

Abstract
Statins are widely used drugs for the treatment of hypercholesterolaemia. A number of recent studies have suggested that statins also have pleiotropic effects on immune responses and statins have proven to be effective in the treatment of autoimmune diseases in animal models. Foxp3+ T regulatory cells are a unique subset of CD4+ T cells that mediate immunosuppression. Foxp3+ T cells develop in the thymus, but can also be induced in peripheral sites in the presence of transforming growth factor-β (TGF-β). We demonstrate here that simvastatin blockade of the mevalonate pathway can mediate induction of mouse Foxp3+ T cells and that simvastatin can synergize with low levels of TGF-β to induce Foxp3+ T cells. The effects of simvastatin are secondary to a blockade of protein geranylgeranylation, are mediated at late time-points after T-cell activation, and are associated with demethylation of the Foxp3 promoter. One major effect of simvastatin was inhibition of the induction of Smad6 and Smad7, inhibitory Smads that inhibit TGF-β signalling. Our results suggest that one mechanism responsible for the immunosuppressive effects of statins is the ability to promote the generation of Foxp3+ T regulatory cells.
Keywords: Foxp3, geranylgeranylation, simvastatin, Smad6, Smad7, transforming growth factor-β, T regulatory cells
Introduction
Regulatory T cells (Tregs) are a unique subset of CD4+ T cells that suppress autoreactivity and maintain immunological homeostasis. Natural Tregs (nTregs) develop in the thymus whereas induced regulatory T cells (iTregs) differentiate in peripheral sites.1In vitro differentiation of iTregs is mediated by T-cell receptor (TCR) -mediated activation together with transforming growth factor-β (TGF-β) and interleukin-2 (IL-2).2 Both types of Tregs constitutively express Foxp3 [forkhead (FKH)-winged helix family protein of transcription regulators], which is the master gene mediating the immunosuppressive function of Tregs.3,4 It is likely that the induction of Foxp3 expression in Tregs with TGF-β is secondary to activation of the enhancer and promoter regions of the Foxp3 gene, as well as being secondary to regulation of histone acetylation and DNA demethylation of the Foxp3 gene.5,6 The role of TGF-β in Treg induction in vivo is unclear because the optimal concentrations of TGF-β used to induce Foxp3 expression in vitro are unlikely to be present in vivo.
Statins are widely used drugs for the treatment of hypercholesterolaemia. They function as competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), which is the rate-determining enzyme of the mevalonate pathway. More recent studies have also suggested that statins can mediate immunosuppressive functions and have proven effective in the treatment of autoimmune diseases or graft-versus-host disease in animal models.7–9 A number of mechanisms have been proposed to explain the immunosuppressive effects of the statins including inhibition of antigen presentation by inhibition of the induction of MHC class II expression, and blocking of T helper type 1 (Th1) cell differentiation by inhibiting TCR-specific phosphorylation of Stat4 in Th1 differentiation.7,10 Suppression of Th1 differentiation by statins in the experimental autoimmune encephalomyelitis mouse model was mediated by inhibition of protein geranylgeranylation, one of the main downstream metabolic branches of the mevalonate pathway.10 Statins may also interfere with the interaction between T cells and antigen-presenting cells by inhibiting the functions of the β-integrin, lymphocyte function-associated antigen 1 (CD11a/CD18).11
Although direct effects of statins on Treg function have not been reported, a number of studies have suggested that Tregs play an important role in the control of pathology in atherosclerosis and atherosclerotic plaques have been reported to contain a lower percentage of Foxp3+ Tregs compared with normal tissue.12,13 Recently, a study has reported that the number of Foxp3+ T cells is elevated in the peripheral blood mononuclear cellsof patients who take statins.14 However, it is still unclear if statins directly increase the number of Foxp3+ cells or indirectly modulate the trafficking of Tregs into blood or to sites of immunopathology.
Taken together, the previous studies raise the possibility that blockade of the mevalonate pathway by statins might not only be critical for the inhibition of Th1 differentiation, but also may modulate the function of nTregs or promote the differentiation of iTregs. Here, we have evaluated the effects of simvastatin blockade of the mevalonate pathway on the induction of Foxp3-expressing iTregs in vitro. We demonstrate that simvastatin itself can mediate induction of Foxp3+ T cells and can also synergize with low levels of TGF-β in the induction of functional Foxp3+ Tregs. The effects of simvastatin are secondary to a blockade of protein geranylgeranylation, are mediated 24 hr after TCR stimulation, and are associated with TCR-specific DNA demethylation of the Foxp3 promoter and TCR-specific induction of Smad6 and Smad7 proteins. The implications of these results for the use of simvastatin as an immunosuppressive drug will be discussed.
Materials and methods
Mice
DO11.10 TCR transgenic RAG2 deficient (−/−), 5CC7 TCR transgenic RAG2−/−, and B10.A mice were obtained from Taconic Farms (Germantown, NY). The Foxp3-GFP-Knock-in (Foxp3gfp) mice were provided by Dr V. Kuchroo (Harvard Medical School, Boston, MA). All the mice were maintained under pathogen-free conditions in the National Institute of Allergy and Infectious Disease animal facility. Mice were used between 4 and 8 weeks of age.
Reagents
Recombinant human IL-2 and recombinant mouse TGF-β were purchased from Peprotech (Rocky Hill, NJ). Simvastatin, geranylgeranyl pyrophosphate and farnesyl pyrophosphate were purchased from Alexis Biochemicals (Plymouth Meeting, PA) and mevalonate, FTI-276 (farnesyl transferase inhibitor), and GGTI-2133 (geranylgeranyltransferase I inhibitor) were purchased from Sigma (St Louis, MO). Allophycocyanin-conjugated anti-Foxp3 (FJK-16s), fluorescein isothiocyanate-conjugated anti-CD4 (L3T4), anti-CD3ε antibody (145-2C11) and anti-CD28 antibody were purchased from eBioscience, Inc. (San Diego, CA). Anti-phospho-Smad3 antibody and anti-Smad3 antibody were purchased from Cell Signaling Technology (Danvers, MA). Anti-Smad6/7 (N-19) antibody was purchased from Santacruz Biotechnology (Santa Cruz, CA). For neutralization of TGF-β, anti-TGF antibody (1D11) was obtained from R&D Systems (Minneapolis, MN).
Cell isolation and in vitro culture
CD4+ T cells were purified from mouse lymph nodes or spleen using magnetic beads (Miltenyi Biotec, Auburn, CA). Foxp3gfp CD4 T cells were isolated by fluorescence-activated cell sorting (FACSAria). Foxp3+ Tregs were induced by stimulating CD4+ Foxp3− T cells (1 × 106) with plate-bound anti-CD3 (1–2 μg, 145-2C11) and plate-bound anti-CD28 antibody (1–2 μg) in the presence of a given concentration of TGF-β1 and/or simvastatin for 72 hr in RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), l-glutamine (2 mm), HEPES (10 mm), non-essential amino acids (0.1 mm), sodium pyruvate (1 mm) and 2-mercaptoethanol (50 μm). In some studies, responder cells were labelled with 2 μm carboxyfluorescein succinimidyl ester (CFSE) for 8 min at room temperature before induction of Foxp3 expression.
In vitro suppression assays were performed by first inducing Foxp3 expression in purified CD4+ Foxp3− T cells isolated from Foxp3gfp mice. Three days after activation, converted Foxp3+ cells were isolated from activated cell mixtures using FACS sorting, and then mixed with CD4+Foxp3− responder cells, γ-irradiated T-depleted splenocytes, and soluble anti-CD3 (1 μg/ml) for 4–5 days. Cell proliferation was assayed by [3H]thymidine uptake as previously described.2
Flow cytometric analysis
To measure intracellular staining of Foxp3, cultured cells were washed with FACS staining buffer (2% fetal bovine serum in phosphate-buffered saline) twice, fixed in 4% paraformaldehyde solution (electron microscope-grade) for 10 min, and then permeabilized in Triton X-100 solution overnight. Permeabilized cells were stained with fluorescent conjugated anti-Foxp3 antibody diluted in permeabilization buffer for 3 hr and then washed in permeabilization buffer twice. Acquisition of FACS data was performed with a FACSCalibur (Beckton-Dickinson, San Jose, CA) and FlowJo software (Tree star, Ashland, OR) was used for FACS analysis. All plots are drawn on standard log scale.
Western blotting
Cells pellets were incubated in modified RIPA buffer (10 mm Tris–HCl, 150 mm NaCl, 0.5% Nonidet P-40, 0.1% deoxycholate, and 1 × protease inhibitor cocktail, Roche, Indianapolis, IN) on ice for 20 min. Protein was quantified using the Bradford method (Pierce, Rockford, IL). Protein samples (4–6 μg) were run on 4–12% bis-tris sodium dodecyl sulphate–polyacrylamide gel electrophoresis (Invitrogen, Carlsbad, CA), and then transferred onto polyvinylidene fluoride membranes (Invitrogen). Non-fat dried milk solution (5% in Tris-buffered saline with Tween-20) was used for blocking. Blocked membranes were incubated with anti-Smad3 (1 : 1000), anti-Smad6/7 (1 : 4000) overnight at 4°. Anti-rabbit immunoglobulin G antibody-HRP (1 : 10 000) was used as a secondary antibody for 2 hr at room temperature. Western bands were visualized using an enhanced chemiluminescence detection kit (West-Pico, Pierce). Relative amounts of loading proteins were normalized to the levels of tubulin on the same membrane.
Quantitative reverse transcription–polymerase chain reaction
Total RNA from CD4+ T cells was isolated using an RNeasy mini-prep kit (Qiagen, Valencia, CA). Total RNA (1 μg) was reverse transcribed to first-strand complementary DNA by incubation with oligo-dT primer for 40 min in the presence of SuperScript II reverse transcriptase (Invitrogen). For measuring the messenger RNA level of Foxp3, Taqman Gene Expression Assay (Applied Biosystems, Foster City, CA) was used. Quantitative polymerase chain reaction (PCR) was performed on a 7900HT sequence detection system (Applied Biosystems).
DNA methylation analysis
All of the protocols and primer design for the DNA methylation analysis of the Foxp3 promoter region were described previously.6 Briefly, genomic DNA was purified using a DNeasy mini-prep kit (Qiagen). Genomic CpGs of the Foxp3 promoter region were converted with bisulphite using the MethylDetector Kit (Active Motif, Carlsbad, CA). Bisulphite-converted CpG of the Foxp3 promoter region was PCR amplified with nested primers (outer primer forward, 5′-TTTTGTGATTTGATTTATTTTTTTT-3′; outer primer reverse, 5′-ATACTA-ATAAACTCCTAACACCCACC-3′; inner primer forward, 5′-TATATTTTTAGATGATTTGTAAAGGGTAAA-3′; and inner primer reverse, 5′-ATCAACCTAACTTATAAAAAACTACCACAT-3′). The PCR products were cloned using a TOPO TA cloning kit (Invitrogen). Sequencing of PCR clones was performed by Macrogen USA Corp (Rockville, MD).
Results
Simvastatin itself and synergistically with TGF-β induces Foxp3 expression
To analyse the potential direct effects of statins on the induction of Foxp3+ Treg cells in vitro, we used a well-characterized system2 in which purified CD4+ T cells from TCR transgenic RAG−/− mice that are free of contaminating Foxp3+ T cells are stimulated in vitro with plate-bound anti-CD3/CD28 in the presence and absence of TGF-β. Addition of simvastatin alone resulted in the induction of Foxp3 expression in 5–10% of the T cells. Simvastatin and low concentrations of TGF-β synergized in the induction of Foxp3 expression. Not only was the percentage of Foxp3-expressing cells increased in the presence of simvastatin, but the mean level of expression of Foxp3 as measured by the mean fluorescence intensity of the positive cells was also increased (Fig. 1a). Most importantly the synergistic effects of simvastatin were completely blocked by the addition of mevalonate, a downstream metabolite of HMGCR. The ability of simvastatin to induce Foxp3 expression alone or in combination with TGF-β was dependent on both the presence of a TCR signal and IL-2 (data not shown).
Figure 1.
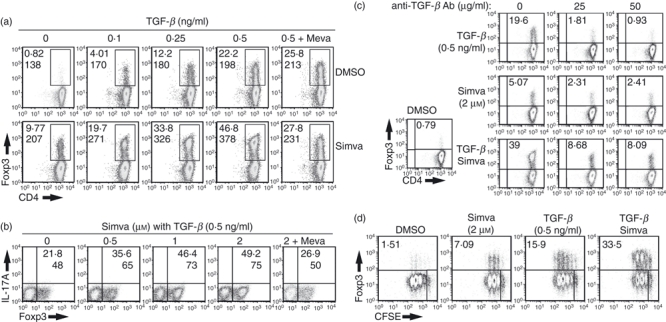
Simvastatin induces Foxp3 expression in the absence or presence of transforming growth factor-β (TGF-β). (a) CD4+Foxp3− T cells isolated from T-cell receptor (TCR) transgenic RAG−/− mice were stimulated for 72 hr with plate-bound anti-CD3/CD28 (4 and 2 μg/ml, respectively), 100 U/ml interleukin-2 (IL-2), in the presence of vehicle alone, TGF-β, simvastatin (2 μm), separately or together. Mevalonate (200 μm) was added to some cultures as indicated. Expression of Foxp3 was measured by intracellular staining. (b) Same protocol as in panel (a) except that a neutralizing anti-TGF-β monoclonal antibody (mAb) was added at the concentrations indicated. (c) Same protocol as in panel (a), except that the concentration of simvastatin was varied. (d) The CD4+ Foxp3− T cells were labelled with carboxyfluorescein succinimidyl ester (CFSE), stimulated for 72 hr under the in vitro activating condition used in (a). CFSE dilution and Foxp3 expression were measured by fluorescence-activated cell sorting analysis. The numbers in black indicate the percentage of Foxp3+ cells and numbers in italics represent the mean fluorescence intensity (MFI) of the Foxp3+ cells. These results are representative of four different experiments.
One possible explanation for the induction of Foxp3 expression by simvastatin alone is that the drug induced the production of TGF-β from the T cells or synergized with the low levels of TGF-β present in the fetal calf serum used in the cell cultures. We therefore attempted to block any T-cell-derived or serum-derived TGF-β by adding a high concentration of a neutralizing anti-TGF-β monoclonal antibody (mAb) to the Foxp3 induction cultures. As a positive control, we tested the ability of this mAb to neutralize the biological activity of 0.5 ng/ml of exogenous TGF-β. When 50 μg of the mAb was added to the cultures in the presence of 0.5 ng/ml of TGF-β, the inducing effects of the TGF-β on Foxp3 expression were almost completely abolished. However, this same concentration of mAb reduced by only 50% the inducing effects of simvastatin alone and only partially abolished the synergistic effects of simvastatin in the presence of TGF-β. We conclude that some of the effects of simvastatin on Foxp3 induction are likely to be TGF-β-independent. Synergistic enhancement of Foxp3 expression by simvastatin occurred only at suboptimal concentrations of TGF-β (0.1–1 ng/ml), and was not observed at the optimal concentration of TGF-β (5 ng/ml) used in our previous studies2 (data not shown).
The synergistic effects of simvastatin were observed at concentrations as low as 0.5 μm and were maximal at a concentration of 2 μm (Fig. 1c). As studies of the effects of statins in other experimental models have suggested that the actions of this class of drugs are related to their anti-proliferative and pro-apoptotic effects on both T cells and tumours, it was important to rule out that the capacity of simvastatin to induce Foxp3 expression was not secondary to an inhibition of responder T-cell proliferation. However, simvastatin either alone or in combination with TGF-β had only a slight inhibitory effect on the proliferation of CFSE-labelled CD4+ T cells stimulated with anti-CD3/CD28 in our induction cultures (Fig. 1d). Furthermore, the addition of simvastatin did not induce apoptosis and had no effect on the cell cycle of Foxp3− T cells (Fig. S1). Hence, the effects of simvastatin are directly mediated by enhancing the conversion of Foxp3− to Foxp3+ T cells.
Foxp3+ T cells induced by the combination of simvastatin/TGF-β are suppressive in vitro
To address whether Foxp3+ T cells induced in vitro in the presence of simvastatin and TGF-β were suppressive, Foxp3− T cells were isolated from the spleen and lymph nodes of Foxp3gfp mice and activated with plate-bound CD3/CD28 antibody in the presence of TGF-β alone or the combination of simvastatin and TGF-β. The induced GFP+ cells were sorted by FACS, added to Foxp3− responder cells and T-depleted spleen cells as antigen-presenting cells, and were stimulated with soluble anti-CD3. The Foxp3+ cells induced in the presence of simvastatin/TGF-β were as suppressive as the Foxp3+ T cells induced with TGF-β alone (Fig. 2). The addition of simvastatin therefore did not modulate the function of the induced Foxp3+ T cells.
Figure 2.

Simvastatin-induced Foxp3-expressing CD4+ T cells exhibit normal suppressive function. Fluorescence-activated cell-sorted CD4+ GFP− T cells were isolated from Foxp3gfp mice, stimulated with plate-bound CD3/CD28 antibodies and interleukin-2 (IL-2) in the presence of transforming growth factor-β (TGF-β) alone or with TGF-β and simvastatin. After the initial 72 hr of culture, converted GFP+ cells were re-sorted and mixed with CD4+ Foxp3− responder cells, T-cell-depleted splenocytes, and soluble anti-CD3 antibody at the indicated cell : cell ratios (x-axis). [3H]Thymidine incorporation was assayed 96 hr after stimulation. Results are presented as counts/min (CPM) ± SEM of triplicate cultures. Similar results were observed in two other experiments.
The synergistic effects of simvastatin on the induction of Foxp3 expression are mediated primarily by an inhibition of protein geranylgeranylation
Simvastatin blocks all downstream pathways of the mevalonate pathway including cholesterol biosynthesis, synthesis of farnesyl bisphosphate, and geranylgeranyl bisphosphate (Fig. 3a). To determine which downstream pathway primarily mediates the synergistic effects of simvastatin on Foxp3 induction, we added simvastatin or downstream pathway-specific inhibitors together with TGF-β to the Foxp3 induction assay (Fig. 3b). As shown above, simvastatin enhanced the induction of Foxp3-expressing cells in the presence of a low concentration of TGF-β. In contrast, the addition of an inhibitor of farnesylation had no effect on the induction of Foxp3 expression whereas the inhibitor of geranylgeranylation mimicked the effects of simvastatin. This result clearly demonstrates that the synergistic effects of simvastatin on the induction of Foxp3 are secondary to inhibition of protein geranylgeranylation.
Figure 3.
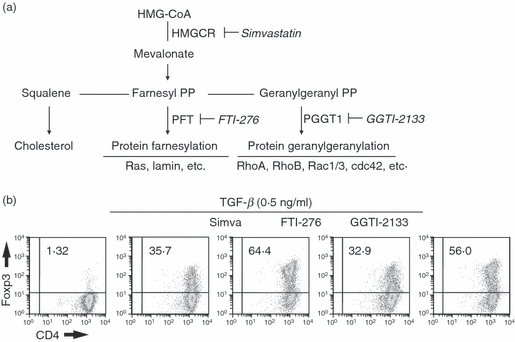
The synergistic effects of simvastatin on transforming growth factor-β (TGF-β) -induced Foxp3 induction are mediated by blocking protein geranylgeranylation. (a) Schematic diagram of the mevalonate pathway is shown. Names in bold indicate metabolic enzymes, names in italics are specific inhibitors of those enzymes. (b) CD4+ Foxp3− T cells were stimulated as in Fig. 1 in the presence of simvastatin or inhibitors of the downstream branches of the mevalonate pathway (20 μm of FTI-276 or 20 μm of GGTI-2133) for 72 hr. Foxp3 expression was measured by intracellular staining. Data are representative of three other experiments.
The synergistic effects of simvastatin on the induction of Foxp3+ Tregs are mediated at a late stage of the T-cell activation cascade
We performed a kinetic study as an initial approach to the analysis of the mechanisms by which simvastatin enhances the induction of Foxp3+ Tregs. When analysed 24 hr after T-cell stimulation, cells cultured with simvastatin alone did not express Foxp3 and no differences were observed, at this time-point between the percentage of Foxp3+ T cells induced by TGF-β and the percentage induced by the combination or TGF-β and simvastatin (Fig. 4a). In contrast, after 48 and 72 hr of stimulation, the synergistic effects of simvastatin on the induction of Foxp3+ T cells were apparent. To further understand the delayed inducing effects of simvastatin, we added simvastatin at different time-points after the initiation of TCR stimulation with TGF-β or added simvastatin at culture initiation and then blocked its action at different time-points by the addition of mevalonate. All cultures were analysed for the expression of Foxp3+ cells after 72 hr of stimulation (Fig. 4b). The maximal inducing effects of simvastatin could be observed even when it was added as late as 24 hr after the initiation of the cultures, but its synergistic activity was completely abolished when it was added after 48 hr (Fig. 4b). Similarly, the neutralization of the effects of simvastatin with mevalonate was only observed when mevalonate was added during the first 24–32 hr of the culture. This study suggested that simvastatin mediated its activity between 24 and 48 hr after T-cell activation. We confirmed this result by adding simvastatin at 24 hr and neutralizing its effects with mevalonate at 48 hr (Fig. 4c). The magnitude of the enhancement of the induction of Foxp3-expressing cells was similar in cells pulse-exposed to simvastatin only between 24 and 48 hr after activation to that in cells that had been exposed for the entire 72-hr culture period.
Figure 4.

The synergistic effects of simvastatin on transforming growth factor-β (TGF-β) -induced Foxp3 induction are mediated at a late stage of T-cell activation. (a) CD4+ Foxp3− T cells were stimulated with plate-bound anti-CD3/28, interleukin-2 (IL-2), in the presence of TGF-β, simvastatin, or both together. Fluorescence-activated cell sorting analysis of Foxp3 expression was performed at the indicated hours after stimulation. Similar results were observed in one other experiment. (b) CD4+ Foxp3− T cells were stimulated with plate-bound anti-CD3/CD28, IL-2, in the presence of TGF-β alone (0.2 ng/ml, closed squares) or together with simvastatin (2 μm, open squares). After initiation of T-cell stimulation, simvastatin (2 μm, closed symbols) or mevalonate (200 μm, open symbols) were added at the indicated time-points (x-axis). After 72 hr, all cultures were simultaneously harvested and the percentage of Foxp3-expressing cells was determined by intracellular staining. Similar results were observed in one other experiment. (c) CD4+ Foxp3− T cells were stimulated as in Fig. 1(d) in the presence of vehicle alone or TGF-β. Simvastatin (2 μm) was added for the entire 72 hr of culture (continuous) or after 24 hr and then neutralized at 48 hr with mevalonate (200 μm, pulsed). Foxp3 expression was measured by intracellular staining at 72 hr. Similar results were observed in two other experiments.
Effects of simvastatin on Foxp3 transcription and DNA methylation in the Foxp3 promoter
To address whether synergistic action of simvastatin on TGF-β-mediated induction is controlled at the transcriptional level, we assayed the Foxp3 messenger RNA (mRNA) levels in cells treated with TGF-β alone or with the combination of TGF-β and simvastatin (Fig. 5a). Up-regulation of Foxp3 mRNA was observed after 24 hr of culture in the TGFβ only treated group compared to cells cultured with vehicle alone and no enhancement of Foxp3 mRNA was seen in cultures with simvastatin. In contrast, marked enhancement of Foxp3 mRNA levels were seen after 48 and 72 hr in cultures containing both TGF-β and simvastatin, whereas levels of Foxp3 mRNA in cultures with TGF-β alone were slightly diminished.
Figure 5.

Simvastatin increases Foxp3 messenger RNA and inhibits DNA methylation of the Foxp3 promoter in the presence of transforming growth factor-β (TGF-β). (a) CD4+ Foxp3− T cells were stimulated as in Fig. 1(d) in the presence of dimethylsulphoxide, TGF-β (0.5 ng/ml) or simvastatin (2 μm) together. After the indicated times, the cultures were harvested and the total RNA extracted. Foxp3 mRNA levels were determined by quantitative RT-PCR and expression levels normalized to 18S ribosomal RNA. Similar results were observed in one other experiment. (b) Foxp3+ and Foxp3- cells were FACS-sorted from lymph node cells of male Foxp3gfp male mice (FACS profile). The methylation status of each CpG site is displayed as closed circles (methylated) and opened circles (demethylated). Each group of circles is arranged in the order of the position of the CpG sites (number on top) and represents an individual cloned sample. (c) Cultures were stimulated as in (a), and genomic DNA was extracted at 72 hr. Methylation status of the Foxp3 promoter region was analysed by disulphite modified CpG sequencing. Blue bold numbers indicate the percentage of methylated sites in each sample after 72 hr. Foxp3 expression was measured at 72 hr of activation by intracellular staining. Similar results were observed in three other experiments.
This result together with the results of the time–course study strongly suggest that the effects of simvastatin are not related to enhancement of the initial signals induced by TGF-β and raise the possibility that simvastatin might regulate epigenetic control of Foxp3 transcription. Recent studies6,15 have identified two or three CpG islands within the promoter and enhancer regions of the Foxp3 gene that regulate the induction of Foxp3 transcription and the stabilization of Foxp3 expression. We focused on one site in the Foxp3 promoter that contains six CpGs within a 173-base-pair sequence of the mouse Foxp3 promoter that are located close to the proximal transcription start site. To verify if this candidate site is specific for Foxp3 promoter activity, Foxp3− and Foxp3+ CD4+ cells were isolated from Foxp3gfp male mice, and methylation profiles of both were analysed by bisulphite-modified sequence reading. As expected, CpGs in this region of the promoter of Foxp3+ cells were all unmethylated, whereas approximately 50% of CpGs of Foxp3− cells were methylated (Fig. 5b). These data suggest that demethylation of this CpG island of the Foxp3 promoter region correlates with Foxp3 expression. The methylation status of this region was evaluated in Foxp3− T cells that were activated for 72 hr in the presence of TGF-β alone, simvastatin alone, and the combination of TGF-β/simvastatin (Fig. 5c). After 72 hr, 48% and 42% of the CpGs of dimethylsulphoxide-treated or simvastatin-treated cells were methylated, respectively. However, this region in TGF-β-treated cells was less methylated (26%) than in dimethylsulphoxide-treated or simvastatin-treated cells and the lowest level of methylation (16%) was observed in the cells treated with simvastatin/TGF-β. Seventy-two hours after activation, the extent of demethylation correlated well with the level of Foxp3 expression detected by FACS analysis (bottom boxes in Fig. 5c). These results suggest that the synergistic action of simvastatin on TGF-β-mediated induction of Foxp3 may be mediated by co-operative control of methylation of the Foxp3 promoter.
Simvastatin inhibits TCR-mediated Smad6/7 induction in a TGF-β-specific manner
To directly examine the effects of simvastatin on TGF-β-mediated signal transduction, we measured phosphorylation of Smad3. Significant phosphorylation of Smad3 was observed 24 hr after activation of cells cultured in the presence of TGF-β, but not simvastatin alone, and the levels of Smad3 phosphorylation were not modulated when the cultures were stimulated with both TGF-β and simvastatin (Fig. 6a). In addition, the total amount of Smad4 was comparable in all treatment groups. The lack of an effect of simvastatin on Smad3 phosphorylation is consistent with its late time of action and raised the possibility that simvastatin might block steps in the negative-feedback regulation of TGF-β signalling. Smad6 and Smad7 are the major inhibitory Smad proteins in the negative feedback regulation of the TGF-β signalling pathway. In contrast to Smad3 phosphorylation, we could not detect Smad7 by Western blot analysis 24 hr after T-cell activation and only low levels of Smad6 were observed. The levels of Smad6/7 increased after 48 hr and were maximal at 72 hr after activation (Fig. 6b). Importantly, when combined with TGF-β, simvastatin markedly inhibited the induction of Smad6 at 48 and 72 hr, and completely blocked Smad7 induction at both 48 and 72 hr. Simvastatin alone also decreased levels of Smad6 and completely blocked Smad7 expression at 72 hr. As TGF-β has been reported to play an important role in Foxp3+ Treg homeostasis,16 we also examined the expression of Smad6/7 in nTregs that were activated under conditions similar to those used in our iTreg induction cultures. Foxp3− and Foxp3+ CD4+ T cells were FACS-sorted from Foxp3gfp mice and activated with anti-CD3/CD28 and IL-2 in the absence or presence of TGF-β for 72 hr (Fig. 6c). Smad6 was only expressed at low levels and Smad7 was not detected in either population in the absence of stimulation. Following stimulation, Smad6/7 could be detected in Foxp3− cells in the presence or absence of TGF-β, whereas Smad6/7 could not be detected in Foxp3+ cells cultured under any conditions. As the expression pattern of Smad6/7 in stimulated nTregs is similar to that seen in TGF-β/simvastatin-generated iTregs, it appears likely that one of the primary mechanisms responsible for the synergistic effects of simvastatin on TGF-β-mediated induction of Foxp3 is the inhibition or down-regulation of Smad6/7 expression.
Figure 6.

Simvastatin has no effect on transforming growth factor-β (TGF-β) -induced phosphorylation of Smad3/4, but blocks TGF-β-induced expression of Smad6 and Smad7. (a) CD4+ Foxp3− T cells were stimulated as in Fig. 1(d) in the presence of dimethylsulphoxide, TGF-β (5 ng/ml), simvastatin (2 μm, Simva) or TGF-β and simvastatin. Proteins were extracted with lysis buffer and analysed by Western blot for phospho-Smad3 (pSmad3) as well as total Smad3 and Smad4. (b) Total proteins from cells stimulated as in (a) were extracted at the indicated times and relative changes of Smad6 and Smad7 expression were determined by Western blotting. (c) CD4+ Foxp3− and CD4+ Foxp3+ cells were isolated from lymph nodes of female Foxp3gfp mice by fluorescence-activated cell sorting. Western blot analysis of the expression of Smad6 and Smad7 was performed on resting cells or following activation for 72 hr with plate-bound anti-CD3 and interleukin-2 in the presence or absence of TGF-β. Similar results were observed in three other experiments.
Discussion
Statins are widely used drugs in the treatment of hypercholesterolaemia and have proven to be extremely useful in the prevention of cardiovascular diseases. Studies since 2000 have also demonstrated that statins have pleiotropic effects on immune responses. They were initially shown to prevent and reverse relapsing and remitting experimental autoimmune encephalomyelitis in the mouse model by inducing a shift from a Th1 to a Th2 cytokine profile.7 Similarly, in acute graft-versus-host disease in the mouse, the effects of statins were mediated through induction of Th2 cells with increased IL-4 production and reduced tumour necrosis factor-α and interferon-γ production.8 Subsequent studies have claimed that statins can act on many distinct cell types in the immune system as well as vascular endothelial cells.17 Most recently, statins have been shown to modulate the production of IL-17 by inducing the expression of suppressors of cytokine signalling (SOCS) 3 and SOCS7 in monocytes resulting in inhibition of the transcription of IL-6 and IL-23 and by inhibiting the transcription factor RORγT in CD4+ T cells.18 Very few studies have addressed the effects of statins on nTregs or on the developments of iTregs in peripheral sites. One study claimed that culture of human peripheral blood mononuclear cells in the presence of atorvastatin, but not mevastatin or pravastatin, increased the number of Foxp3+ T cells and claimed that the effects of atorvastatin were mediated by conversion of Foxp3− to Foxp3+ T cells.14 The results of this study are difficult to interpret because conversion of Foxp3− to Foxp3+ T cells requires that the responsive T cell be stimulated through their TCR and TCR stimulation was not used in this paper.2,19
The goal of our studies was to examine the potential effects of statins on the conversion of mouse Foxp3− T cells to Foxp3+ Tregs. We used an in vitro model system in which highly purified Foxp3− T cells, obtained from TCR transgenic mice on a RAG−/− background, were cultured in the absence of antigen-presenting cells in the presence of a TCR stimulus, CD28-mediated co-stimulation and IL-2. Under these conditions the addition of simvastatin alone had a modest effect on the induction of Foxp3+ T cells that was partially independent of the presence of TGF-β. Importantly, simvastatin exerted a potent synergistic effect on Foxp3 induction when combined with low concentrations of TGF-β. All of the effects of the statin could be reversed by the addition of mevalonate, the product of HMGCR. To address which downstream metabolic pathway is the major target for the synergistic induction of Foxp3 by simvastatin, we added a farnesyltransferase inhibitor or a geranylgeranyltransferase inhibitor instead of simvastatin. No effects of the farnesyltransferase inhibitor were seen in cultures with low doses of TGF-β, whereas the geranylgeranyltransferase inhibitor was as effective as simvastatin in functioning synergistically with TGF-β to induce Foxp3. To rule out the contribution of cholesterol biosynthesis in the synergistic effects of simvastatin, we added squalene, which is a downstream metabolite of cholesterol biosynthesis in cells treated with simvastatin, but squalene failed to reverse the synergistic induction of Foxp3 by simvastatin (data not shown). The major effects of simvastatin on Foxp3 induction involve the geranylgeranylation pathway. Similar conclusions were recently reported by Kagami et al.20
One possible mechanism of action of simvastatin on the induction of Foxp3 might be mediated by epigenetic modulation of the Foxp3 gene. Two CpG islands have been identified in the Foxp3 gene, one in the proximal promoter and the second in the first intronic enhancer region.6,15 The site in the intronic enhancer region is also called the Treg-specific demethylated region and plays a major role in maintaining the stability of Foxp3 expression.15,21 In contrast, methylation of the proximal promoter region is controlled by TGF-β-mediated signals.6 When we analysed the differential effects of simvastatin treatment on these two sites, the CpGs of the intronic enhancer region were highly methylated in conventional activated T cells, TGF-β-treated T cells, or simvastatin plus TGF-β co-treated cells, and no differences were detected among these groups (data not shown). However, the demethylation status of promoter region correlated with the level of expression of Foxp3 as determined by FACS analysis. Hence, the effects of simvastatin treatment are mediated only by way of TGF-β-susceptible DNA methylation sites rather than other methylation target sites. A correlation therefore exists between the effects of simvastatin on Foxp3 expression and control of the methylation status of the Foxp3 promoter.
Kagami et al.20 have shown that inhibition of protein geranylgeranylation induces SOCS3 expression and attenuates Th17 cell differentiation through the inhibition of STAT3 (signal transducer and activator of transcription 3) signalling. Although inhibition of Th17 differentiation was accompanied by the reciprocal enhancement of Foxp3 differentiation in their studies, we do not believe that induction of SOCS3 expression is the primary mechanism by which simvastatin enhances TGF-β-mediated Foxp3 expression. One of the most striking findings in our studies was that simvastatin could mediate its enhancing effects when added as long as 24 hr after culture initiation. In contrast, the presence of TGF-β was required for the entire 48 hr culture (data not shown). This result contrasts with the effects of simvastatin on SOCS3 induction that were maximal after 24 hr of stimulation. When we examined the effects of simvastatin on the early events in the TGF-β signal transduction cascade, we did not observe any augmentation of Smad3 phosphorylation. In contrast, the major effects of simvastatin were associated with a decreased induction of Smad6/7, inhibitory Smads that inhibit TGF-β signalling by blocking the phosphorylation of Smad2/3. We favour the view that simvastatin can directly block the induction of Smad6/7 expression, as the drug also inhibited the induction of Smad6/7 at 72 hr in the presence of a TCR signal alone in the absence of TGF-β. Alternatively, it is possible that the effects of simvastatin on Smad6/7 expression are mediated indirectly via a direct effect on Foxp3 expression as Fantini et al.22 have demonstrated that transfection of Foxp3 is capable of blocking TGF-β-induced Smad7 expression by acting directly on the Smad7 promoter. This mechanism is consistent with our findings that Smad6/7 cannot be induced in Foxp3+ nTregs following TGF-β signalling.
Although it is difficult to extrapolate from our in vitro model systems to the in vivo situation, our results that simvastatin can markedly enhance the induction of Foxp3 expression in the presence of low concentrations of TGF-β strongly suggest that some of the beneficial effects of simvastatin include the generation of Tregs in the inflammatory milieu of the atherosclerotic plaque. Further analysis of the mechanism of action of simvastatin will require identification of the targets of geranylgeranylation at different time-points after T-cell activation. Ras, Rho, CDC42 and many different GTPases are important for early signal transduction after engagement of the TCR and may play a role in induction of SOCS3. However, our findings suggest that the effects of simvastatin are on proteins synthesized 24 hr after TCR stimulation. At the very least, our study strongly implies that an analysis of TCR-specific protein prenylation is a potential pathway for pharmacological manipulation of Tregs in vivo.
Acknowledgments
This study was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health (Bethesda, MD).
Disclosures
The authors have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Simvastatin does not induce cell death or alter the cell cycle of Foxp3− cells.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Shevach EM. From vanilla to 28 flavors: multiple varieties of T regulatory cells. Immunity. 2006;25:195–201. doi: 10.1016/j.immuni.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–6. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 3.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 4.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 5.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 6.Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204:1543–51. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Youssef S, Stuve O, Patarroyo JC, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 8.Zeiser R, Youssef S, Baker J, Kambham N, Steinman L, Negrin RS. Preemptive HMG-CoA reductase inhibition provides graft-versus-host disease protection by Th-2 polarization while sparing graft-versus-leukemia activity. Blood. 2007;110:4588–98. doi: 10.1182/blood-2007-08-106005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hori A, Kanda Y, Goyama S, et al. A prospective trial to evaluate the safety and efficacy of pravastatin for the treatment of refractory chronic graft-versus-host disease. Transplantation. 2005;79:372–4. doi: 10.1097/01.tp.0000151001.64189.1d. [DOI] [PubMed] [Google Scholar]
- 10.Dunn SE, Youssef S, Goldstein MJ, Prod’homme T, Weber MS, Zamvil SS, Steinman L. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med. 2006;203:401–12. doi: 10.1084/jem.20051129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weitz-Schmidt G, Welzenbach K, Brinkmann V, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. 2001;7:687–92. doi: 10.1038/89058. [DOI] [PubMed] [Google Scholar]
- 12.Ait-Oufella H, Salomon BL, Potteaux S, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–80. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 13.de Boer OJ, van der Meer JJ, Teeling P, van der Loos CM, van der Wal AC. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS ONE. 2007;2:e779. doi: 10.1371/journal.pone.0000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mausner-Fainberg K, Luboshits G, Mor A, Maysel-Auslender S, Rubinstein A, Keren G, George J. The effect of HMG-CoA reductase inhibitors on naturally occurring CD4+CD25+ T cells. Atherosclerosis. 2008;197:829–39. doi: 10.1016/j.atherosclerosis.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 15.Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–54. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 17.Kou R, Sartoretto J, Michel T. Regulation of Rac1 by simvastatin in endothelial cells: differential roles of AMP-activated protein kinase and calmodulin-dependent kinase kinase-beta. J Biol Chem. 2009;284:14734–43. doi: 10.1074/jbc.M808664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Jin J, Peng X, Ramgolam VS, Markovic-Plese S. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol. 2008;180:6988–96. doi: 10.4049/jimmunol.180.10.6988. [DOI] [PubMed] [Google Scholar]
- 19.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kagami S, Owada T, Kanari H, et al. Protein geranylgeranylation regulates the balance between Th17 cells and Foxp3+ regulatory T cells. Int Immunol. 2009;21:679–89. doi: 10.1093/intimm/dxp037. [DOI] [PubMed] [Google Scholar]
- 21.Polansky JK, Kretschmer K, Freyer J, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–63. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 22.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25– T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–53. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.