CD19 and CD1c, respectively [magnetic-activated cell sorting (MACS); Miltenyi Biotec, Auburn, CA] according to the manufacturer’s protocols. The purity of primary cell preparations routinely exceeded 90%. Cells were cultured in the presence or absence of the CatG-specific inhibitor I (10 μm; Calbiochem, San Diego, CA; Compound 7 in29) or E64d (10 μm; Calbiochem) for 4·5, 24 or 72 hr at 37°, and either analysed by flow cytometry or prepared for western blotting by lysis in 10 mm Tris (pH 7·5), 150 mm NaCl, 0·5% NP-40, and CatG-specific inhibitor (1 μm), followed by adjustment for equal total protein content (quantified by the Bradford assay).
Protein purification
Purification of full-length native HLA-DR molecules was performed essentially as described previously.26,27 Briefly, B-LCLs were lysed in 10 mm Tris (pH 7·8), 140 mm NaCl, and 0·5% NP-40. The lysate was pre-cleared by centrifugation and filtration and passed over an anti-DR (L243)-sepharose immunoaffinity column (L243: IgG2a anti-DR). The column was washed extensively (50 mm Na-phosphate, 150 mm NaCl and 1% octylglucoside, pH 8) and eluted at high pH (100 mm glycine-NaOH and 1% octylglucoside, pH 11). Soluble HLA-DR was purified from insect cell supernatants by a similar method, except that detergents were omitted. Soluble DM molecules were purified by affinity purification using the FLAG epitope on the DMA C-terminal end, as described previously.26 The identity and purity of the isolated molecules were tested using sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie Blue or silver staining (not shown).
In vitro processing
CatG from human sputum or from neutrophils was purchased from Sigma-Aldrich (St Louis, MO); CatL and CatB were purchased from Caltag (Burlingame, CA) or R & D Systems (Minneapolis, MN). Full-length or soluble MHC II or DM molecules (100 μg/ml) were incubated with different isolated cathepsins (50–100 ng protein) in reaction buffer [phosphate-buffered saline (PBS), pH 7·2, 2·5 mm dithiothreitol (DTT) or 0·1 m citrate, pH 5·0–6·0, and 2·5 mm DTT] at 37° for various times (routinely 2 hr). Digestion products were resolved by SDS-PAGE and analysed by silver staining.
N-terminal sequencing and matrix-assisted laser desorption-ionisation time-of-flight (MALDI-TOF) mass spectrometry
Soluble HLA-DR1 expressed in Schneider cells and purified26 was used for digestion with CatG. The digested products were separated by SDS-PAGE followed by transfer to an Immulon-PSQ membrane (Millipore, Billerica, MA). The membrane was stained with Coomassie Blue and air-dried. The bands were cut out and submitted for N-terminal sequencing to the Protein and Nucleic Acid Facility (Stanford University School of Medicine). Soluble HLA-DR1 expressed in Escherichia coli (a kind gift from L. Stern, Biochemistry and Molecular Pharmacology, University of Massachusetts, Worcester, MA) was used for digestion with CatG and stained with Gelcode Blue (Pierce, Rockford, IL). Prominent CatG cleavage products were excised, reduced with DTT and alkylated with iodoacetamide. Duplicate gel pieces for each band were digested with either Arg-C or Glu-C (Sigma-Aldrich) and peptides were extracted using established protocols.30 Protease digests were subjected to reverse-phase high-performance liquid chromatography (HPLC) separation and the HPLC eluant was spotted to MALDI target plates for MALDI-TOF/TOF mass spectrometry (MS) (Applied Biosystems 4700, Foster City, CA) analysis. Peptides were identified by tandem mass spectrometry (MS/MS) analysis utilizing the Mascot search engine.
Fluorescence resonance energy transfer (FRET) assay for peptide/MHC II binding
Recombinant soluble HLA-DR1 molecules were loaded with 100-fold excess of a 7-amino-4-methylcoumarin-3-acetic acid (AMCA)-labelled variant of the influenza A hemagglutinin (HA) 307-319 peptide (AMCA-HA) (a kind gift from L. Stern) in PBS overnight at 37°. Free AMCA-HA was removed by centrifuging the binding reactions through spin columns (Sephadex G50 Superfine; BioRad, Hercules, CA) according to the manufacturer’s instructions. Binding stoichiometry was determined by absorption spectrophotometry at 280 and 350 nm, as described previously.31 HLA-DR molecules were 70–90% loaded with AMCA-HA. HLA-DR1/AMCA-HA complexes were incubated with 50 ng of CatG in CatG digestion buffer (PBS, pH 7·4, and 0·05% Tween-20). Fluorescence measurements were performed after 0 and 14 hr of incubation of reaction mixtures in black 96-well polypropylene plates (Greiner Bio-One, Monroe, NC) at 37°, using a FluoroMax 96-well-plate fluorimeter (Molecular Dynamics, Sunnyvale, CA). FRET was measured by monitoring excitation at 280 nm and emission at 450 nm, and normalized to total AMCA fluorescence (excitation at 350 nm and emission at 450 nm). Complete digestion of HLA-DR1 during the reaction was verified by SDS-PAGE analysis and silver staining of recovered reaction mixtures.
Western blot
Samples were boiled after the addition of 8 × Laemmli SDS-PAGE sample buffer with 5% v/v 2-mercaptoethanol, run on 12% SDS-PAGE gels, and transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Freiburg, Germany). Membranes were blocked for 1 hr in blocking buffer (1× PBS, 0·05% Tween 20 and 1× Rotiblock; Roth, Karlsruhe, Germany) and incubated for an additional hour with the HLA-DR-specific antiserum CHAMP32 diluted in blocking buffer. After washing in PBS with 0·05% Tween 20, horseradish peroxidase (HRP)-conjugated secondary antibody (donkey anti-rabbit immunoglobulin; GE Healthcare) was diluted 1 : 5000 in blocking buffer and incubated for 1 hr. Following additional washes, HRP activity was revealed using an enhanced chemiluminescence (ECL) detection kit (GE Healthcare) and visualized using Hyperfilm ECL (GE Healthcare).
Mice
Cathepsin G−/− (CG−/−) mice on a C57BL/6 background were received from the laboratory of C. Pham (Department of Internal Medicine, Washington University School of Medicine, Saint Louis, MO). C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were bred and maintained at the Stanford University Research Animal Facility. The handling of all mice followed guidelines and requirements established by the National Institutes of Health and Stanford University animal research committee.
Splenocyte isolation
Mice were killed with compressed CO2 gas and by cervical dislocation, and spleens were removed. Single-cell suspensions were prepared by mechanical disruption of the spleen through a 70-μm filter. Spleens were then treated with 1 × red blood cell (RBC) lysis buffer [1·68 m NH4Cl, 0·10 m potassium bicarbonate and 1 mm ethylenediaminetetraacetic acid (EDTA)], washed twice, and used directly for analysis.
Flow cytometry
The following antibodies were purchased from BD Biosciences (San Jose, CA): anti-mouse I-Ab phycoerythrin (PE), anti-mouse CD11b PE-Cy7, anti-mouse CD11c allophycocyanin (APC), and anti-mouse CD19 APC-Cy7. Anti-mouse CD3 Pacific Blue, anti-mouse CD45R (B220) fluorescein isothiocyanate (FITC), and anti-mouse F4/80 PerCP-Cy5.5 were purchased from eBiosciences (San Diego, CA). Before staining, cell preparations were blocked with 3·3 μg/ml anti-mouse CD16/CD32 (Fc Block; BD Biosciences) in PBS containing 0·5% bovine serum albumin (BSA) and 0·1% NaN3 for 15 min. For intracellular staining, cells were permeabilized with Cytofix/Cytoperm (BD Biosciences) for 20 min on ice, and washed with 1 × Perm/Wash Buffer (BD Biosciences). After staining, data were acquired using a Digital LSR II from BD Biosciences and analysed using FlowJo software (Tree Star, Inc., Ashland, OR).
Results
Cleavage of HLA-DR molecules by CatG
In order to identify lysosomal proteases capable of initiating MHC II degradation, we screened a panel of cathepsins for their ability to proteolyse purified, detergent-solubilized human HLA-DR3, isolated from B-LCLs. Initially, based on the notion that molecules with loosely bound peptides might be more susceptible to proteolysis, we used HLA-DR3 molecules isolated from the HLA-DM-deficient cell line 9.5.3. More than 70% of HLA-DR3 molecules isolated from the 9.5.3 cell line are loaded with CLIP.33 Degradation was monitored by SDS-PAGE and silver staining. Digestion of HLA-DR3 molecules with CatG at neutral pH generated two proteolytic intermediates, migrating at 15 and 18 kDa (Fig. 1a), which subsequent work showed to be derived from the DR β chain (see below). The degradation of the β chain of HLA-DR3 was blocked (Fig. 1a) by addition of the CatG inhibitor,29 confirming that the observed β chain fragments were cleavage products generated specifically by CatG and not by contaminating proteases. No other cathepsin tested (D, L, S, H, and B) degraded HLA-DR3 at either neutral or endosomal pH (Fig. 1b and data not shown), although CatB and CatL degraded HLA-DM at pH 5·0 (see below) and CatD, H and S were active on myelin basic protein (MBP) and/or model substrates (data not shown). Thus, native HLA-DR3 molecules are susceptible to at most a small subset of lysosomal proteases, including CatG, in vitro.
Figure 1.
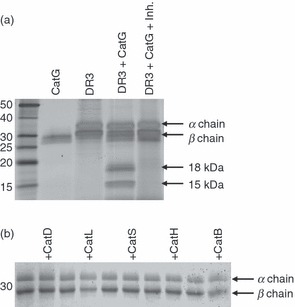
Specific proteolysis of human leukocyte antigen (HLA)-DR mediated by cathepsin G (CatG) in vitro. (a) HLA-DR3 (DR3), affinity purified from DR3 transfectants of the HLA-DM-negative B-cell line 5.2.4, was incubated for 2 hr at 37° and pH 7 with CatG in the presence or absence of a CatG inhibitor. The degradation products were visualized by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining. Data are representative of five independent experiments. (b) Lack of DR3 proteolysis by other cathepsins. DR3 molecules purified from the HLA-DM-deficient B-cell line 9.5.3 were incubated with the indicated cathepsins at pH 5 for 2 hr before SDS-PAGE and Coomassie Blue staining. Data are representative of two independent experiments. Inh., CatG inhibitor.
Peptide binding does not abrogate CatG cleavage
HLA-DR molecules purified from DM-deficient cells, as well as insect cell-derived HLA-DR molecules, are mostly occupied by loosely bound peptides, and some fraction of these HLA-DR molecules may lack bound peptides. In order to test whether CatG susceptibility of HLA-DR was linked to occupancy of the peptide binding groove, we compared CatG cleavage of HLA-DR3 molecules purified from DM-null (5.2.4-DR3) and DM-expressing (8.1.6) B-LCLs. CatG treatment of 5.2.4-derived DR3 and 8.1.6-derived DR3 molecules resulted in similar fragmentation patterns, as visualized by Western blotting. Of the two fragments seen by silver staining, only the 18-kDa fragment is immunoreactive with the antiserum used (Fig. 2a). In addition, we tested whether the stable interaction between HLA-DR1 and the influenza haemagglutinin (306–318) peptide34 influences CatG susceptibility. Soluble insect cell-derived DR1 (sDR1) was loaded to 80% saturation with AMCA-labelled influenza hemagglutinin-derived (AMCA-HA) peptide, free peptide was removed, and the resultant AMCA-HA/sDR1 complexes were digested with CatG in the presence or absence of a CatG inhibitor. The persistence of the AMCA-HA/sDR1 complex was then monitored by fluorescence resonance energy transfer (FRET), which occurs between tryptophan residues of sDR1 and the AMCA fluorophore attached to the HA peptide when the two are in close physical proximity. Occupancy (80%) with the AMCA-HA peptide did not prevent the complete cleavage of the HLA-DR β chain by CatG, with generation of an 18-kDa fragment (Fig. 2b), as detected by SDS-PAGE. Strikingly, there was only minimal loss of binding of the AMCA-HA peptide to HLA-DR1 upon digestion with CatG, and this slight loss was unaffected by the CatG inhibitor (Fig. 2c). Thus, peptide-loaded HLA-DR molecules are susceptible to CatG proteolysis, and cleavage of the β chain does not disrupt the integrity of the antigen-binding groove occupied by the peptide.
Figure 2.
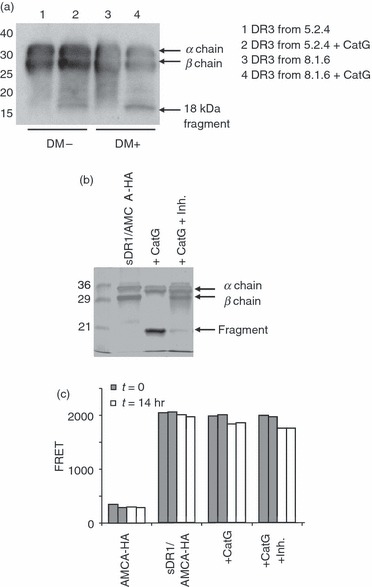
Peptide occupancy does not abrogate cathepsin G (CatG) susceptibility of human leukocyte antigen (HLA)-DR molecules. (a) HLA-DR molecules purified from DR3-transfectants of the HLA-DM-negative B-lymphoblastoid cell line (B-LCL) 5.2.4 and from the DR3-expressing, HLA-DM-positive B-LCL 8.1.6 were digested with CatG, separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membrane and immunoblotted with CHAMP anti-DR rabbit antiserum. Similar results were obtained in two experiments. Note that the CHAMP antiserum does not react with the 15-kDa fragment seen in Fig. 1a, and reacts less strongly with the 18-kDa fragment than with intact DRβ; the latter observation may explain the differences between Figs 1a and 2a with respect to the relative band intensities of the 18-kDa fragment and intact DRβ. (b) Digestion of 7-amino-4-methylcoumarin-3-acetic acid-hemagglutinin (AMCA-HA)-bound soluble DR1 (sDR1) molecules by CatG, in the presence or absence of CatG inhibitor, was monitored by SDS-PAGE analysis and silver staining. These are the same reactions as used for fluorescence resonance energy transfer (FRET) in (c). (c) Duplicate FRET analysis of sDR1/AMCA-HA complexes after incubation with or without CatG and CatG inhibitor (5 μm = 100 × inhibitor dissociation constant) for 0 or 14 hr. Free AMCA-HA peptide was included as a negative control.
CatG cleavage site on the HLA-DR β chain
To determine the exact CatG cleavage site within the HLA-DR β chain, we performed N-terminal sequencing as well as peptide mapping of the digestion products of purified soluble HLA-DR1 (sDR1). For these experiments we used sDR1 expressed in either insect cells or E. coli. Neither of these have a transmembrane domain and E. coli purified sDR1 is not glycosylated, which led to the fragments being smaller on gels (10 and 15 kDa). sDR1 expressed in insect cells (not shown) was used for identification of the N-terminal sequence of both fragments by Edman degradation (underlined italic sequence, Fig. 3a). The first residue of the larger fragment corresponds to the glycine (G) in position 1 of the mature protein. The first residue of the smaller fragment was identified as glutamine (Q) at position 110. In order to define the boundaries of both fragments, we also digested sDR1 expressed in E. coli (Fig. 3a), which is not glycosylated and was therefore used for MALDI-TOF analysis. The two bands were excised from a gel and digested with trypsin, Staphylococcus aureus V8 protease, or Arg-C protease. All peptides of these digests identified by mass spectrometry are indicated in black text in Fig. 3a. The peptide SFTVQRRVEPKVTVYPSKTQPL (underlined in Fig. 3a) was identified from a V8 digest and the peptide RVEPKVTVYPSKTQPL was identified from an Arg-C digest of the larger fragment, indicating that CatG did not cleave after the arginine (R), but did cleave after leucine 109 (L109). Based on the masses of the two fragments and on the fact that their sequences were contiguous, these fragments appear to represent the complete β chain, which therefore has only a single CatG cleavage site. The cleavage site, between HLA-DRβ L109 and glutamine 110 (Q110, L/Q), is located on a loop between fx1 and fx2 of the membrane-proximal, immunoglobulin-like domain, as indicated on the crystal structure of HLA-DR (Fig. 3b).
Figure 3.
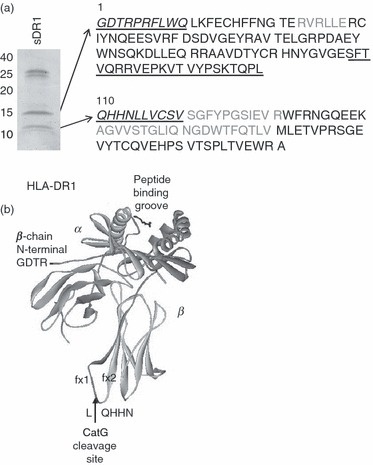
Mapping of the cathepsin G (CatG) cleavage site within the human leukocyte antigen (HLA)-DR β chain. (a) Gelcode Blue-stained sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) gel of Escherichia coli-derived, soluble recombinant HLA-DR1 molecules digested with CatG. The two cleavage products of ∼10 and 15 kDa were submitted for tryptic, V8, and Arg-C digestion and matrix-assisted laser desorption-ionisation time-of-flight (MALDI-TOF) analysis. Peptides identified by MALDI-TOF are indicated in black. Insect cell-derived soluble DR1 (sDR1) was also digested with CatG (not shown) and the two cleavage products were submitted for N-terminal Edman degradation. Sequences identified by Edman degradation are indicated on the sequence of HLA-DR1 in italic underlined text. The first residue of the 15-kDa fragment (as identified by Edman degradation) corresponds to the first residue of the mature protein (glycine). The first residue of the 10-kDa fragment (as identified by Edman degradation) corresponds to the glutamine (Q) at position 110. (b) Crystal structure of HLA-DR1 depicting the CatG cleavage site between fx1 and fx2 of the lower β chain loop. The upper dark domain of the β chain corresponds to the 15-kDa fragment, and the lower lighter domain of the β chain corresponds to the 10-kDa fragment.
HLA-DR polymorphism and susceptibility to CatG cleavage
To explore whether HLA-DR β chain polymorphism might influence CatG susceptibility, we first compared the amino acid sequences of several HLA-DR β chains [DRB1*0101 (DR1), DRB1*1501 (DR2b), DRB1*0301 (DR3), DRB1*0401, and DRB1*0404] and found conservation of the L/Q cleavage site (Fig. 4a). We then subjected various recombinant soluble HLA-DR allelic variants to digestion with CatG and used HLA-DR-specific rabbit serum (CHAMP) to measure residual levels of DRβ and detect the 18-kDa DRβ fragment (Fig. 4b). As predicted from sequence alignment, CatG degraded the β chain of all HLA-DR molecules tested. As the mouse MHC II allele I-Ag7 has a conservative asparagine (N) for Q substitution at position β110 and the mouse MHC II allele I-Ek has a more dramatic glutamic acid (E) for Q substitution at this position (Fig. 4a), we also tested these alleles. CatG digested I-Ag7, but not I-Ek (Fig. 4c), indicating that the Q to E change in I-Ek influences the ability of CatG to cleave at that site. Published sequences suggest that HLA-DR, -DQ and -DP alleles are susceptible to CatG (http://www.ebi.ac.uk/imgt/hla/)35 and I-A, but not I-E, alleles are susceptible to CatG. The sequence of DMβ predicted that this protein would be resistant to CatG cleavage on the fx1/fx2 loop. Insect cell-derived soluble DM (sDM) was resistant to proteolysis by CatG, at both pH 5 and pH 7, but was cleaved by the lysosomal cysteine proteases CatL and CatB at pH 5 (Fig. 4d). We concluded that CatG is capable of initiating proteolysis of many MHC II alleles (but not sDM) at a specific β chain cleavage site in vitro.
Figure 4.
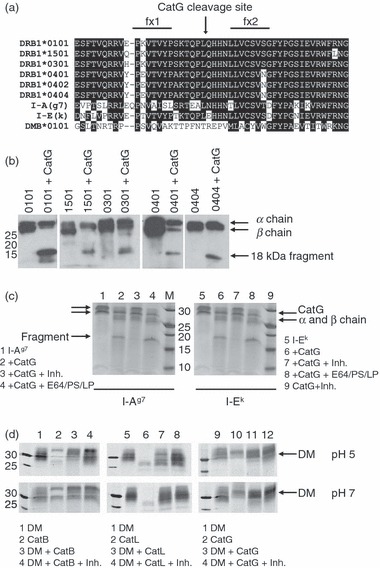
Cathepsin G (CatG) cleavage of multiple major histocompatibility complex (MHC) II allelic variants. (a) Sequence alignment of MHC II β chains and human leucocyte antigen (HLA)-DMβ (*0101 allele) in the region of CatG cleavage. All polymorphisms found within this region in the ImMunoGeneTics (IMGT)/HLA database (http://www.ebi.ac.uk/imgt/hla/) are shown.35 Identical and conserved amino acids are displayed as white text on a black background. The CatG cleavage site (L/Q) is indicated by an arrow. Also included are two mouse alleles: I-Ag7 and I-Ek. (b) Various HLA-DR alleles were expressed as soluble recombinant molecules in insect cells, purified, digested with CatG for 2 hr, analysed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE), and visualized by immunoblotting using the anti-DR antiserum CHAMP. Soluble HLA-DR 0404 β chain was epitope tagged, resulting in co-migration of α and β chains. The experiment was repeated with similar results. (c) Digestion of mouse MHC II (I-Ag7 and I-Ek) with CatG in the presence or absence of the CatG inhibitor or with inhibitors [the cysteine protease inhibitors E64 and leupeptin (LP), and the aspartyl protease inhibitor pepstatin (PS)] used during the purification of I-Ek. The β chain of I-Ag7 includes a linker and tethered peptide, resulting in the β chain migrating more slowly than the α chain. (d) Digestion of soluble recombinant human HLA-DM (sDM) molecules with selected cathepsins at pH 5 or 7. Data are representative of three independent experiments.
CatG cleavage of mutant HLA-DR molecules
Given the evidence that DM is able to preserve MHC II binding sites and is thought to rescue MHC II molecules from degradation,36,37 we hypothesized that DM/MHC II complexes might be resistant to CatG. Stable, covalent complexes of HLA-DR and DM are not available, and sDR molecules in reversible complexes formed by engineering DM and HLA-DR with complementary leucine-zippers28 remain CatG susceptible (not shown). To address whether DM and CatG interaction sites might overlap, we tested the CatG susceptibility of a series of purified, full-length mutant HLA-DR molecules, carrying substitutions that had previously been shown to disrupt DM interaction. Two mutations related to the DM interface on HLA-DR conferred some resistance to CatG (Fig. 5a). The mutation in one resistant mutant (DR βD152N) results in addition of an aberrant glycan on the DM interaction face of HLA-DR. The second resistant mutant introduces a positively charged lysine for a glutamic acid (βE187K). Although the amount of input DR was somewhat variable, this is unlikely to have confounded our results, because the resistant mutant DR molecules were not present in excessive amounts (thus the lack of inhibition was not a result of substrate inhibition), nor in quantities too small to allow detection of β-chain degradation (as confirmed by overexposure of the blots shown). The positions of the mutations and the CatG cleavage site are indicated in Fig. 5b on the crystal structure of HLA-DR1. The former mutation probably sterically inhibits CatG access to its cleavage site, while the latter may introduce charge repulsion of the highly cationic CatG at a region of HLA-DR involved in CatG binding. HLA-DR molecules with mutations in other regions remained susceptible (Fig. 5a and data not shown). Together these results implicate the membrane-proximal portion of the DM interface on HLA-DR in CatG binding and suggest, but do not prove, that DM binding may protect MHC II molecules from CatG digestion.
Figure 5.
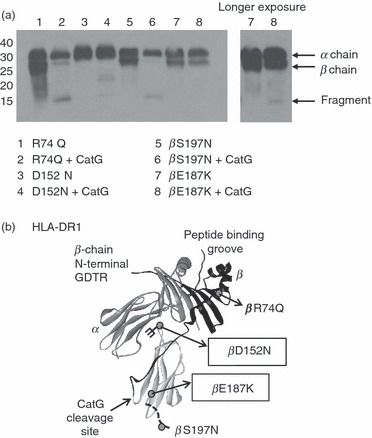
Human leukocyte antigen (HLA)-DR mutations at the DM interaction surface interfere with cathepsin G (CatG) cleavage. (a) HLA-DR3 molecules with the indicated mutations were incubated with CatG at pH 7 and visualized by western blot with CHAMP anti-DR antiserum. Data are representative of two independent experiments. (b) Three-dimensional structure of HLA-DR1 with the CatG cleavage site as well as the location of mutations of HLA-DR [molecules used in (a)] indicated. Grey circles on the crystal structure indicate mutations; branched structure indicates the addition of a glycan. Mutations that diminished susceptibility of HLA-DR to CatG are boxed.
Is CatG an unlocking enzyme for MHC II?
To test whether other proteases might further degrade the 15- or 18-kDa fragments after CatG makes the initial cut, HLA-DR3 was pre-digested with CatG and then incubated with CatL or CatB. In this sequential digest, neither CatL nor CatB could further digest the 15- and 18-kDa fragments (Data S1), although these enzymes were active as reflected in their ability to degrade MBP. Similarly, we were unable to detect further proteolysis of CatG-treated HLA-DR using CatS, D, X, H, or AEP (data not shown).
MHC II molecules resist CatG cleavage in vivo
In order to examine whether endogenous CatG might contribute to MHC II proteolysis in living APCs, we first looked for inverse correlations between changes in MHC II levels and CatG levels in human primary APCs following in vitro stimulation. Indeed, CatG, which is expressed by mDC1s,19 was down-regulated upon lipopolysaccharide (LPS) stimulation (Data S2), a manipulation known to increase surface MHC II levels and shut down MHC II turnover.4 Similarly, decreased levels of CatG correlated with increased MHC II levels in primary human B cells after stimulation with interleukin (IL)-4 (Data S2).
Next, to test whether CatG is causally involved in MHC II turnover, we examined whether addition of CatG to APCs modulates DR expression. Previously, we demonstrated that B-LCLs do not express CatG, but can acquire CatG added to culture media and target it to endocytic compartments.38 To evaluate the impact of CatG on the steady-state levels of HLA-DR molecules, we incubated B-LCLs with or without CatG for 4·5 hr. The cells were harvested, and HLA-DR levels were compared by immunoblotting. Levels of both alpha and beta chains of HLA-DR were unchanged when incubated with purified CatG or with CatG and the CatG inhibitor (Data S3). Furthermore, CatG added exogenously to a B-LCL did not alter surface levels of HLA-DR molecules, as quantified by flow cytometry with L243 (Data S4) and the anti-DR antibody Tü36 (data not shown). Importantly, the B-LCL used in these experiments, 9.5.3, is DM-deficient; thus, these negative results were not explained by DM-mediated protection from CatG.
In order to examine whether endogenous CatG expression in certain primary human APC types, such as mDC1 and B cells,19 contributes to MHC II turnover, we tested whether the CatG inhibitor,29 which inhibits intracellular CatG in intact cells,21 causes accumulation of DR in such APCs. No increase in total DR levels in primary B cells was seen by western blot, however, after 4·5, 24 and 72 hr of CatG inhibition (Fig. 6a–c). Similarly, total DR levels were unchanged in mDC1s when CatG was subjected to prolonged inhibition (24 and 72 hr; data not shown). Moreover, analysis of cell surface levels of HLA-DR by flow cytometry demonstrated that the CatG inhibitor did not affect expression of surface HLA-DR in either B cells or mDC1s (Data S5). These results argued against a causal relationship between the inversely correlated levels of endogenous CatG and MHC II expression.
Figure 6.
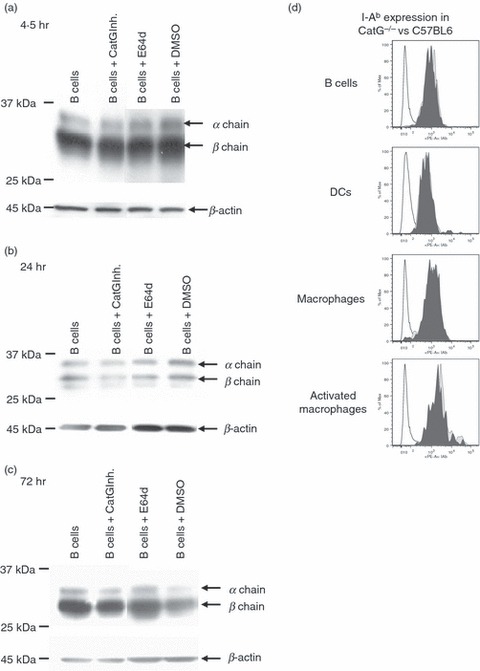
Human leukocyte antigen (HLA)-DR levels in primary antigen-presenting cells (APCs) are unaffected by the addition or ablation of cathepsin G (CatG). (a–c) Influence of the CatG inhibitor upon HLA-DR levels in primary human B cells. Primary B cells, magnetic activated cell sorting (MACS)-purified from peripheral blood mononuclear cells (PBMC), were cultured with or without CatG inhibitor (10 μm) or E64d (10 μm) for (a) 4·5 hr, (b) 24 hr or (C) 72 hr. Cells were lysed in 1% Nonidet P-40 (NP-40) with 1 μm CatG inhibitor to avoid post-lysis degradation by cell-derived CatG. Matched amounts of total protein were resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted for HLA-DR (CHAMP) and β-actin (loading control). Cells were incubated with the cell-permeable cysteine protease inhibitor E64d to inhibit CatB, H, S, and X. Dimethyl sulphoxide (DMSO) alone (vehicle) was used as a control. Data are representative of two independent experiments, each using cells from different healthy donors for each of the three time-points (n = 6 donors in total). (d) Analysis of I-Ab cell surface expression of splenocyte-derived APCs from CatG−/− compared with C57BL6 control mice. Freshly isolated splenic B cells (B220+, CD19+), resting macrophages (B220−, CD11c−, CD11b+, F480+), activated macrophages (B220−, C D19−, CD3−, CD11c+, CD11b+, F480+), and dendritic cells (CD19−, F480−, CD3−, CD11bint, CD11c+) were analysed by flow cytometry. CD3+ cells served as a negative control for major histocompatibility complex (MHC) II expression. Data are from one experiment of three with similar results.
Lastly, we considered the possibility that physiological regulation of MHC II levels by CatG might be detectable in CatG-deficient mice. Splenocytes were removed from both CatG-deficient mice and C57BL6 control mice, and cell surface and total expression of MHC II (I-Ab) was analysed by flow cytometry. Levels of surface (Fig. 6d) or total (not shown) I-Ab in B cells, DCs, and resting or activated macrophages did not differ between CatG-deficient and control mice. Analysis of peritoneal macrophages also revealed no differences in I-Ab expression between CatG−/− and C57BL6 control mice (data not shown). We concluded that, by several criteria, CatG lacks the ability to modulate steady-state MHC II levels in vivo and in live, cultured APCs.
Discussion
Our findings provide information on the mechanisms by which MHC II molecules resist endosomal proteolysis, a key biochemical requirement for their function in presentation of peptides captured in endocytic compartments. In their native conformation, purified, detergent-solubilized MHC II molecules failed to be degraded by most lysosomal proteases tested (cathepsins D, L, S, H, and B). The resistance of MHC II molecules to these proteases thus is an inherent property of the folded MHC II ectodomains. In contrast, purified MHC II molecules were susceptible to proteolytic attack by CatG at a single cleavage site, which is broadly, but not universally, conserved amongst MHC II molecules. However, using several independent approaches, we were unable to detect any involvement of CatG in the turnover of MHC II molecules embedded in membranes of live APCs. These results show, on the one hand, that proteolytic resistance of MHC II molecules is not absolute, allowing some scope for regulated turnover; on the other hand, they suggest that the CatG cleavage site is inaccessible in the membrane-embedded native MHC II protein, in vivo.
The resistance of MHC II molecules to many endosomal proteases is structurally plausible: the immunoglobulin superfamily domain fold, which is adopted by the membrane-proximal domains, is well known to be highly protease-resistant, and the peptide-loaded antigen-binding groove is highly compact. Initiation of HLA-DR proteolysis by CatG in vitro involved site-specific cleavage between leucine (L) and glutamine (Q) within fx1 and fx2 of the lower loop of the β domain, which may be one of very few sites with sufficient flexibility to allow proteolytic attack. These findings are reminiscent of previous studies, in which CatG was demonstrated to initiate cleavage within the flexible hinge regions of immunoglobulins.39 The membrane-proximal location of the cleavage site, away from the antigen binding groove, is consistent with our observation that CatG cleaves peptide-loaded MHC II molecules, and that peptide binding is retained by CatG-cleaved DR molecules. The fact that peptide-loaded molecules are substrates for CatG supports the notion that CatG is capable of initiating proteolysis of MHC II molecules in their native conformation.
Previous studies have shown that chymotrypsin also digests the β chain of HLA-DR4 molecules in vitro into two fragments (of around 15 and 13 kDa), utilizing a cleavage site between L and Q.40 These results are consistent with our own, as CatG is known to have a chymotrysin-like activity, although digestion patterns of other substrates by these two proteases are not always identical.38 The finding that cleavage of MHC II occurs after L is consistent with published data on CatG specificity, the preferred P1 amino acids for CatG cleavage being Y, F, R, L, and K.41,42
Both in vitro and ex vivo data initially suggested, but did not prove, that CatG might be involved in physiological MHC II turnover. The DR loop that harbours the cleavage site is physically close to the DM interaction site of DR, and a subset of adjacent mutations that impair DM interaction also confer resistance to CatG-mediated proteolysis. DM is known to stabilize empty MHC II molecules against degradation during endosomal peptide exchange, and this protective effect might be attributable to protection of DM-associated empty DR molecules from CatG cleavage. We were unable to reproduce this effect with DM/DR complexes formed in vitro (data not shown), but this negative result might reflect the fact that these are reversible, non-covalent complexes. Furthermore, the inverse relationships between changes in CatG activity and MHC II levels during immune cell activation were consistent with a role for CatG in MHC II turnover. Previous work has shown that CatG accumulates in endocytic compartments of primary APCs and contributes to endosomal processing of autoantigens,38,43 so its subcellular location would be compatible with participation of CatG in endosomal MHC II turnover.
However, three independent experiments failed to provide positive evidence that would implicate CatG in MHC II turnover in APCs. First, pharmacological inhibition of CatG for extended periods of time in primary human APCs failed to cause accumulation of HLA-DR molecules or of large degradation intermediates. In some preliminary experiments, we noticed that endogenous CatG activity appeared to cause DR degradation following detergent lysis of cells (data not shown); however, inclusion of the CatG inhibitor in the lysis buffer prevented this artifact, and this precaution was adopted in the experiments shown here. Similarly, genetic ablation of CatG in mice had no effect on steady-state levels of murine MHC II molecules. Collectively, our data suggest that CatG acts enzymatically upon detergent-solubilized, but not upon membrane-embedded native MHC II molecules. We considered two possible explanations for the lack of CatG cleavage in live APCs. One possibility is that the resistance of MHC II molecules to endosomal CatG cleavage reflected the neutral, rather than endosomal, pH optimum of CatG cleavage of MHC II. If true, this would reflect unique features of MHC II molecules as a substrate, because we have shown previously that MBP and other antigens are processed by CatG in endocytic compartments of APCs, and are susceptible to cleavage at acidic and neutral pH in vitro21,38,43 (T. Burster, unpublished data). We note that a dihistidine motif is adjacent to the CatG cleavage site of MHC II molecules (Fig. 4a), which might regulate CatG access in a pH-dependent fashion.
However, the pH dependence does not explain why even high concentrations of CatG added to B-LCLs at neutral pH failed to cleave DR molecules at the cell surface. The simplest interpretation of the latter result is that the CatG cleavage site of MHC II molecules is sterically inaccessible when the MHC II molecules are embedded in endosomal or cell surface membranes. The steric hindrance could, in principle, come from the proximity of the membrane itself, or from noncovalent associations with other proteins, both of which would be disrupted by detergent lysis. Partial steric masking may also explain why, in most experiments, full-length DR embedded in detergent micelles was digested less completely than soluble recombinant DR ectodomains.
Our results do not prove that CatG is never involved in MHC II degradation in vivo. For instance, CatG might conceivably act on MHC II molecules that have partially lost their native conformation at the end of their useful life. However, our findings do suggest that MHC II molecules have evolved resistance to endosomal proteolysis by a combination of mechanisms. The inherent resistance of MHC II ectodomains to many cathepsins is likely to be important. Other protease cleavage sites, such as the CatG cleavage site studied here, may be cryptic, either because of charge characteristics that impair proteolytic attack in acidic endosomal compartments, or because they are sterically inaccessible at APC membranes, or both. Steric inaccessibility of the CatG cleavage site may be particularly important in allowing antigen presentation to be maintained in inflamed tissues, in which CatG is abundantly released into the extracellular space by activated neutrophils. Whether cryptic protease cleavage sites contribute to regulated turnover of MHC II molecules remains to be determined.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG; BU1822/1-1) to TB, SFB 518, GRK 1041-2, and Else Kröner-Fresensius-Stiftung to BOB, funding from Sidney Sussex College and the Arthritis Research Campaign (ref. 18543) to RB, and grants from the NIH and the Child Health Research Program (Stanford University) to EDM. We gratefully acknowledge A. Guzzetta for mass spectrometry and L. Stern for providing HLA-DR1 molecules made in E. coli and the CHAMP anti-DR antisera. Other purified MHC II molecules, HLA-DR2b, murine I-Ag7 and I-Ek, were kindly provided by K. Wucherpfennig, L. Teyton and M. Davis, respectively. CatG−/− mice were kindly provided by C. Pham.
Glossary
Abbreviations
- AMCA
7-amino-4-methylcoumarin-3-acetic acid
- APC
antigen-presenting cell
- B-LCL
B-lymphoblastoid cell line
- Cat
cathepsin
- CLIP
class II-associated Ii peptides
- DC
dendritic cell
- FRET
fluorescence resonance energy transfer
- HA
hemagglutinin
- HLA
human leukocyte antigen
- Ii
MHC II-associated invariant chain
- MBP
myelin basic protein
- mDC
myeloid dendritic cell
- MHC
major histocompatibility complex
- PBMC
peripheral blood mononuclear cells
- pDC
plasmacytoid dendritic cell
- TCR
T-cell receptor
Disclosures
The authors do not have any conflicting interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Sequential digest of HLA-DR3.
Data S2. Reciprocal regulation of CatG activity and DR levels in primary human APC.
Data S3. Effects of CatG addition on MHC II levels in intact APC (Western blot).
Data S4. Effects of CatG addition on cell surface MHC II levels in intact APC.
Data S5. Effects of CatG inhibition on cell surface MHC II levels using primary intact APC.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Rocha N, Neefjes J. MHC class II molecules on the move for successful antigen presentation. EMBO J. 2008;27:1–5. doi: 10.1038/sj.emboj.7601945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drozina G, Kohoutek J, Jabrane-Ferrat N, Peterlin BM. Expression of MHC II genes. Curr Top Microbiol Immunol. 2005;290:147–70. doi: 10.1007/3-540-26363-2_7. [DOI] [PubMed] [Google Scholar]
- 3.Villadangos JA, Cardoso M, Steptoe RJ, van Berkel D, Pooley J, Carbone FR, Shortman K. MHC class II expression is regulated in dendritic cells independently of invariant chain degradation. Immunity. 2001;14:739–49. doi: 10.1016/s1074-7613(01)00148-0. [DOI] [PubMed] [Google Scholar]
- 4.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–7. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 5.Pierre P, Turley SJ, Gatti E, et al. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–92. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 6.De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, Gatti E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci USA. 2008;105:3491–6. doi: 10.1073/pnas.0708874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bikoff EK, Germain RN, Robertson EJ. Allelic differences affecting invariant chain dependency of MHC class II subunit assembly. Immunity. 1995;2:301–10. doi: 10.1016/1074-7613(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 8.Elliott EA, Drake JR, Amigorena S, Elsemore J, Webster P, Mellman I, Flavell RA. The invariant chain is required for intracellular transport and function of major histocompatibility complex class II molecules. J Exp Med. 1994;179:681–94. doi: 10.1084/jem.179.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viville S, Neefjes J, Lotteau V, Dierich A, Lemeur M, Ploegh H, Benoist C, Mathis D. Mice lacking the MHC class II-associated invariant chain. Cell. 1993;72:635–48. doi: 10.1016/0092-8674(93)90081-z. [DOI] [PubMed] [Google Scholar]
- 10.Villadangos JA, Bryant RA, Deussing J, et al. Proteases involved in MHC class II antigen presentation. Immunol Rev. 1999;172:109–20. doi: 10.1111/j.1600-065x.1999.tb01360.x. [DOI] [PubMed] [Google Scholar]
- 11.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, Mellins ED. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev. 2005;207:242–60. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 12.Nelson CA, Petzold SJ, Unanue ER. Peptides determine the lifespan of MHC class II molecules in the antigen-presenting cell. Nature. 1994;371:250–2. doi: 10.1038/371250a0. [DOI] [PubMed] [Google Scholar]
- 13.Wolf PR, Tourne S, Miyazaki T, Benoist C, Mathis D, Ploegh HL. The phenotype of H-2M-deficient mice is dependent on the MHC class II molecules expressed. Eur J Immunol. 1998;28:2605–18. doi: 10.1002/(SICI)1521-4141(199809)28:09<2605::AID-IMMU2605>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 14.Koonce CH, Wutz G, Robertson EJ, Vogt AB, Kropshofer H, Bikoff EK. DM loss in k haplotype mice reveals isotype-specific chaperone requirements. J Immunol. 2003;170:3751–61. doi: 10.4049/jimmunol.170.7.3751. [DOI] [PubMed] [Google Scholar]
- 15.Guerra CB, Busch R, Doebele RC, Liu W, Sawada T, Kwok WW, Chang MD, Mellins ED. Novel glycosylation of HLA-DRalpha disrupts antigen presentation without altering endosomal localization. J Immunol. 1998;160:4289–97. [PubMed] [Google Scholar]
- 16.Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, Ploegh HL. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell. 1990;61:171–83. doi: 10.1016/0092-8674(90)90224-3. [DOI] [PubMed] [Google Scholar]
- 17.Blum JS, Cresswell P. Role for intracellular proteases in the processing and transport of class II HLA antigens. Proc Natl Acad Sci USA. 1988;85:3975–9. doi: 10.1073/pnas.85.11.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12:201–10. doi: 10.1016/s1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- 19.Stoeckle C, Sommandas V, Adamopoulou E, et al. Cathepsin G is differentially expressed in primary human antigen-presenting cells. Cell Immunol. 2009;255:41–5. doi: 10.1016/j.cellimm.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Burster T, Macmillan H, Hou T, Boehm BO, Mellins ED, Cathepsin G. Roles in antigen presentation and beyond molecular immunology. Mol Immunol. 2009;47:658–65. doi: 10.1016/j.molimm.2009.10.003. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reich M, Lesner A, Legowska A, Sienczyk M, Oleksyszyn J, Boehm BO, Burster T. Application of specific cell permeable cathepsin G inhibitors resulted in reduced antigen processing in primary dendritic cells. Mol Immunol. 2009;46:2994–9. doi: 10.1016/j.molimm.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 22.Pious D, Dixon L, Levine F, Cotner T, Johnson R. HLA class II regulation and structure. Analysis with HLA-DR3 and HLA-DP point mutants. J Exp Med. 1985;162:1193–207. doi: 10.1084/jem.162.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mellins E, Kempin S, Smith L, Monji T, Pious D. A gene required for class II-restricted antigen presentation maps to the major histocompatibility complex. J Exp Med. 1991;174:1607–15. doi: 10.1084/jem.174.6.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doebele RC, Busch R, Scott HM, Pashine A, Mellins ED. Determination of the HLA-DM interaction site on HLA-DR molecules. Immunity. 2000;13:517–27. doi: 10.1016/s1074-7613(00)00051-0. [DOI] [PubMed] [Google Scholar]
- 25.Doebele RC, Pashine A, Liu W, Zaller DM, Belmares M, Busch R, Mellins ED. Point mutations in or near the antigen-binding groove of HLA-DR3 implicate class II-associated invariant chain peptide affinity as a constraint on MHC class II polymorphism. J Immunol. 2003;170:4683–92. doi: 10.4049/jimmunol.170.9.4683. [DOI] [PubMed] [Google Scholar]
- 26.Sloan VS, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller DM. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 1995;375:802–6. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 27.Busch R, Reich Z, Zaller DM, Sloan V, Mellins ED. Secondary structure composition and pH-dependent conformational changes of soluble recombinant HLA-DM. J Biol Chem. 1998;273:27557–64. doi: 10.1074/jbc.273.42.27557. [DOI] [PubMed] [Google Scholar]
- 28.Busch R, Pashine A, Garcia KC, Mellins ED. Stabilization of soluble, low-affinity HLA-DM/HLA-DR1 complexes by leucine zippers. J Immunol Methods. 2002;263:111–21. doi: 10.1016/s0022-1759(02)00034-0. [DOI] [PubMed] [Google Scholar]
- 29.Greco MN, Hawkins MJ, Powell ET, et al. Nonpeptide inhibitors of cathepsin G: optimization of a novel beta-ketophosphonic acid lead by structure-based drug design. J Am Chem Soc. 2002;124:3810–1. doi: 10.1021/ja017506h. [DOI] [PubMed] [Google Scholar]
- 30.Hellman U, Wernstedt C, Gonez J, Heldin CH. Improvement of an “In-Gel” Digestion Procedure for the Micropreparation of Internal Protein Fragments for Amino Acid Sequencing. Anal Biochem. 1995;224:451–5. doi: 10.1006/abio.1995.1070. [DOI] [PubMed] [Google Scholar]
- 31.Joshi RV, Zarutskie JA, Stern LJ. A three-step kinetic mechanism for peptide binding to MHC class II proteins. Biochemistry. 2000;39:3751–62. doi: 10.1021/bi9923656. [DOI] [PubMed] [Google Scholar]
- 32.Carven GJ, Chitta S, Hilgert I, et al. Monoclonal antibodies specific for the empty conformation of HLA-DR1 reveal aspects of the conformational change associated with peptide binding. J Biol Chem. 2004;279:16561–70. doi: 10.1074/jbc.M314315200. [DOI] [PubMed] [Google Scholar]
- 33.Mellins E, Cameron P, Amaya M, Goodman S, Pious D, Smith L, Arp B. A mutant human histocompatibility leukocyte antigen DR molecule associated with invariant chain peptides. J Exp Med. 1994;179:541–9. doi: 10.1084/jem.179.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pashine A, Busch R, Belmares MP, Munning JN, Doebele RC, Buckingham M, Nolan GP, Mellins ED. Interaction of HLA-DR with an acidic face of HLA-DM disrupts sequence-dependent interactions with peptides. Immunity. 2003;19:183–92. doi: 10.1016/s1074-7613(03)00200-0. [DOI] [PubMed] [Google Scholar]
- 35.Robinson J, Waller MJ, Parham P, de Groot N, Bontrop R, Kennedy LJ, Stoehr P, Marsh SGE. IMGT/HLA and IMGT/MHC: sequence databases for the study of the major histocompatibility complex. Nucleic Acids Research. 2003;31:311–4. doi: 10.1093/nar/gkg070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kropshofer H, Arndt SO, Moldenhauer G, Hammerling GJ, Vogt AB. HLA-DM acts as a molecular chaperone and rescues empty HLA-DR molecules at lysosomal pH. Immunity. 1997;6:293–302. doi: 10.1016/s1074-7613(00)80332-5. [DOI] [PubMed] [Google Scholar]
- 37.Denzin LK, Hammond C, Cresswell P. HLA-DM interactions with intermediates in HLA-DR maturation and a role for HLA-DM in stabilizing empty HLA-DR molecules. J Exp Med. 1996;184:2153–65. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burster T, Beck A, Tolosa E, et al. Cathepsin G, and not the asparagine-specific endoprotease, controls the processing of myelin basic protein in lysosomes from human B lymphocytes. J Immunol. 2004;172:5495–503. doi: 10.4049/jimmunol.172.9.5495. [DOI] [PubMed] [Google Scholar]
- 39.Ryan MH, Petrone D, Nemeth JF, Barnathan E, Bjorck L, Jordan RE. Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol Immunol. 2008;45:1837–46. doi: 10.1016/j.molimm.2007.10.043. [DOI] [PubMed] [Google Scholar]
- 40.Kaufman JF, Strominger JL. The extracellular region of light chains from human and murine MHC class II antigens consists of two domains. J Immunol. 1983;130:808–17. [PubMed] [Google Scholar]
- 41.Wysocka M, Legowska A, Bulak E, Jaskiewicz A, Miecznikowska H, Lesner A, Rolka K. New chromogenic substrates of human neutrophil cathepsin G containing non-natural aromatic amino acid residues in position P(1) selected by combinatorial chemistry methods. Mol Divers. 2007;11:93–9. doi: 10.1007/s11030-007-9063-7. [DOI] [PubMed] [Google Scholar]
- 42.Polanowska J, Krokoszynska I, Czapinska H, Watorek W, Dadlez M, Otlewski J. Specificity of human cathepsin G. Biochim Biophys Acta. 1998;1386:189–98. doi: 10.1016/s0167-4838(98)00085-5. [DOI] [PubMed] [Google Scholar]
- 43.Burster T, Beck A, Tolosa E, et al. Differential processing of autoantigens in lysosomes from human monocyte-derived and peripheral blood dendritic cells. J Immunol. 2005;175:5940–9. doi: 10.4049/jimmunol.175.9.5940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.