

Abstract
Rationale
Tribbles 3 (TRB3) is an intracellular pseudokinase that modulates the activity of several signal transduction cascades. TRB3 has been reported to inhibit the activity of Akt protein kinases. TRB3 gene expression is highly regulated in many cell types, and amino acid starvation, hypoxia, or endoplasmic reticulum (ER) stress promotes TRB3 expression in non-cardiac cells.
Objective
The objective of this work was to examine TRB3 expression and function in cultured cardiac myocytes and in mouse heart.
Methods and Results
Agents that induced ER stress increased TRB3 expression in cultured cardiac myocytes while blocking insulin-stimulated Akt activation in these cells. Knockdown of TRB3 in cultured cardiac myocytes reversed the effects of ER stress on insulin signaling. Experimental myocardial infarction led to increased TRB3 expression in murine heart tissue in the infarct border zone suggesting that ER stress may play a role in pathological cardiac remodeling. Transgenic mice with cardiac-specific overexpression of TRB3 were generated and they exhibited normal contractile function but altered cardiac signal transduction and metabolism with reduced cardiac glucose oxidation rates. Transgenic TRB3 mice were also sensitized to infarct expansion and cardiac myocyte apoptosis in the infarct border zone after myocardial infarction.
Conclusions
These results demonstrate that TRB3 induction is a significant aspect of the ER stress response in cardiac myocytes and that TRB3 antagonizes cardiac glucose metabolism and cardiac myocyte survival.
Keywords: ER Stress, Akt, TRB3, myocardial infarction, signal transduction
Introduction
Many cellular proteins are produced by ribosomes that coat the endoplasmic reticulum (ER), and stress of the rough ER occurs when proteins within its lumen are misfolded.1, 2 This can occur as a result of reduced calcium levels, insufficient concentrations of molecular chaperones, altered protein glycosylation machinery, altered redox status, and other factors within the ER lumen. ER stress leads to the initiation of the unfolded protein response (UPR), an adaptive mechanism that initially promotes organelle recovery.1,2 However, if ER stress is prolonged or if the UPR is unsuccessful in promoting recovery, apoptotic cell death can ensue.2 The UPR promotes global translational blockade, but also leads to the selective transcription and translation of several genes.1, 2
In response to protein misfolding, the glucose regulated protein 78 (GRP78, also called BiP) chaperone is released by the ER transmembrane proteins PERK (a protein kinase), IRE-1 (an endoribonuclease), and activating transcription factor 6 (ATF6), so that it can directly bind to the misfolded proteins within the ER lumen. 1, 2 Release of GRP78 leads to altered activity of its transmembrane binding partners, and this triggers various aspects of the UPR. Release of GRP78 allows PERK to become activated through dimerization, and active PERK phosphorylates the ribosomal elongation factor eIF-2α, resulting in the inhibition of most protein translation, with the exception of certain proteins such as GRP78 and ATF4.1, 2 Release of GRP78 allows IRE-1 to splice an mRNA encoding X-box-binding protein-1 (XBP1), creating a new open reading frame that encodes an active transcription factor. Finally, release of GRP78 allows ATF6 to migrate to the Golgi where S1P and S2P cleave the protein releasing the N-terminal cytosolic portion of the protein, called N-ATF6 which has a transcriptional activation domain, DNA-binding domain, and nuclear localization sequence. XBP1active and N-ATF6 bind to ER stress response elements in various ER stress response genes, including GRP78, CHOP (CHOP10, GADD153, DDIT3, C/EBPζ) and XBP1.1, 2
ER stress occurs in cardiac myocytes and cardiac tissue in response to various stressors, including ischemia, inflammation and exposure to alcohol.3-5 Indeed, experimental autoimmune cardiomyopathy induced by injection of a peptide corresponding to a portion of the β1 adrenergic receptor was associated with cardiac ER stress as measured by induction of GRP78 and CHOP gene expression.4 Furthermore, autoimmune cardiomyopathy was associated with inhibition of the phosphatidylinositol-3′ kinase (PI3K)/Akt protein kinase signaling cascade.4 Similarly, treatment of animals with alcohol resulted in the development of a cardiomyopathy associated with ER stress and inhibition of Akt activity.5 In earlier work with other cell types, the relationship between ER stress and Akt activation appears to be biphasic: acute Akt activation occurs during the initial phase of the UPR to promote cell survival, and this is followed by delayed Akt inhibition that contributes to apoptosis.6,7 Indeed, Akt and target of rapamycin (TOR) activation was transiently observed 2 and 4 hours after treatment of cultured MCF-7 breast cancer and H1299 lung cancer cells with the glycosylation inhibitor tunicamycin or the sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA) inhibitor thapsigargin, and acute Akt activation was essential to promote cell survival in the acute phase.6 Similar transient Akt activation followed by Akt inhibition was observed in cultured primary glial cells treated tunicamycin or thapsigargin.7
Tribbles 3 (TRB3) is a pseudokinase that modulates several signaling pathways, including the PI3K/Akt cascade, the nuclear factor κ-light-chain-enhancer of activated B cells cascade, and the ATF4/CHOP pathway.8-10 TRB3 binds to and inhibits the kinase activity of Akt family members, and Akt proteins are well-described activators of protein translation via TOR.8,9 TRB3 also binds to and inhibits the transcriptional activity of ATF4 and CHOP.10 Previous work in non-cardiac mammalian cells suggested that agents that induce ER stress, such as thapsigargin, tunicamycin, the long chain fatty acid palmitate, and hypoxia all induce the expression of TRB3.11-16 In this work we investigated whether ER stress in cardiac myocytes and intact heart tissue induces the expression of TRB3 and the physiological consequences of altering TRB3 levels.
Material and Methods
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org which includes specific details about cell culture and chemicals, siRNA treatment of HL-1 cells, isolation of RNA and quantitative real-time PCR, western blotting, murine experimental myocardial infarction, generation of TRB3 transgenic mice, murine echocardiography, and isolated working heart analysis of cardiac metabolism. HL-1 cells were generously provided by Dr. William W. Claycomb (Louisiana State University Health Sciences Center, New Orleans, LA). All animal protocols in this study were approved by the Animal Studies Committee of the Division of Comparative Medicine at Washington University in St. Louis.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
ER Stress Inhibits Akt Activation and Promotes TRB3 Expression in Cultured Cardiac myocytes
In previous work, alcohol exposure and autoimmune myocarditis were found to be associated with cardiac ER stress and inhibition of Akt signaling.4,5 We initially wished to confirm whether agents that induce ER stress in cardiac myocytes would inhibit Akt signaling. Cultured HL-1 murine atrial cardiac myocytes were treated with thapsigargin and then stimulated with insulin.17,18 HL-1 cells were treated with thapsigargin (2 μM), and 24 hours later cells were treated with insulin (10 nM) or control buffer and Akt activation was examined by immunoblotting with an anti-phospho-Akt antibody (Figure 1A,B). Thapsigargin-treated HL-1 cells were resistant to insulin-stimulated Akt activation. Furthermore, thapsigargin-treated HL-1 cells exhibited increased protein levels for GRP78 and CHOP, two markers of ER stress (Figure 1A).
Figure 1.
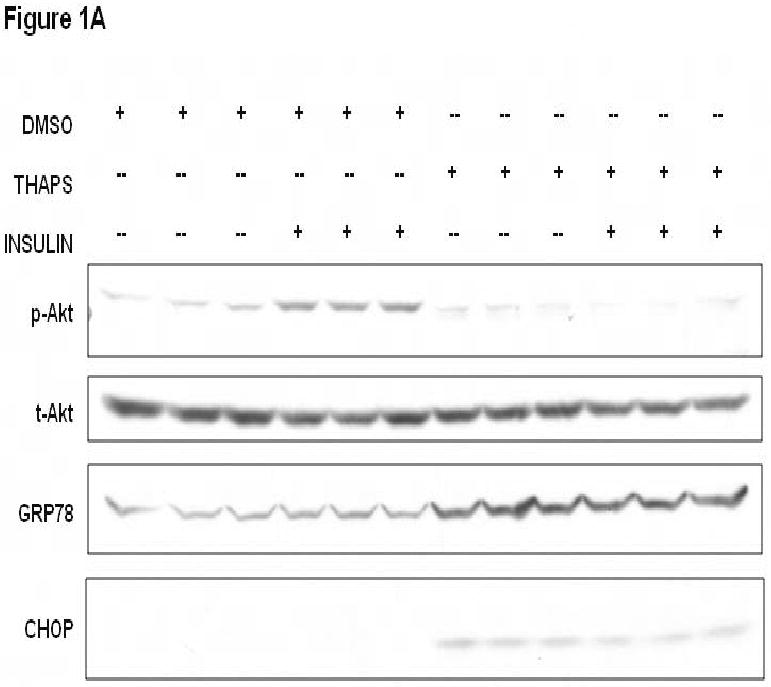



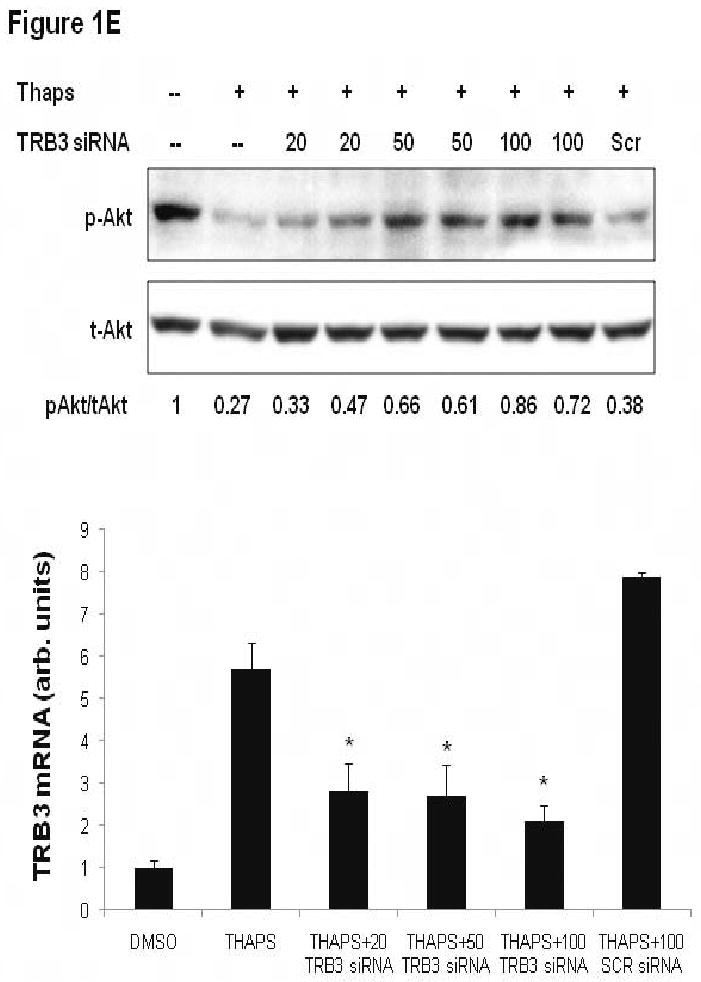
Cardiomyoycte ER stress inhibits Akt activation due to TRB3 induction. A. Treatment of HL-1 atrial myocytes with thapsigargin results in ER stress and reduced insulin-stimulated Akt activation. HL-1 cells were pre-treated with thapsigargin (THAPS; 2 μM) or DMSO for 24 hours. HL-1 cells were treated with insulin (10 nM) or control buffer for 10 minutes. Protein lysates were generated from HL-1 cells and proteins were separated by SDS-PAGE followed by immunoblotting with primary antibodies directed against phospho-Akt1/2 (p-Akt), total-Akt1/2 (t-Akt), GRP78, and CHOP. B. Densitometric analysis of Akt activation in HL-1 cells incubated with thapsigargin or DMSO and then stimulated with insulin (Ins) or buffer as depicted in 1A. Phospho-Akt levels were normalized by total Akt levels for each sample. *, P<0.001 versus HL-1 cells that were not treated with thapsigargin or insulin by Student's t-test; ˆ, P=0.004 versus HL-1 cells treated with DMSO and insulin by Student's t-test. C. TRB3 protein levels increase in HL-1 in response to ER stress. HL-1 cells were treated with DMSO or thapsigargin (THAPS; 2 μM) for 24 hours and protein lysates were obtained. Protein lysates were separated by SDS-PAGE followed by immunoblotting with an anti-TRB3 primary antibody. The anti-TRB3 antibody specifically recognizes a 42 kilodalton band or a doublet (depending on the resolution of the gel). Blots were re-probed with an anti-β-actin antibody to control for protein loading. The TRB3 protein levels were measured by computerized densitometry and were normalized by β-actin protein levels. The TRB3/actin levels are indicated below each lane in arbitrary units. The mean TRB3/actin level was 1.0 ± 0.19 for control DMSO-treated cells, and was 1.88 ± 0.14 for thapsigargin-treated cells (P=0.021 by Student's t-test). D. Agents that promote ER stress cause induction of TRB3 mRNA in HL-1 cardiac myocytes. HL-1 cells were treated with tunicamycin (2 μg/ml), thapsigargin (2 μM) or DMSO (control) for 24 hours. RNA was purified from HL-1 cells and TRB3 and GAPDH mRNA levels were analyzed by quantitative real-time PCR. *, P<0.05 by Kruskal-Wallis one way ANOVA on ranks. ˆ, P<0.05 by Kruskal-Wallis one way ANOVA on ranks. E. ER stress-mediated blockade of Akt activation is dependent on TRB3. HL-1 cells analyzed in this figure were treated with thapsigargin (THAPS; 2 μM) or DMSO for 24 hours. Some cells were also pre-treated with TRB3 siRNA at the indicated doses (20, 50 or 100 nM) or with scrambled siRNA (100 nM) for 24 hours prior to the addition of thapsigargin. In the upper panel, cells were serum starved for 6 hours and then insulin (10 nM) was added to all HL-1 cells for 10 minutes and protein lysates were obtained for SDS-PAGE followed by immunoblotting. Immunoblots depict the levels of activated Akt (p-Akt), total Akt (t-Akt), and GRP78. Under the blots, a computerized densitometry analysis of phospho-Akt levels normalized by total Akt protein levels for each lane is provided in arbitrary units. In the lower panel, HL-1 cells were cultured in parallel with those used for the immunoblot experiment, and were treated with TRB3 siRNA (20, 50 or 100 nM) or with scrambled siRNA (100 nM) for 24 hours. RNA was purified from HL-1 cells and TRB3 and GAPDH mRNA levels were analyzed by quantitative real-time PCR. *, P<0.05 by one way ANOVA (Holm-Sidak method) versus scrambled siRNA.
To determine whether the impaired insulin-stimulated Akt activation in thapsigargin-treated HL-1 cells was correlated with TRB3 induction, TRB3 protein levels were determined by immunoblotting (Figure 1C). HL-1 cells treated with thapsigargin (2 μM) for 24 hours exhibited a nearly 2-fold increase in TRB3 protein levels. Furthermore, HL-1 cells treated with thapsigargin (2 μM) or the glycosylation inhibitor tunicamycin (2 μg/ml) for 24 hours exhibited significantly increased TRB3 mRNA levels when determined by quantitative real-time PCR (Figure 1D).
To determine whether TRB3 induction in response to agents that cause ER stress was responsible for defective insulin signaling, we performed knock down experiments. HL-1 cells were transfected with a TRB3 siRNA that is effective at reducing TRB3 levels in murine cultured cardiac myocytes. Insulin-stimulated phosphorylation of Akt1/2 was attenuated by pretreatment with thapsigargin, but the inhibitory effect of thapsigargin was reversed by knockdown of TRB3 by use of siRNA at a dosage of 50 or 100 nM (Figure 1E). Treatment of HL-1 cells with 100 nM scrambled siRNA did not reverse the effect of thapsigargin on insulin signaling (Figure 1E).
To further evaluate whether TRB3 induction in response to ER stress specifically affected Akt signaling, we performed rescue experiments with adenoviral-mediated Akt2 overexpression in HL-1 cells. Cultured HL-1 cells were infected with recombinant adenoviruses encoding β-galactosidase (ad-βGal, control) or wild type Akt2 (ad-Akt2) and 48 hours later were treated with thapsigargin for 24 hours. Insulin-stimulated phosphorylation of glycogen synthase kinase 3β, an indicator of Akt activation, was increased in thapsigargin-treated HL-1 cells infected with ad-Akt2 compared to those infected with ad-GFP (Online Figure I).
Provocative stimuli promote TRB3 expression in cardiac tissue in vivo
In vitro experiments described above showed that agents that promote ER stress in cultured cardiac myocytes induce TRB3 gene expression. In previous in vivo work, several mouse models of cardiomyopathy were found to be associated with cardiac ER stress. For example, transgenic mice with cardiac-specific overexpression of monocyte chemoattractant protein 1 (MCP-1) developed a form of ischemic cardiomyopathy that was associated with myocardial ER stress and TRB3 expression.19 In another mouse model system, experimental myocardial infarction resulted in ER stress in the infarct border zone.3 Increased levels of GRP78 protein were detected in the infarct border zone of mice subjected to experimental myocardial infarction 4 days earlier.3 Finally, pressure overload by transverse aortic constriction in mice resulted in cardiac ER stress, as measured by increased levels of GRP78 and CHOP.20
To evaluate whether provocative stimuli that promote cardiac ER stress in vivo also induce TRB3 expression, we performed experimental myocardial infarction surgery in mice.21-23 Anesthetized and ventilated wild type C57BL/6J_mice were subjected to ligation of the left anterior descending coronary artery or to a sham operation, and then left ventricular tissue was obtained 4 and 24 hours after surgery. GRP78 and CHOP mRNA levels did not change 4 hours after myocardial infarction, but exhibited a trend towards increased levels after 24 hours (Figure 2A, B). Analysis of TRB3 mRNA levels from cardiac samples similarly revealed that TRB3 mRNA did not increase significantly 4 hours after myocardial infarction, but increased by 2.5-fold 24 hours after myocardial infarction when compared to sham tissue (P<0.05) (Figure 2C).
Figure 2.



ER stress markers and TRB3 expression is induced in cardiac tissue after myocardial infarction. Wild type C57BL/6J mice were subjected to experimental myocardial infarction by ligation of the left anterior descending coronary artery. A. Total left ventricle was isolated 4 and 24 hours after MI or sham operation for RNA isolation. Quantitative real-time PCR analysis of GRP78 was performed. B. Total left ventricle was isolated 4 and 24 hours after MI or sham operation for RNA isolation. Quantitative real-time PCR analysis of CHOP was performed. C. Total left ventricle was isolated 4 and 24 hours after MI or sham operation for RNA isolation. Quantitative real-time PCR analysis of TRB3 was performed. *, P<0.05 by one way ANOVA (Holm-Sidak method) versus sham operation. D. Infarct border zone (defined as one-quarter circumference on either side of the infarct edge) and remote left ventricular tissue was isolated 24 hours after MI surgery for RNA isolation. In addition, the entire left ventricle was isolated 24 hours after a sham operation for RNA isolation. Quantitative real-time PCR analysis of TRB3 was performed. *, P<0.05 by one way ANOVA (Holm-Sidak method) versus remote left ventricle.
To evaluate the location of increased TRB3 expression after myocardial infarction, infarct border zone and remote left ventricular tissue was dissected from heart 24 hours after experimental myocardial infarction. Analysis of TRB3 mRNA levels demonstrated that TRB3 mRNA levels were significantly increased in the infarct border zone but not in the remote myocardium (Figure 2D).
To evaluate whether nutrient deprivation and hypoxia was responsible for the induction of TRB3 in the infarct border zone of mice, we performed additional in vitro experiments with cultured cardiac myocytes. Culturing cardiac myocytes under hypoglycemic conditions for 24 hours resulted in a dramatic increase in TRB3 mRNA levels (Online Figure II). Indeed, culturing cardiac myocytes in 0.5 mM glucose resulted in an 8.4-fold increase in TRB3 mRNA levels compared to cells cultured in 25 mM glucose. Furthermore, subjecting cultured cardiac myocytes to an in vitro model of ischemia for 2 hours (hypoxia and hypoglycemia) followed by an in vitro model of reperfusion for 2 hours (normoxia and normoglycemia) resulted in a 2.3-fold increase in TRB3 mRNA levels (Online Figure III).
Generation of transgenic mice with cardiac-specific overexpression of TRB3
In vitro experiments showed that ER stress induced TRB3 expression in cultured cardiac myocytes, and that TRB3 blocked cardiac myocyte insulin signaling in vitro. To evaluate the ability of TRB3 to modulate cardiac metabolism in vivo, transgenic mice with cardiac-specific overexpression of TRB3 were generated. A DNA construct was made that included the α–myosin heavy chain promoter linkered to a myc-1 epitope-tagged cDNA encoding murine TRB3 corresponding to Genbank NM_175093. This DNA construct was injected into the male pro-nucleus of one-cell murine embryos in order to generate transgenic mice. Multiple lines were obtained and lines designated 1×-TRB3 (low expression), 2×-TRB3 (medium expression), and 4×-TRB3 (high expression) based on differential genomic DNA expression by PCR were selected for initial evaluation. Mice from all lines appeared grossly normal at birth and were fertile. Overexpression of TRB3 in transgenic mice was confirmed by immunoblotting of cardiac protein lysates (Figure 3A).
Figure 3.
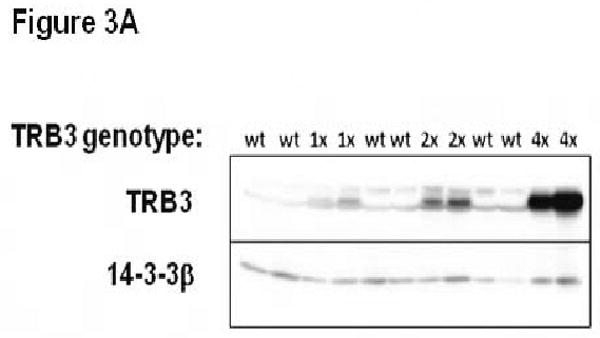

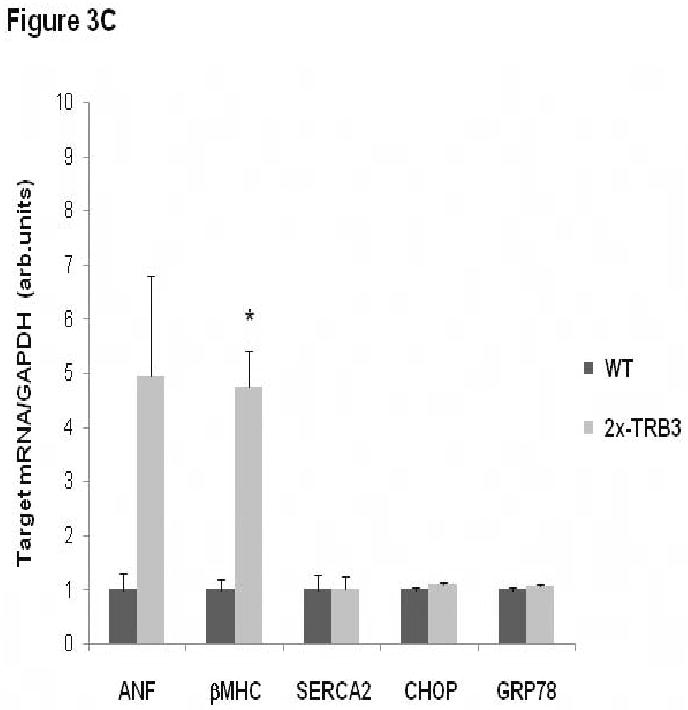
Analysis of transgenic mice with cardiac-specific overexpression of TRB3. A. Increased TRB3 protein levels in 1×-TRB3, 2×-TRB3, and 4×-TRB3 transgenic heart tissue. Left ventricular tissue was isolated from12 week old transgenic mice and nontransgenic C57BL/6J littermates. Ventricular protein lysates were analyzed by immunoblotting for TRB3 protein content. Blots were re-probed with anti-14-3-3β primary antibody to control for protein loading. B. Reduced insulin-stimulated intracellular signal transduction in 2×-TRB3 transgenic myocardium. 2×-TRB3 mice and their wild type (WT) littermates were anesthetized and treated with insulin (0.1 units/kg body weight) or vehicle by jugular vein injection. Five minutes later, hearts were isolated and ventricular tissue was used to generate protein lysates. Proteins were separated by SDS-PAGE and immunoblotting was performed to evaluate the phosphorylation status of Akt and GSK-3β. Blots were re-probed with anti-total Akt1/2 primary antibody to control for protein loading. Depicted in the figure are the computerized densitometry results that indicate the relative phospho-Akt or phospho-GSK3β/total-Akt protein level for each sample. *, P<0.001 versus wild type (WT) cardiac tissue not treated with insulin by Student's t-test; ˆ, P=0.014 versus WT cardiac tissue treated with insulin by Student's t-test; **, P<0.001 versus WT cardiac tissue not treated with insulin by Student's t-test;ˆˆ, P=0.004 versus WT cardiac tissue treated with insulin by Student's t-test. C. Increased expression of β-MHC in 2×-TRB3 cardiac tissue. RNA was purified from ventricular tissue isolated from 12-16-week-old 2×-TRB3 mice and their nontransgenic wild type littermates (WT). ANF, β-MHC, SERCA2, GRP78 and CHOP gene expression was analyzed by quantitative real-time PCR. *, P=0.006 by Student's t-test versus WT heart.
To evaluate cardiac signal transduction in transgenic mice, anesthetized 2×-TRB3 mice were administered 0.1 units/kg body weight insulin by jugular vein injection. After 5 minutes, hearts were excised and left ventricular lysates obtained. Analysis of Akt1/2 and GSK-3β phosphorylation status by immunoblotting revealed that insulin-stimulated signaling pathway activation was reduced in 2×-TRB3 hearts (Figure 3B).
Morphometric analysis of 4-month-old and 12-month-old 2×-TRB3 hearts demonstrated that these mice do not spontaneously develop cardiac hypertrophy (Online Table I). The biventricular weight-to-body weight ratio was nearly identical in 4-month-old 2×-TRB3 mice (4.02 ± 0.14 mg/g) when compared to wild type littermates (4.01 ± 0.14 mg/g, P=0.871). Similarly, the biventricular weight-to-body weight ratio was similar in 12-month-old 2×-TRB3 mice (3.78 ± 0.17 mg/g) when compared to wild type littermates (4.00 ± 0.25 mg/g, P=0.477). Similar relationships were observed for the left ventricular-to-body weight ratios (Online Table I).
Echocardiographic evaluation of awake 12-week-old 2×-TRB3 and 4×-TRB3 mice demonstrated that they had normal left ventricular contractile function when compared to wild type mice as measured by fractional shortening (Online Table II). Furthermore, the LV mass index, which normalizes calculated LV mass by body weight, did not show a significant difference between 12-week-old 2×-TRB3, 4×-TRB3 and wild type mice (Online Table II).
Although 2×-TRB3 transgenic mice do not spontaneously develop cardiac hypertrophy and have normal left ventricular contractile function, we evaluated genetic markers of cardiac stress in these animals. Cardiac ventricular tissue was obtained from wild type and 2×-TRB3 12-16-week-old mice and was examined by quantitative real-time PCR for the expression of atrial natriuretic factor (ANF), β-myosin heavy chain (β-MHC) and sarcoplasmic/endoplasmic reticulum calcium-ATPase 2 (SERCA2) (Figure 3C). The expression of ANF was increased 5-fold (P=0.11), the expression of β-MHC was increased by 4.7-fold (P=0.006), and the expression of SERCA2 was unchanged in 2×-TRB3 cardiac tissue (Figure 3C). The expression of the ER stress marker genes GRP78 and CHOP were unchanged in 2×-TRB3 cardiac tissue (Figure 3C).
Given the ability of TRB3 overexpression to antagonize insulin-stimulated cardiac Akt signaling, we evaluated cardiac metabolism in transgenic mice. An ex vivo working heart analysis was performed on 2×-TRB3 and 4×-TRB3 12-week-old mice and their nontransgenic littermates. Peak systolic pressure, cardiac output and cardiac work levels were similar between wild type and transgenic genotypes of mice, confirming the echocardiographic finding that transgenic mice have normal cardiac systolic function at baseline (Online Tables III and IV). Analysis of cardiac metabolism revealed that both 2×-TRB3 and 4×-TRB3 mice had significantly reduced glucose oxidation rates, but normal palmitate oxidation rates when compared to nontransgenic littermates (Figure 4A, B). These results demonstrate that overexpression of TRB3 in heart inhibits glucose metabolism.
Figure 4.

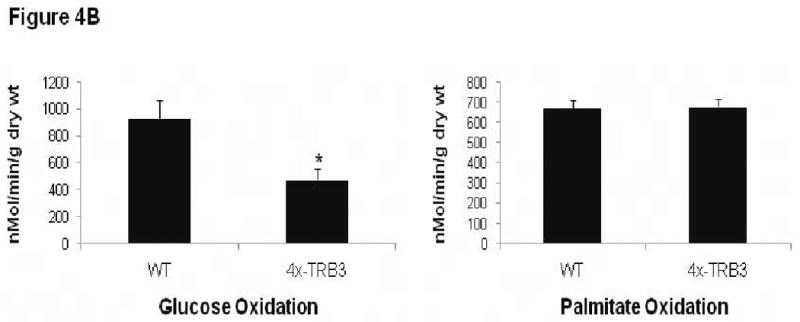
Altered cardiac metabolism in 2×- and 4×-TRB3 transgenic mice determined by ex vivo working heart analysis. A. Analysis of cardiac glucose and palmitate oxidation rates in 2×-TRB3 transgenic hearts in the ex vivo working mode. 12-week-old 2×-TRB3 mice (n=5) and their nontransgenic wild type (WT) littermates (n=7) were used for these studies. Hearts were isolated from mice and were analyzed ex vivo in working mode. *, P=0.048 by Student's t-test versus WT heart. B. Analysis of cardiac glucose and palmitate oxidation rates in 4×-TRB3 transgenic hearts in the ex vivo working mode. 12-week-old 4×-TRB3 mice (n=4) and their nontransgenic wild type (WT) littermates (n=6) were used for these studies. Hearts were isolated from mice and were analyzed ex vivo in working mode. *, P=0.040 by Student's t-test versus WT heart.
Evaluation of remodeling after experimental myocardial infarction in 2×-TRB3 mice
The cardiac ER stress response, including TRB3 gene induction, is activated in response to myocardial ischemia. TRB3 is an Akt inhibitor and we previously demonstrated that akt2-/- mice exhibit increased pathological cardiac remodeling after experimental MI.22 To determine whether cardiac-specific overexpression of TRB3 would sensitize myocardium to pathological cardiac remodeling, we performed experiment MI surgery with 2×-TRB3 mice and their wild type littermates at 12 weeks of age. One day after MI surgery, mice were evaluated by transthoracic echocardiography and this confirmed that the initial infarct sizes were similar for the two genotypes (Figure 5A). Seven days after MI surgery, the hearts were isolated for histological examination of scar area and cardiac myocyte apoptosis.
Figure 5.




Pathological cardiac remodeling is increased after myocardial infarction in 2×-TRB3 mice. At 12 weeks of age, 2×-TRB3 mice (n=9) and their nontransgenic littermates (n=13) were subjected to experimental myocardial infarction surgery by ligation of the left anterior descending coronary artery. A. Evaluation of the initial infarct size by transthoracic echocardiography one day after surgery. The initial infarct size was evaluated in 2×-TRB3 and nontransgenic mice one day after surgery. The segmental wall motion score index (SWMSI) was employed to evaluate initial infarct size. B. Increased LV scar area in 2×-TRB3 mice 7 days after MI. LV sections from 2×-TRB3 and nontransgenic littermates were stained with Masson's trichrome to determine infarct size. C. Computerized analysis of LV infarct size in 2×-TRB3 (n=9) and nontransgenic littermate (n=13) mice determined 7 days after MI. *, P=0.009 versus nontransgenic littermates by Student's t-test. D. Increased apoptotic cardiac myocytes in infarct border zone of 2×-TRB3 mice. LV sections from 2×-TRB3 and nontransgenic littermates obtained 7 days after MI were analyzed by TUNEL in the infarct border zones (defined as one-quarter circumference and either side of the scar edge). *, P=0.012 versus nontransgenic LV sections by Rank Sum test.
Histological examination of LV sections obtained 7 days after MI surgery showed that the infarct scar area was significantly greater in 2×-TRB3 mice when compared to wild type littermates (Figure 5B, C). Indeed, the scar area as a percentage of total LV area was 25.0 ± 1.8% in wild type mice, but was increased to 35.9 ± 3.7% in 2×-TRB3 mice (P=0.009). Furthermore, cardiac myocyte apoptosis in the infarct border zone was evaluated by TUNEL, and this demonstrated that the number of apoptotic cardiac myocytes was significantly greater in 2×-TRB3 mice (Figure 5D). Therefore, 2×-TRB3 mice are sensitized to the development of pathological cardiac remodeling after MI.
Discussion
ER stress occurs in response to nascent protein misfolding in the ER lumen and a complex variety of cellular responses to this stress initially act to promote ER recovery.1,2 If the initial mechanisms that are designed to promote ER functional recovery are inadequate, however, prolonged activation of stress responses lead to apoptosis of the cell. TRB3 is a pseudokinase that inhibits the activity of several signal transduction cascades, including the Akt pathway and the ATF4/CHOP pathway.8-10 Inhibition of all Akt signaling impacts the cellular response to insulin and other growth factors, in part, by reducing glucose uptake and metabolism, inhibiting protein synthesis, and promoting apoptosis.
In this work we investigated the role of TRB3 in the UPR of cardiac myocytes. We found that various chemical inducers of ER stress, including tunicamycin and thapsigargin, strongly up-regulated TRB3 expression in cultured cardiac myocytes. Furthermore, nutrient deprivation and hypoxia also induced the expression of TRB3 in cardiac myocytes. Inducers of ER stress blocked Akt activation in cultured cardiac myocytes. Experimental myocardial infarction, a physiological stimulus of ER stress, led to increased TRB3 expression in the infarct border zone of the left ventricle after 24 hours. In previous work, another group performed a comprehensive analysis of gene expression at several time points after myocardial infarction in mice.24 Inspection of the microarray data from that study shows that TRB3 and CHOP mRNA levels rose at 12 and 24 hours after MI in the ischemic/infarcted area. This rise in TRB3 was exactly paralleled by increased CHOP mRNA levels. To determine the physiological consequences of increased TRB3 gene expression in vivo, we generated transgenic mice with cardiac-specific overexpression of TRB3 and showed that these mice had reduced cardiac insulin-stimulated signal transduction and reduced cardiac glucose oxidation rates. Furthermore, we demonstrated that these transgenic mice were sensitized to pathological cardiac remodeling after myocardial infarction.
Cardiac insulin signaling results in Akt2 activation that is known to promote GLUT4 transporter translocation to the plasma membrane resulting in increased glucose uptake by cardiac myocytes.22 Insulin signaling also leads to Akt2-mediated inactivation of the FoxO1 transcription factor that regulates cardiac metabolism.22 The UPR-mediated induction of TRB3 is predicted to block insulin-mediated glucose uptake and oxidation. It is not entirely clear why this might be beneficial to promote ER recovery although it is possible that reduced glucose oxidation might generate less reactive oxygen species or other toxins. Alternatively, TRB3 induction may be a purely deleterious aspect of the UPR after ER recovery is deemed to be impossible. TRB3 induction as a component of the UPR is predicted to lower the apoptotic threshold of cardiac myocytes by blocking Akt activity.
Despite the fact that the acute UPR is designed to reduce protein synthesis, and that TRB3 is induced as a component of the UPR, we observed that 2×-TRB3 and 4×-TRB3 transgenic mice did not exhibit cardiac atrophy at any time point. Therefore, it appears that TRB3 induction is not a significant mechanism to reduce protein synthesis in the myocardium during the UPR.
Since the UPR promotes cellular survival through reparative mechanisms in its early phase, and promotes cell death through apoptosis in its later phases, it is difficult to predict the overall consequences of antagonizing the overall ER stress pathway in cardiac tissue. Previous work suggests that several factors induced in response to ER stress are cardioprotective.25,26 For example ATF6 and regulator of calcineurin 1 (RCAN1) are cardioprotective factors that are induced in response to cardiac ER stress.25,26 The TRB3 knockout mouse has no obvious phenotype at baseline, but this is not entirely surprising given that TRB3 is largely an inducible factor in the ER stress pathway.27 Further work is required to delineate the physiological effects of various aspects of the UPR in the heart.
Novelty and Significance.
What is known?
The intracellular Akt protein kinases regulate cardiac myocyte growth, survival and glucose metabolism.
Cardiac myocyte endoplasmic reticulum (ER) stress occurs in response to several provocative stimuli that result in protein misfolding within the ER.
ER stress in cardiac myocytes and other cell types leads to activation of a complicated response mechanism that initially promotes ER recovery, but if unsuccessful, promotes cell death.
What new information does this article contribute?
Activation of the ER stress response in cultured cardiac myocytes and murine myocardium results in increased expression of the Tribbles 3 (TRB3) pseudokinase.
Activation of the ER stress response in cultured cardiac myocytes inhibits Akt protein kinase activity in a TRB3-dependent fashion.
Cardiac-specific overexpression of TRB3 in transgenic mice results in abnormal cardiac glucose metabolism and insulin signaling, and also results in increased sensitivity to ER stress pathway-mediated cardiac myocyte death.
Summary
Akt protein kinases play a critical role in cardiac myocyte growth, survival and metabolism. Many proteins are produced by ribosomes that coat the ER, and ER stress occurs when lumenal proteins are misfolded. Studies of ER stress in non-cardiac cells have shown that ER stress results in the induction of TRB3, a pseudokinase that inhibits Akt family members. This study demonstrated that cardiac ER stress leads to TRB3 induction, Akt inhibition and cardiac myocyte death. In response to various stimuli that are known to promote ER stress, cultured cardiac myocytes exhibited reduced Akt activity dependent on increased expression of TRB3. Experimental myocardial infarction in mice resulted in the induction of TRB3 and other ER stress markers in the infarct border zone. Cardiac-specific overexpression of TRB3 in mice was associated with reduced glucose metabolism and insulin signaling. Furthermore, TRB3 overexpression sensitized animals to pathological cardiac remodeling after myocardial infarction, with increased apoptosis in the border zone. We conclude that myocardial infarction results in cardiac ER stress with induction of TRB3, and that TRB3 antagonizes Akt activity and promotes cell death. Agents that inhibit TRB3 expression or activity may lead to reduced pathological cardiac remodeling in patients.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (HL61567, HL057278, HL076670, and HL091913) and The Longer Life Foundation (A.J. Muslin). B.J. DeBosch was supported by the Cardiovascular Physiology Training Grant T32-HL07873.
Non-standard Abbreviations
- ANF
atrial natriuretic factor
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- CHOP
C/EBP homologous protein
- GRP78
glucose regulated protein 78
- ER
endoplasmic reticulum
- βMHC
β-myosin heavy chain
- SERCA2
sarcoplasmic/endoplasmic reticulum calcium-ATPase 2
- TRB3
Tribbles 3
- UPR
unfolded protein response
Footnotes
Disclosures
None.
References
- 1.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 3.Thuerauf DJ, Marcinko M, Hude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 4.Mao W, Iawai C, Liu J, Sheu SS, Fu M, Liang CS. Darbepoetin alfa exerts cardioprotective effect in autoimmune cardiomyopathy via reduction of ER stress and activation of the PI3K/Akt and STAT3 pathways. J Mol Cell Cardiol. 2008;45:250–260. doi: 10.1016/j.yjmcc.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li SY, Ren J. Cardiac expression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: Role of insulin signaling and ER stress. J Mol Cell Cardiol. 2008;44:992–1001. doi: 10.1016/j.yjmcc.2008.02.276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J Biol Chem. 2004;279:49420–49429. doi: 10.1074/jbc.M407700200. [DOI] [PubMed] [Google Scholar]
- 7.Hosoi T, Hyoda K, Okuma Y, Nomura Y, Ozawa K. Akt up- and down-regulation in response to endoplasmic reticulum stress. Brain Res. 2007;1152:27–31. doi: 10.1016/j.brainres.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 8.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 9.Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 10.Jousse C, Deval C, Maurin AC, Parry L, Chérasse Y, Chaveroux C, Lefloch R, Lenormand P, Bruhat A, Fafournoux P. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J Biol Chem. 2007;282:15851–15861. doi: 10.1074/jbc.M611723200. [DOI] [PubMed] [Google Scholar]
- 11.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ord D, Ord T. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochem Biophys Res Commun. 2005;330:210–218. doi: 10.1016/j.bbrc.2005.02.149. [DOI] [PubMed] [Google Scholar]
- 13.Corcoran CA, Luo X, He Q, Jiang C, Huang Y, Sheikh MS. Genotoxic and endoplasmic reticulum stresses differentially regulate TRB3 expression. Cancer Biol Ther. 2005;4:1063–1067. doi: 10.4161/cbt.4.10.2205. [DOI] [PubMed] [Google Scholar]
- 14.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bowers AJ, Skully S, Boylan JF. SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene. 2003;22:2823–2835. doi: 10.1038/sj.onc.1206367. [DOI] [PubMed] [Google Scholar]
- 17.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–H829. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 19.Azfer A, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Physiol Heart Circ Physiol. 2006;291:H1411–H1420. doi: 10.1152/ajpheart.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 21.Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38:617–623. doi: 10.1016/j.yjmcc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 22.DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem. 2006;281:32841–32851. doi: 10.1074/jbc.M513087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau JM, Jin X, Ren J, Avery J, DeBosch BJ, Treskov I, Lupu TS, Kovacs A, Weinheimer C, Muslin AJ. The 14-3-3tau phosphoserine-binding protein is required for cardiomyocyte survival. Mol Cell Biol. 2007;27:1455–1466. doi: 10.1128/MCB.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harpster MH, Bandyopadhyay S, Thomas DP, Ivanov PS, Keele JA, Pineguina N, Gao B, Amarendran V, Gomelsky M, McCormick RJ, Stayton MM. Earliest changes in the left ventricular transcriptome postmyocardial infarction. Mamm Genome. 2006;17:701–715. doi: 10.1007/s00335-005-0120-1. [DOI] [PubMed] [Google Scholar]
- 25.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 26.Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA, Glembotski CC. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283:14012–14021. doi: 10.1074/jbc.M709776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okamoto H, Latres E, Liu R, Thabet K, Murphy A, Valenzeula D, Yancopoulos GD, Stitt TN, Glass DJ, Sleeman MW. Genetic deletion of Trb3, the mammalian Drosophila tribbles homolog, displays normal hepatic insulin signaling and glucose homeostasis. Diabetes. 2007;56:1350–1356. doi: 10.2337/db06-1448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.