

Abstract
Multiple sclerosis (MS) is a neurological disorder that affects more than a million people world-wide. The aetiology of MS is not known and there is no medical treatment available that can cure MS. Experimental autoimmune encephalomyelitis (EAE) is a T-cell-mediated autoimmune disease model of MS. The pathogenesis of EAE/MS is a complex process involving activation of immune cells, secretion of inflammatory cytokines and destruction of myelin sheath in the central nervous system (CNS). Peroxisome proliferator-activated receptors (PPARs) are nuclear hormone receptor transcription factors that regulate cell growth, differentiation and homeostasis. PPAR agonists have been used in the treatment of obesity, diabetes, cancer and inflammation. We and others have shown that PPARγ, α and δ agonists inhibit CNS inflammation and demyelination in the EAE model of MS. In this study we show that the PPARδ agonists GW501516 and L165041 ameliorate MOGp35-55-induced EAE in C57BL/6 mice by blocking interferon (IFN)-γ and interleukin (IL)-17 production by T helper type 1 (Th1) and Th17 cells. The inhibition of EAE by PPARδ agonists was also associated with a decrease in IL-12 and IL-23 and an increase in IL-4 and IL-10 expression in the CNS and lymphoid organs. These findings indicate that PPARδ agonists modulate Th1 and Th17 responses in EAE and suggest their use in the treatment of MS and other autoimmune diseases.
Keywords: experimental autoimmune encephalomyelitis/multiple sclerosis, interleukin-12 family cytokine, inflammation, peroxisome proliferator-activated receptor, T-helper type 1 (Th1)/Th17 response
Introduction
Multiple sclerosis (MS) is a neurological disorder that affects more than one million people world-wide, of whom 30% develop paralysis and become wheelchair-bound for the rest of their lives.1 Although the aetiology of MS is not known, it is generally viewed as a myelin-reactive T-cell-mediated autoimmune disease characterized by the destruction of myelin sheath and axonal loss in the central nervous system (CNS).2 Experimental autoimmune encephalomyelitis (EAE) is a CD4+ T-cell-mediated autoimmune disease model that is commonly used to test the efficacy of potential therapeutic agents for the treatment of MS.3–6 The pathogenesis of EAE and MS is a complex process involving activation of antigen-presenting cells, differentiation of neural antigen-specific T helper type 1 (Th1)/Th17 cells and secretion of inflammatory cytokines in the CNS. Interleukin (IL)-12 is a 70 kiloDalton (kD) heterodimeric cytokine, composed of p35 and p40 subunits, that plays a crucial role in the differentiation of encephalitogenic Th1 cells and the pathogenesis of EAE and MS.7,8 MS patients show elevated levels of IL-12 in brain lesions, cerebrospinal fluid (CSF) and circulation.9 The development of EAE was also found to be associated with an increase in the expression of IL-12 in the CNS and lymphoid organs.10,11 While the administration of recombinant IL-12 exacerbates EAE, targeted disruption or pharmacological inhibition of IL-12 production and IL-12 signalling prevents Th1 differentiation and the pathogenesis of EAE.11–14 Earlier studies demonstrated that IL-23, a heterodimeric cytokine composed of IL-12p40 and IL-23p19 subunits, induces the differentiation of Th17 cells and mediates the pathogenesis of EAE.15,16 Many recent studies have also demonstrated the critical role of IL-23 in the differentiation of Th17 cells in other models.15,17 While Th1 and Th17 cells play distinct roles at the initiation and effector phases of EAE,18,19 the anti-inflammatory cytokines such as IL-4 and IL-10 produce recovery.20
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear receptor transcription factors that regulate cell growth, differentiation and homeostasis. PPARα, β/δ and γ are three isoforms with distinct tissue distribution and function.21 Upon activation with specific ligands, PPARs undergo conformational changes leading to heterodimerization with retinoid X receptor (RXRα) and bind to promoter regions of target genes.22 PPARs regulate lipid and glucose metabolism and PPARγ agonists are effective in the treatment of type 2 diabetes. Recent studies have shown that PPAR agonists inhibit immune and inflammatory responses,23,24 including CNS inflammation and demyelination in EAE.3,23–27 We demonstrated previously that PPARγ agonists 15d-PGJ2 and ciglitazone ameliorate EAE by blocking IL-12 signalling and Th1 differentiation.3 Further analyses showed that PPARγ-deficient heterozygous mice develop an exacerbated EAE in association with an augmented neural antigen-specific Th1 response, suggesting a physiological role for PPARγ in the regulation of inflammation and demyelination in EAE.28
While PPARα is predominantly expressed in adipose tissue and muscle, PPARδ is expressed ubiquitously in the heart, the gastrointestinal tract and the CNS,29 and PPARδ-deficient mice show developmental defects in the brain.30 Fatty acids such as bromopalmitate and the synthetic drugs L165041 and GW501516 are PPARδ selective agonists that can modulate fatty acid metabolism and insulin sensitivity. PPARδ agonists inhibit inflammatory bowel disease and atherosclerosis in animal models.31–33 PPARα and PPARδ agonists also inhibit clinical and pathological symptoms of EAE.34,35 While PPARα and γ agonists ameliorate EAE by modulating proinflammatory cytokines, the specific mechanisms by which PPARδ agonists inhibit inflammation in EAE are not well defined. In this study we show that in vivo treatment with PPARδ agonists inhibits neural antigen-induced Th1 and Th17 responses in EAE, suggesting their use in the treatment of MS and other autoimmune diseases.
Materials and methods
Animals
C57BL/6 mice were obtained from Harlan (Indianapolis, IN). PPARδ−/− mice were a kind gift from Dr Gonzalez (National Institute of Health, Bethesda, MD/Pennsylvania State University, University Park, PA). The mice were housed and maintained in the animal facility at the Methodist Research Institute (Indianapolis, IN). All animal protocols used in the experiments were approved by the Institutional Animal Care and Use Committee.
Reagents
The 21-amino acid peptide (MEVGWYRSPFSRVVHLYRNGK) corresponding to mouse MOGp35-55 (96·81% pure) was obtained from Genemed Synthesis Inc. (San Francisco, CA). PPARδ selective agonists L165041 (C22H26O7; 4-[3-(2-propyl-3-hydroxy-4-acetyl)phenoxy]propyloxyph-enoxy-acetic acid) and GW501516 (C21H18F3NO3S2; 2-[2-methyl-4-([4-methyl-2-[4-(trifluoromethyl)phenyl)-1,3-thiazol-5- 1-yl]methylsulfanyl]phenoxy] acetic acid were obtained from Calbiochem (San Diego, CA) and Alexis Biochem (San Diego, CA), respectively. WST-1 was purchased from Roche Diagnostics (Indianapolis, IN). [3H]thymidine was purchased from GE Healthcare BioSciences (Piscataway, NJ) and carboxyfluorescein diacetate N-succinimidyl ester (CFSE) from Invitrogen (Carlsbad, CA). Recombinant mouse IL-12, interferon (IFN)-γ, IL-17, IL-23, IL-4, IL-6, IL-10 and transforming growth factor (TGF)-β were purchased from R&D Systems (Minneapolis, MN). Antibodies to IFN-γ, IL-17, IL-23p19, IL-4, IL-10 and CD28 were purchased from eBioscience (San Diego, CA), while anti-IL-12p40 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Biotin-conjugated antibodies specific to IL-17 were purchased from BD Pharmingen (Franklin Lakes, NJ), IFN-γ from Endogen (Woburn, MA), IL-12/23p40 from Biolegend (San Diego, CA), IL-12p70 from Mabtech (Mariemont, OH) and IL-4 and anti-IL-10 from eBioscience. Anti-CD4 magnetic beads, magnetic antibody cell sorting (MACS) LS columns and the magnet for T-cell purification were purchased from Myltenyi Biotec (Auburn, CA). The antibodies specific to CD3-efluor 450, CD4-phycoerythrin (PE), IL-17A-fluorescein isothiocyanate (FITC), IFN-γ-FITC, IL-4-allophycocyanin (APC) and streptavidin-PE-conjugated secondary antibodies were obtained from eBioscience. Monensin (GolgiStop) was purchased from BD Biosciences (San Jose, CA) and TRIzol reagent was obtained from Invitrogen. The TaqMan reverse transcription kit, Universal Master Mix, and fast optical 96-well reaction plates were purchased from Applied Biosystems (Foster City, CA). The quantitative reverse transcription–polymerase chain reaction (qRT-PCR) primers for IL-12p35 (99999066-m1), IL-12p40 (Mm99999067-m1), IL-23p19 (Mm00518984-m1), IL-27p28 (Mm00461165-m1), T-bet (Mm00450960-m1), IL-10 (Mm00439616-m1), IL-4 (Mm00445259-m1), IL-17 (Mm00439619-m1) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Mm99999915-g1) were purchased from Applied Biosystems. The IFN-γ (241036) qRT-PCR primer set was purchased from Qiagen (Valencia, CA).
Induction and treatment of EAE
EAE was induced in 4–6-week-old female C57BL/6 mice by subcutaneous (s.c.) immunization with 100 μg of MOGp35-55 peptide in 150 μl of emulsion of complete Freund’s adjuvant (Difco, Detroit, MI) at the lower dorsum on days 0 and 7. The mice also received 100 ng of pertussis toxin in 100 μl of phosphate-buffered saline (PBS) intraperitoneally (i.p.) on days 0 and 2. The mice were treated (i.p.) with 25 or 100 μg of GW501516 or L165041 in 25 μl of dimethyl sulphoxide (DMSO) every other day from day 0 to day 30 following immunization. The control mice received only 25 μl of DMSO. Four- to six-week-old male PPARδ−/− mice were also induced to develop EAE as described above and treated (i.p.) with 100 μg of L165041 or GW501516 in 25 μl of DMSO every other day from day 0 to day 30. Clinical signs of EAE were scored every day from day 0 to day 30 in a blinded manner as follows: 0, normal; 0·5, stiff tail; 1, limp tail; 1·5, limp tail with inability to right; 2, paralysis of one limb; 2·5, paralysis of one limb and weakness of one other limb; 3, complete paralysis of both hind limbs; 4, moribund; 5, death. The mean clinical scores (MCS) were calculated by adding every day clinical score for all mice in a group and then dividing by the total number of mice. The maximum mean clinical score (MMCS) was the MCS at the peak of disease. The average mean clinical score (AMCS) was calculated by adding the MCS for all days (from day 0 to day 30) and then dividing by 30. The mean clinical score more than 1 (MCS > 1) was obtained by counting the number of days with MCS more than 1. The mean clinical score on day 30 (MCS day 30) was obtained by averaging the clinical scores of all animals on day 30. Disease incidence (DI) denotes the number of mice with a score of 0·5 or greater in each group. The area under the curve (AUC) was calculated using GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA).
Histological analysis
To assess the pathology of CNS inflammation and demyelination, mice induced to develop EAE and treated with PPARδ agonists were killed on day 30 by CO2 asphyxiation and perfused by intracardiac infusion of 4% paraformaldehyde and 1% glutaraldehyde in PBS. Spinal cord samples were removed and fixed in 10% formalin in PBS. Transverse sections of cervical, upper thoracic, lower thoracic and lumbar regions were stained with Luxol Fast Blue or haematoxylin and eosin. Inflammation and demyelination in the CNS were assessed under a microscope in a blinded manner. The spinal cord sections were viewed as anterior, posterior and two lateral columns (four quadrants). Each quadrant displaying the infiltration of mononuclear cells or loss of myelin was assigned a score of one inflammation or one demyelination, respectively. Thus, each animal has a potential maximum score of 16 and the pathological score for each group is expressed as the percentage positive quadrants.
T-cell proliferation assay
The effect of PPARδ agonists on T-cell proliferation/survival was measured using WST-1, [3H]thymidine uptake and CFSE assays. To examine the ex vivo response, C57BL/6 mice were immunized with MOGp35-55 and treated with 25 and 100 μg of PPARδ agonists or 25 μl of DMSO. The spleen cells were isolated from all groups on day 14 and cultured in 96-well tissue culture plates in RPMI medium (2 × 105/200 μl/well) with 0, 2·5, 5 and 10 μg/ml MOGp35-55 antigen in the absence of PPARδ agonists in culture. [3H]thymidine (0·5 μCi/well) was added at 72 hr and the cells were harvested at 96 hr using a Tomtec Harvester 96 (Hamden, CT). [3H]thymidine uptake was measured using a Wallac Microbeta Trilux counter (Perkin Elmer, Waltham, MA). WST-1 reagent (10 μl/well) was added after 96 hr and the optical density (OD) at 450 nm was measured after 4 hr using a titre-plate reader (Alpha Diagnostics, San Antonio, TX). Spleen cells (5 × 106 cells/ml) from all groups were also incubated in PBS with 0·2 μm CFSE (Invitrogen) for 5 min at 37°. Cells were washed and cultured in RPMI medium (2·5 × 106 cells/ml) with 0, 5 and 10 μg/ml MOGp35-55 peptide. After 72 hr, cells were harvested, stained for CD4-PE and re-suspended in PBS/0·1% bovine serum albumin (BSA) containing 10 μg/ml of propidium iodide. Live cells were gated on CD4 and the percentage of proliferating CD4+/CFSE+ cells determined based on CFSE dilution by flow cytometry using an LSR II instrument (BD Biosciences, Franklin Lakes, NJ). To examine the in vitro response, MOGp35-55 immune spleen cells were isolated from DMSO-treated control mice and cultured in 96-well plates in RPMI medium (2 × 105/200 μl/well) with 0 and 5 μg/ml of antigen in the presence of 0, 1, 2·5, 5 and 10 μm PPARδ agonists. The proliferation was measured using [3H]thymidine uptake and WST-1 assays as described above.
Quantitative real-time PCR
The effect of PPARδ agonists on the expression of inflammatory cytokines in EAE was determined by quantitative real-time PCR.36 In brief, brain and spleen samples were isolated from C57BL/6 mice treated with DMSO or PPARδ agonists on day 14 following immunization with MOGp35-55. Spleen cells were cultured in 24-well tissue culture plates in RPMI medium with 10 μg/ml MOGp35-55 for 24 hr. Total RNA was extracted from brain, spleen, and cultured spleen cells using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The RNA was reverse-transcribed into cDNA using the TaqMan reverse transcription kit and the cDNA was amplified using TaqMan Universal Master Mix with probe and primers in the ABI 7900 Fast Sequence Detection System (Applied Biosystems, Foster city, CA). The levels of gene expression normalized to GAPDH were calculated using the relative quantification (delta-delta-Ct) study software (Applied Biosystems) and the results are presented as arbitrary fold change compared with control.
Flow cytometry of intracellular cytokines
The effect of PPARδ agonists on intracellular cytokines in EAE was determined by flow cytometry. To determine the ex vivo response, spleen cells were isolated from C57BL/6 mice treated with DMSO or PPARδ agonists on day 14 following immunization with MOGp35-55 and cultured in RPMI medium (1 × 106/ml) in 12-well plates with 5 μg/ml MOGp35-55 for 36 hr. To determine the in vitro response, MOGp35-55 immune spleen cells from DMSO-treated control mice were cultured in RPMI medium (1 × 106 cells/ml) in 12-well plates with 5 μg/ml MOGp35-55 antigen in the presence of 0, 2·5 and 10 μm PPARδ agonists for 36 hr. GolgiStop was added for the last 4 hr to prevent cytokine secretion. The cells were washed and incubated in PBS/0·1% BSA with anti-CD3-eFluor450 antibody (1 : 50) at 4° for 30 min. Samples for IFN-γ and IL-17 analyses were also stained with anti-CD4-PE antibody (1 : 50) at 4° for 30 min. All samples were then fixed and permeabilized using BD Cytofix/Cytoperm solution (BD Bioscience Pharmingen, San Diego, CA) at room temperature for 15 min. Samples were washed and incubated in BD Perm/Wash buffer with antibodies specific to IL-17-FITC (1 : 50), IFN-γ-FITC (1 : 50), IL-12-biotin (1 : 50), IL-23-biotin (1 : 50) or isotype controls at 4° for 1 hr. Cells were also incubated with IL-10-biotin (1 : 50) and IL-4-APC (1 : 50) at 4° for 1 hr. All biotinylated antibodies were followed with streptavidin-PE secondary antibody (1 : 1000) at 4° for 30 min. Samples were washed and analysed by flow cytometry using an LSR II instrument (BD Biosciences).
Cytokine enzyme-linked immunosorbent assay (ELISA)
To determine the ex vivo cytokine response, spleen cells from MOGp35-55 sensitized C57BL/6 mice treated with PPARδ agonists were cultured in RPMI medium (1 × 106 cells/ml) in 24-well plates in the presence of 0 and 5 μg/ml MOGp35-55 antigen without PPARδ agonists. To determine the in vitro response, MOGp35-55 immune spleen cells from DMSO-treated control mice were cultured in RPMI medium (1 × 106 cells/ml) in 24-well tissue culture plates in the presence of 5 μg/ml MOGp35-55 in combination with 0, 2·5 and 10 μm PPARδ agonists. The culture supernatants were collected after 48 hr and the levels of IFN-γ, IL-17, IL-12p70, IL-23, IL-4 and IL-10 were measured by ELISA as described previously.5 Briefly, 96-well ELISA plates were coated with capture antibodies in 50 μl/well of 0·06 m carbonate buffer (pH 9·6) at 4° overnight. The excess antibodies were washed off and the residual binding sites blocked by the addition of 1% BSA in PBS for 1 hr. The test samples (culture supernatants) and standards were added to the respective wells and incubated at 4° overnight. Plates were washed with PBS containing 0·05% Tween-20 and biotin-conjugated detection antibodies were added. After incubation at room temperature for 1 hr, the plates were washed and avidin-alkaline phosphatase was added, followed by 1 mg/ml of p-nitrophenyl phosphate. After 30 min of incubation at room temperature, the absorbance was read at 405 nm and the concentrations of cytokines in the culture supernatants were calculated from the standard curve.
T helper cell polarization assay
To determine the in vitro effect of PPARδ agonists on T helper cell polarization, spleen cells were isolated from 6- to 8-week-old naïve female C57/BL6 mice and cultured in a 12-well plate (1 × 106 cells/ml/well) in RPMI medium with anti-CD3 (5 μg/ml) and anti-CD28 (2 μg/ml) monoclonal antibodies (mAbs). The cells were treated with recombinant IL-12 (rIL-12) (2 ng/ml) and anti-IL-4 antibody (2 μg/ml) for Th1, rIL-6 (5 ng/ml) and rTGF-β (2 ng/ml) for Th17 and rIL-4 (2 ng/ml), anti-IL-12 (2 μg/ml) and anti-IFN-γ (2 μg/ml) for Th2 polarizing conditions. The cells were cultured with DMSO, L165041 (2·5 μm) or GW501516 (2·5 μm), respectively. After 5 days the cells were washed and cultured in RMPI medium with 5 μg/ml anti-CD3 mAb. After 48 hr the culture supernatants were collected and the levels of IFN-γ, IL-17 and IL-4 were measured by ELISA as described above.
To further determine the in vitro effect of PPARδ agonists on T-cell polarization, CD4+ cells were isolated from 6- to 8-week-old naïve female C57/BL6 mice using MACS magnetic beads. Briefly, spleen cells were washed and incubated (1 × 108 cells) in 490 μl of buffer with 10 μl of biotin-conjugated antibody cocktail at 4° for 10 min. After washing in buffer, the cells were incubated in 900 μl of buffer with 100 μl of streptavidin microbeads at 4° for 15 min. The cells were washed, re-suspended in 500 μl of buffer and passed through an LS column mounted on a MACS magnet. The flow through was collected to remove CD4– cells. The column was separated from the magnet and the CD4+ cells were flushed through the column using fresh buffer. The CD4+ T cells (> 80% pure) were cultured under Th0, Th1 and Th17 polarizing conditions in the absence or presence of PPARδ agonists and analysed as described above.
Statistical analysis
The experiments were repeated more than three times and the values are expressed as mean ± standard deviation (SD). The data were analysed by one-way analysis of variance (ANOVA) using the GraphPad Prism 5.0 software and P values are expressed as * (P < 0·05), ** (P < 0·01) and *** (P < 0·001) in the figures.
Results
PPARδ agonists inhibit EAE in C57BL/6 mice
To determine the therapeutic effects of PPARδ agonists in the treatment of MS, we examined the effect of two PPARδ agonists, L165041 and GW501516, on EAE. As shown in Fig. 1, C57BL/6 mice immunized with MOGp35-55 antigen developed EAE by day 10 and the clinical severity increased to maximum by day 15. Interestingly, in vivo treatment with PPARδ agonists resulted in a significant decrease in the severity of EAE without affecting the day of onset or duration of the disease. While 25- and 100-μg doses of L165041 and a 25-μg dose of GW501516 induced partial inhibition, treatment with 100 μg of GW501516 resulted in complete inhibition of EAE (Fig. 1a,b). Further analyses showed that DMSO-treated control mice developed an MMCS of 2·17 on day 16 with an AMCS of 0·87, MCS > 1 for 17 days, an AUC of 26·19, and an ending MCS of 1·33 on day 30. Interestingly, the mice treated with 25 or 100 μg of L165041 and GW501516 showed a significant decrease in MMCS, AMCS, MCS > 1 and AUC (Table 1). These results show that PPARδ agonists inhibit the clinical signs of EAE.
Figure 1.
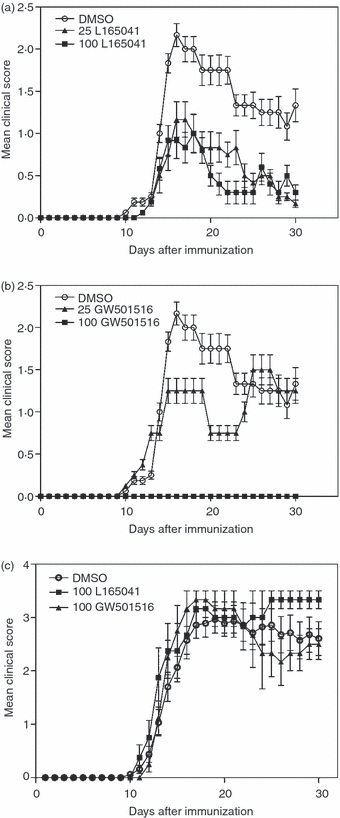
Inhibition of experimental allergic encephalomyelitis (EAE) by peroxisome proliferator-activated receptor δ (PPARδ) agonists. C57BL/6 mice were induced to develop EAE by immunization with MOGp35-55 antigen. The mice were treated intraperitoneally (i.p.) with dimethyl sulphoxide (DMSO) and 25 or 100 μg of L165041 (a) or GW501516 (b) every other day from day 0 to day 30. Clinical signs were scored every day in a blinded manner. The mean clinical score of 10 mice per group from two different experiments is shown. PPARδ−/− mice were induced to develop EAE by immunization with MOGp35-55 antigen and were treated (i.p.) with DMSO or 100 μg of L165041 or GW501516 every other day from day 0 to day 30 (c). Clinical symptoms were scored every day in a blinded manner. The mean clinical score of four mice per group is shown.
Table 1.
Clinical analysis of experimental allergic encephalomyelitis (EAE) in peroxisome proliferator-activated receptor δ (PPARδ) agonist-treated mice
| Analysis | DMSO | 25 μg L165 | 100 μg L165 | 25 μg GW501 | 100 μg GW501 |
|---|---|---|---|---|---|
| MMCS | 2·17 (100%) | 1·17 (54%) | 0·92 (42%) | 1·50 (69%) | 0·0 (0%) |
| AMCS | 0·87 (100%) | 0·37 (42%) | 0·31 (36%) | 0·67 (77%) | 0·0 (0%) |
| MCS > 1 days | 17·00 (100%) | 3·00 (18%) | 1·00 (6%) | 12·00 (71%) | 0·0 (0%) |
| MCS day 30 | 1·33 (100%) | 0·17 (13%) | 0·30 (22%) | 1·25 (94%) | 0·0 (0%) |
| AUC | 26·19 (100%) | 11·42 (44%) | 9·35 (36%) | 20·13 (77%) | 0·0 (0%) |
| DI | 9/10 (90%) | 8/10 (80%) | 6/10 (60%) | 3/10 (30%) | 0/10 (0%) |
C57BL/6 mice induced to develop EAE were treated with dimethyl sulphoxide (DMSO) and 25 or 100 μg of the PPARδ agonist L165041 (L165) or GW501516 (GW501) and clinical signs were scored every day from day 0 to day 30. The data from 10 mice per group were analysed, and the maximum mean clinical score (MMCS), average mean clinical score (AMCS), number of days with mean clinical score more than 1 (MCS > 1 day), mean clinical score on day 30 (MCS day 30), disease incidence (DI) and area under the curve (AUC) are presented. The values in parentheses are percent clinical score considering the DMSO control as 100%.
To determine the specific role of PPARδ in the amelioration of EAE by L165041 and GW501516, we examined the effect of these drugs in PPARδ−/− mice. Six- to eight-week-old male PPARδ−/− mice were treated with 100 μg of PPARδ agonists L165041 and GW501516, every other day from day 0 to day 30 following the induction of EAE. As shown in Fig. 1c, the PPARδ−/− mice developed EAE, with MCS reaching 2·8 on day 14 following immunization with MOGp35-55 antigen. However, treatment with L165041 or GW501516 showed no significant decrease in the onset or clinical severity of EAE compared with the DMSO-treated control (Fig. 1c). These results demonstrate the involvement of PPARδ-dependent mechanisms in the regulation of EAE by L165041 and GW501516, suggesting their use in the treatment of MS.
PPARδ agonists inhibit CNS inflammation and demyelination in EAE
We then examined the effect of PPARδ agonists on the pathology of CNS inflammation and demyelination in EAE. As shown in Fig. 2, DMSO-treated control EAE mice developed extensive myelin loss (demyelination) with infiltration of mononuclear cells (inflammation) in the spinal cord. In contrast, the mice treated with PPARδ agonists showed a significant decrease in both inflammation and demyelination in the spinal cord. The DMSO-treated control group showed 79% spinal cord quadrants positive for demyelination, which decreased to 26% and 21% in mice treated with 25 and 100 μg of L165041 and to 34% and 16% in mice treated with 25 and 100 μg of GW501516, respectively (Fig. 2). The DMSO-treated mice also showed 88% spinal cord quadrants positive for inflammation, which decreased to 24% and 21% in mice treated with 25 and 100 μg of L165041 and to 44% and 13% after treatment with 25 and 100 μg of GW501516, respectively (Fig. 2). These results show that PPARδ agonists inhibit CNS inflammation and demyelination which correlates with the clinical severity of EAE, further suggesting their use in the treatment of MS.
Figure 2.

Inhibition of central nervous system (CNS) inflammation and demyelination by peroxisome proliferator-activated receptor δ (PPARδ) agonists in experimental allergic encephalomyelitis (EAE). C57BL/6 mice induced to develop EAE were treated intraperitoneally (i.p.) with dimethyl sulphoxide (DMSO) and 25 or 100 μg of L165041 or GW501516 every other day from day 0 to day 30. Mice were sacrificed on day 30, and spinal cords were removed and fixed. Transverse sections of cervical, upper thoracic, lower thoracic and lumbar regions were stained with Luxol Fast Blue or haematoxylin and eosin. The pathology of demyelination (left) and inflammation (right) in the spinal cord sections was visualized by microscopy and the representative 50× and 200× pictures from mice treated with DMSO and 100 μg of PPARδ agonist are shown. The number of positive quadrants with inflammation and demyelination was scored for 10 mice per group and is expressed as the percentage of positive quadrants in the graph (*P < 0·05, **P < 0·01, and ***P < 0·001).
PPARδ agonists inhibit EAE without affecting neural antigen-induced T-cell proliferation
To understand the mechanisms underlying the inhibition of EAE by PPARδ agonists, we first examined their effects on neural antigen-induced T-cell proliferation. As shown in Fig. 3, spleen cells from EAE mice treated with DMSO and the PPARδ agonist L165041 or GW501516 showed a dose-dependent increase in viability (Fig. 3a) and proliferation (Fig. 3b) in response to MOGp35-55 antigen in culture. When compared with the DMSO-treated control group, EAE mice treated with PPARδ agonists showed no significant difference in MOGp35-55-induced proliferation ex vivo (Fig. 3a,b, left panels). Furthermore, L165041 or GW501516 failed to inhibit antigen-induced proliferation of MOGp35-55-immune spleen cells from DMSO-treated controls in vitro (Fig. 3a,b, right panels). The CFSE assay has further demonstrated that the CD4+ T cells from EAE mice treated with DMSO and the PPARδ agonist L165041 or GW501516 proliferate (CFSEdilute) similarly in response to MOGp35-55 antigen ex vivo (Fig. 3c). When compared with the DMSO-treated control, PPARδ agonists failed to show any inhibitory effect on neural antigen-induced proliferation of CD4+ T cells ex vivo, suggesting the involvement of other mechanisms in the regulation of EAE by PPARδ agonists.
Figure 3.
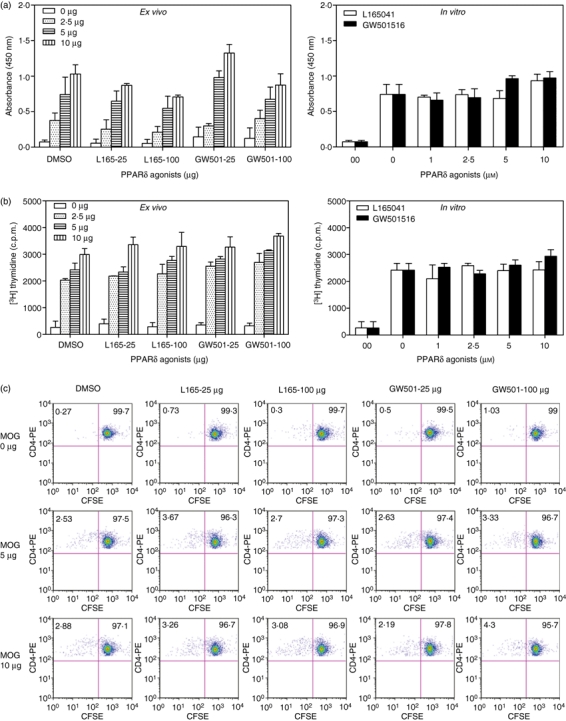
Effect of peroxisome proliferator-activated receptor δ (PPARδ) agonists on neural antigen-specific T-cell proliferation. C57BL/6 mice induced to develop experimental allergic encephalomyelitis (EAE) were treated intraperitoneally (i.p.) with dimethyl sulphoxide (DMSO) and 100 μg of L165041 or GW501516 every other day. On day 14, spleen cells were isolated and cultured with 0, 2·5, 5 and 10 μg/ml MOGp35-55 (ex vivo). Spleen cells from DMSO-treated EAE mice were also cultured with 0 or 10 μg/ml MOGp35-55 in the presence of 0, 1, 2·5, 5 and 10 μg/ml PPARδ agonists (in vitro). The viable cell count was determined using the WST-1 assay (a) and proliferation using the [3H]thymidine uptake assay (b). Spleen cells from EAE mice treated with DMSO and PPARδ agonist were isolated on day 14, stained with diacetate succinimidyl ester (CFSE) and cultured with 0, 5 or 10 μg/ml MOGp35-55. After 72 hr, proliferation was determined by flow cytometry (c). The figure is representative of six independent experiments. c.p.m., counts per minute.
PPARδ agonists inhibit the expression of IFN-γ, IL-17 and T-bet in EAE
To further determine the mechanisms underlying the inhibition of EAE by PPARδ agonists, we then examined the in vivo effects of PPARδ agonists on Th1 and Th17 responses in EAE. As shown in Fig. 4a, the mice induced to develop EAE expressed elevated levels of mRNA for IFN-γ, IL-17 and T-bet in the brain and spleen compared with naïve mice. Interestingly, in vivo treatment with L165041 or GW501516 resulted in a significant decrease in the expression of IFN-γ, IL-17 and T-bet in the brains and spleens from EAE mice. To confirm our findings, we further examined the ex vivo and in vitro effects of PPARδ agonists on IFN-γ and IL-17 production from neural antigen-sensitized spleen cells in culture. In vitro culture of MOGp35-55-immune spleen cells with antigen showed a significant increase in the expression of IFN-γ, IL-17 and T-bet mRNA (Fig. 4a) and secreted protein (Fig. 4b). When compared with the DMSO-treated control group, spleen cells from EAE mice treated ex vivo with L165041 or GW501516 showed a significant decrease in antigen-induced mRNA expression (Fig. 4a) and secretion of IFN-γ and IL-17 in culture (Fig. 4b). Similarly, MOGp35-55-immune spleen cells stimulated with antigen in the presence of PPARδ agonists in vitro also showed a dose-dependent decrease in the secretion of IFN-γ and IL-17 in culture (Fig. 4b).
Figure 4.
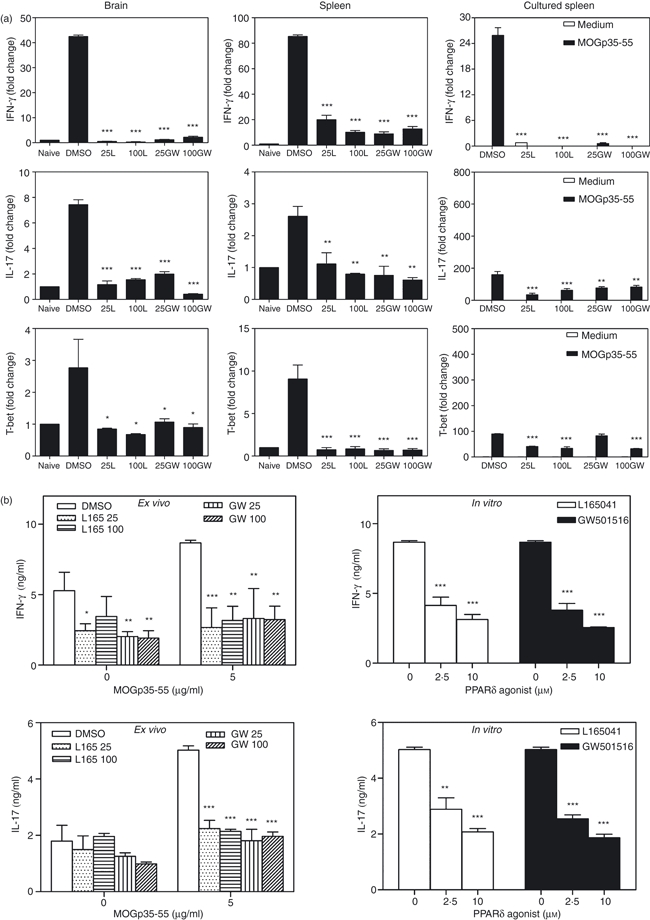
Inhibition of interferon (IFN)-γ, interleukin (IL)-17 and T-bet by peroxisome proliferator-activated receptor δ (PPARδ) agonists in experimental allergic encephalomyelitis (EAE). C57BL/6 mice induced to develop EAE and treated with dimethyl sulphoxide (DMSO) and 25 or 100 μg of the PPARδ agonist L165041 (L) or GW501516 (GW) were sacrificed on day 14. Total RNA was extracted from the brain, the spleen and spleen cells cultured with MOGp35-55. The expression of IFN-γ, IL-17 and T-bet was analysed by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control. The fold changes in the expression of cytokines in EAE were calculated using naïve mice as a control (a). Spleen cells from mice induced to develop EAE and treated with DMSO and 25 or 100 μg of the PPARδ agonist L165041 or GW501516 were cultured in the presence of 0 and 5 μg/ml MOGp35-55 (ex vivo). Spleen cells from DMSO-treated EAE mice were cultured with 0 or 5 μg of MOGp35-55 in the presence of 0, 2·5 and 10 μm PPARδ agonists (in vitro). After 48 hr, culture supernatants were collected and the levels of IFN-γ and IL-17 were determined by enzyme-linked immunosorbent assay (ELISA) (b). The figure is representative of three independent experiments (*P < 0·05, **P < 0·01, and ***P < 0·001).
To further confirm our findings, we examined the ex vivo and in vitro effects of PPARδ agonists on IFN-γ- and IL-17-producing spleen cells in EAE. As shown in Fig. 5, flow cytometric analyses of spleen cells from DMSO-treated EAE mice showed significant numbers of IFN-γ- and IL-17-positive CD3+ CD4+ T cells after stimulation with MOGp35-55 antigen in culture. Interestingly, spleen cells from EAE mice treated ex vivo with PPARδ agonists showed a decrease in CD3+ CD4+ IFN-γ+ and CD3+ CD4+ IL-17+ T cells after stimulation with MOGp35-55 antigen in culture (Fig. 5a). In vitro treatment of spleen cells from DMSO-treated EAE mice with PPARδ agonists also showed a significant reduction in the percentage of CD3+ CD4+ IFN-γ+ and CD3+ CD4+ IL-17+ T cells after stimulation with MOGp35-55 antigen in culture (Fig. 5b). These results suggest that PPARδ agonists ameliorate EAE in association with the inhibition of Th1 and Th17 responses in the CNS and lymphoid organs.
Figure 5.
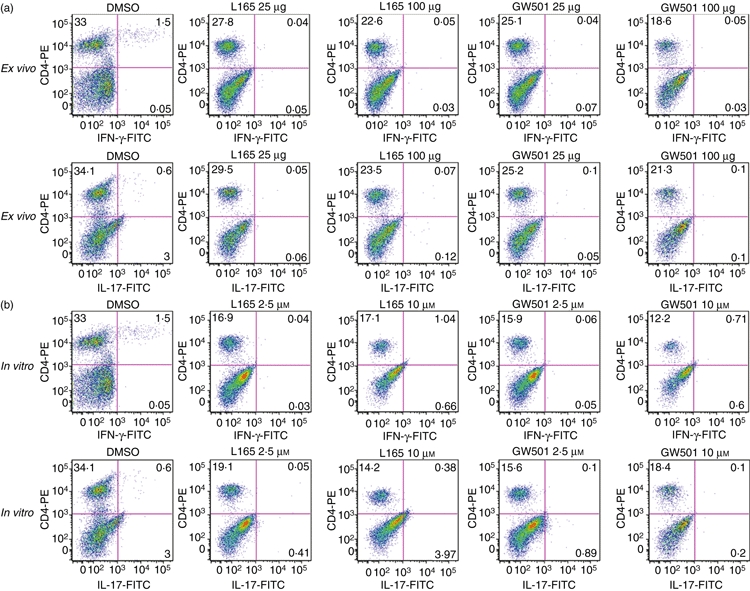
Inhibition of interferon (IFN)-γ- and interleukin (IL)-17-positive T cells by peroxisome proliferator-activated receptor δ (PPARδ) agonists in experimental allergic encephalomyelitis (EAE). C57BL/6 mice induced to develop EAE were treated with dimethyl sulphoxide (DMSO) and 25 or 100 μg of the PPARδ agonist L165041 (L165) or GW501516 (GW501). On day 14 the spleen cells were isolated and cultured with 5 μg/ml of MOGp35-55 (a). Spleen cells from DMSO-treated EAE mice were also cultured with 5 μg/ml MOGp35-55 in the presence of 0, 2·5 and 10 μm PPARδ agonists (b). The cells were harvested after 40 hr and the percentages of CD3 + CD4+ IFN-γ+ and CD3+ CD4+ IL-17+ cells were determined by flow cytometry. The figure is representative of three independent experiments.
PPARδ agonists inhibit the expression of IL-12 and IL-23 in EAE
To determine how PPARδ agonists inhibit Th1 and Th17 responses, we then examined the expression of IL-12 and IL-23 in the CNS and lymphoid organs of mice induced to develop EAE. As shown in Fig. 6a, EAE mice expressed elevated levels of IL-12p35, IL-12p40 and IL-23p19 in the brain and spleen compared with naïve mice. Interestingly, when compared with the DMSO-treated control group, the EAE mice treated with L165041 and GW501516 showed a decrease in the expression of IL-12p35, IL-12p40 and IL-23p19 in the brain and spleen (Fig. 6a). When compared with DMSO controls we found no significant inhibition in MOGp35-55-induced secretion of IL-12p70 from spleen cells of PPARδ agonist-treated mice ex vivo or in vitro (Fig. 6b). The PPARδ agonist-treated mice also showed no significant decrease in MOGp35-55-induced secretion of IL-23, except at 100 μg of GW501516, when compared with DMSO control mice ex vivo. However, splenocytes from DMSO-treated EAE mice showed significant inhibition of MOGp35-55-induced IL-23 secretion when exposed to PPARδ agonists in vitro. Flow cytometry analyses further showed that the percentages of CD3− IL-12+ (Fig. 7a) and CD3− IL-23+ (Fig. 7b) spleen cells were significantly reduced in PPARδ agonist-treated mice compared with the DMSO controls, both ex vivo and in vitro. These results suggest that PPARδ agonists inhibit Th1 and Th17 responses in association with the inhibition of IL-12 family cytokines in EAE.
Figure 6.
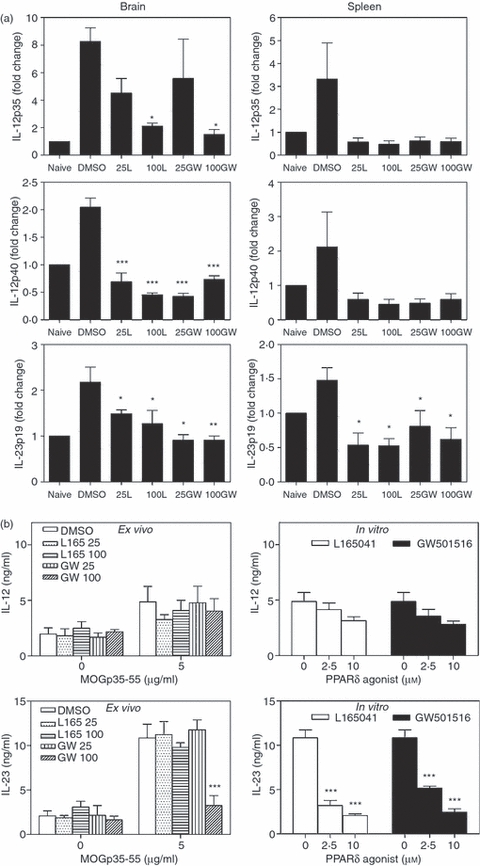
Inhibition of interleukin (IL)-12 and IL-23 expression by peroxisome proliferator-activated receptor δ (PPARδ) agonists in experimental allergic encephalomyelitis (EAE). C57BL/6 mice induced to develop EAE and treated with dimethyl sulphoxide (DMSO) and 25 or 100 μg of the PPARδ agonists L165041 (L) and GW501516 (GW) were sacrificed on day 14. Total RNA was extracted from the brain and the spleen and the expression of IL-12p35, IL-12p40 and IL-23p19 was analysed by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control. The fold changes in the expression of cytokines in EAE were calculated using naïve mice as a control (a). Spleen cells from mice induced to develop EAE and treated with DMSO and 25 or 100 μg of the PPARδ agonist L165041 or GW501516 were cultured in the presence of 0 and 5 μg/ml MOGp35-55 (ex vivo). Spleen cells from DMSO-treated EAE mice were also cultured with 0 and 5 μg/ml MOGp35-55 in the presence of 0, 2·5 and 10 μm PPARδ agonists (in vitro). After 48 hr, culture supernatants were collected and the levels of IL-12 and IL-23 were determined by enzyme-linked immunosorbent assay (ELISA) (b). The figure is representative of three independent experiments (*P < 0·05, **P < 0·01, and ***P < 0·001).
Figure 7.
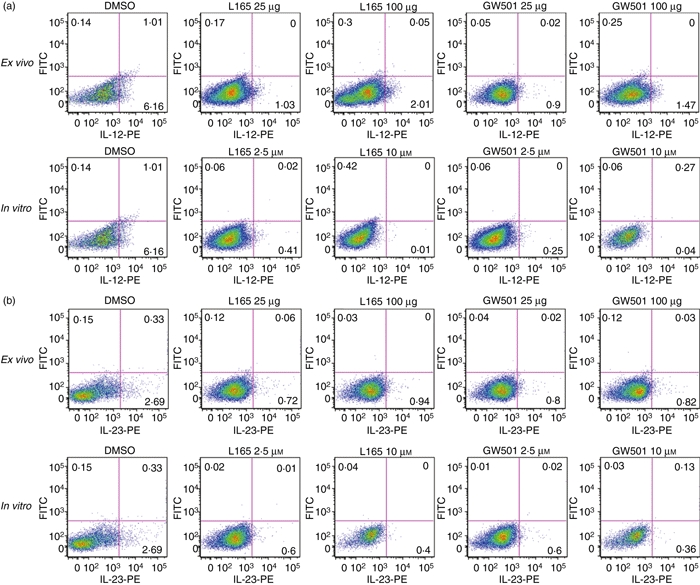
Inhibition of interleukin (IL)-12- and IL-23-positive spleen cells by peroxisome proliferator-activated receptor δ (PPARδ) agonists in experimental allergic encephalomyelitis (EAE). C57BL/6 mice induced to develop EAE and treated with dimethyl sulphoxide (DMSO) and 25 or 100 μg of the PPARδ agonist L165041 (L165) or GW501516 (GW501) were sacrificed on day 14. The spleen cells were isolated and cultured with 5 μg/ml of MOGp35-55 (ex vivo). Spleen cells from DMSO-treated EAE mice were also cultured with 5 μg/ml MOGp35-55 in the presence of 0, 2·5 and 10 μm PPARδ agonists (in vitro). The cells were harvested after 40 hr and the percentage of CD3− IL-12+ (a) and CD3− IL-23+ (b) cells was determined by flow cytometry. The figure is representative of three independent experiments.
PPARδ agonists increase the expression of IL-4 and IL-10 in EAE
To further determine the mechanisms by which PPARδ agonists inhibit EAE, we examined the expression of IL-4 and IL-10 in EAE. As shown in Fig. 8a, EAE mice expressed lower mRNA levels of IL-4 and IL-10 in the brain and spleen compared with naïve mice. Interestingly, in vivo treatment with L165041 and GW501516 increased the mRNA expression of IL-4 and IL-10 in the brain and spleen of EAE mice. Similarly, MOGp35-55-immune spleen cells cultured with antigen showed detectable levels of IL-4 and IL-10 mRNA expression which increased significantly in spleen cells of mice treated with PPARδ agonists (Fig. 8a). Further analyses showed that the spleen cells from EAE mice treated with 100 μg of L165041 and 25 μg of GW501516 secreted elevated levels of IL-4 in response to MOGp35-55 ex vivo when compared with DMSO controls (Fig. 8b). However, no such difference was observed in the secretion of IL-4 following in vitro treatment of MOGp35-55-immune spleen cells with PPARδ agonists. The mice treated with PPARδ agonist also showed a trend towards increased secretion of IL-10 compared with DMSO-treated mice ex vivo (Fig. 8b). The MOGp35-55-immune spleen cells treated in vitro with PPARδ agonists also showed a similar increase in IL-10 secretion. Interestingly, the PPARδ agonist-treated EAE mice showed an increase in the percentage of CD3+ IL-10+ spleen cells compared with the DMSO controls. However, we observed no significant difference in the percentage of CD3+ IL-4+ spleen cells in PPARδ agonist-treated mice compared with DMSO controls (Fig. 8c, ex vivo). The MOGp35-55-immune spleen cells treated in vitro with PPARδ agonists also showed no difference in CD3+ IL-4+ spleen cells and an increase in CD3+ IL-10+ spleen cells only with 10 μm GW101516 (Fig. 8c, in vitro). These results suggest an influence of IL-4 and IL-10 in the regulation of EAE by PPARδ agonists.
Figure 8.
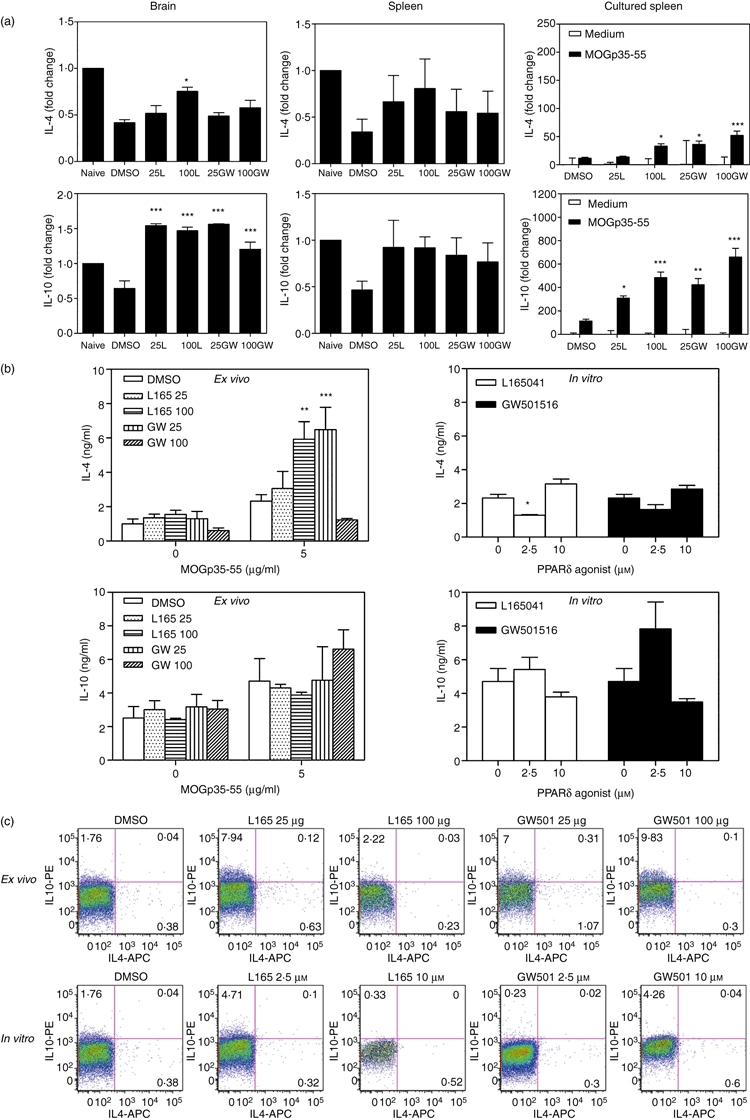
Induction of interleukin (IL)-4 and IL-10 expression by peroxisome proliferator-activated receptor δ (PPARδ) agonists in experimental allergic encephalomyelitis (EAE). C57BL/6 mice induced to develop EAE and treated with dimethyl sulphoxide (DMSO) and 25 or 100 μg of the PPARδ agonists L165041 (L) and GW501516 (GW) were sacrificed on day 14. Total RNA was extracted from the brain, the spleen or spleen cells cultured with MOGp35-55. The expression of IL-4 and IL-10 was analysed by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control. The fold changes in the expression of cytokines in EAE were calculated using naïve mice as a control (a). Spleen cells from mice induced to develop EAE and treated with DMSO and 25 or 100 μg of the PPARδ agonist L165041 or GW501516 were cultured with 0 or 5 μg/ml MOGp35-55 (ex vivo). Spleen cells from DMSO-treated EAE mice were also cultured with 0 or 5 μg/ml MOGp35-55 in the presence of 0, 2·5 and 10 μm PPARδ agonists (in vitro). After 48 hr, culture supernatants were collected and the levels of IL-4 and IL-10 determined by enzyme-linked immunosorbent assay (ELISA) (b). The cells cultured as described above were harvested after 40 hr and the percentage of CD3+ IL-4+ and CD3+ IL-10+ cells determined by flow cytometry. The figure is representative of three independent experiments (*P < 0·05, **P < 0·01, and ***P < 0·001).
PPARδ agonists inhibit Th1 and Th17 polarization in vitro
To further determine the mechanisms underlying the regulation of EAE by PPARδ agonists, we examined their influence on Th1 and Th17 differentiation in vitro. As shown in Fig. 9, the spleen cells and purified CD4+ T cells differentiated into Th1, Th17 and Th2 cells under appropriate polarizing conditions, as evidenced by IFN-γ, IL-17 and IL-4 secretion, respectively, following secondary stimulation. Interestingly, addition of the PPARδ agonist L165041 or GW501516 resulted in a significant decrease in IFN-γ and IL-17 secretion from spleen and CD4+ T cells under Th1 and Th17 polarizing conditions, respectively (Fig. 9). The addition of L165041 also resulted in an increase in IL-4 secretion from spleen cells under Th2 polarizing conditions (Fig. 9). These results suggest that PPARδ agonists influence the differentiation of Th1 and Th17 cells.
Figure 9.
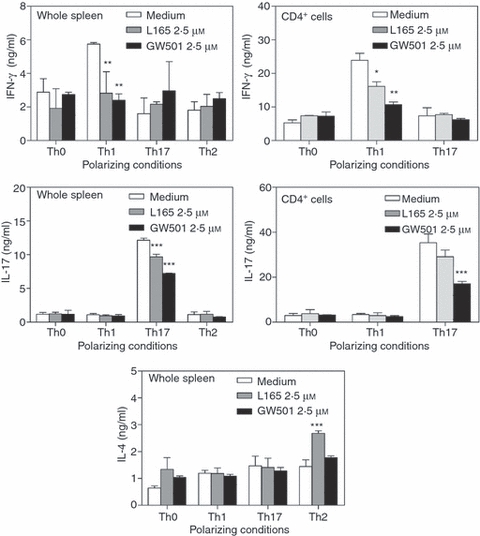
Inhibition of T helper type 1 (Th1) and Th17 differentiation by peroxisome proliferator-activated receptor δ (PPARδ) agonists. Whole spleen cells and CD4+ T cells were isolated from naïve C57BL/6 mice and cultured under Th0, Th1, Th2 and Th17 polarizing conditions in the presence of dimethyl sulphoxide (DMSO) and a 0 or 2·5 μm concentration of the PPARδ agonist L165041 or GW501516. The cells were harvested after 5 days and cultured in fresh medium with anti-CD3 monoclonal antibody (mAb) for secondary stimulation. After 48 hr the culture supernatants were collected and the levels of interferon (IFN)-γ, interleukin (IL)-17 and IL-4 measured by enzyme-linked immunosorbent assay (ELISA). The figure is representative of three independent experiments (*P < 0·05, **P < 0·01, and ***P < 0·001).
Discussion
Several groups have independently demonstrated the beneficial effects of PPAR agonists in attenuating EAE, suggesting their use in the treatment of MS.3,26,34,35 While the selective agonists of PPARα, δ and γ ameliorate EAE, their mode of action has not been fully elucidated. The pathogenesis of EAE/MS is a complex process involving the activation of immune cells, secretion of inflammatory cytokines and differentiation of Th1 and Th17 cells in the CNS.15,37,38 We and other have shown previously that PPARγ agonists inhibit CNS inflammation and demyelination by modulating neural antigen-specific Th1 and Th17 cells in EAE.3,39 In this study we found that PPARδ selective agonists L165041 and GW501516 inhibit EAE by modulating the development of Th1 and Th17 responses and the production of IFN-γ and IL-17 in the CNS and lymphoid organs. Our findings here are comparable to those showing PPARγ agonist-induced responses in EAE,3,25–27 but different from an earlier report showing no inhibition of IFN-γ production by PPARδ agonist GW0742 in EAE.35 Although the exact mechanisms are not known, our findings highlight the fact that PPARδ agonists ameliorate EAE in association with the inhibition of Th1 and Th17 responses and suggest their use in the treatment of MS and other autoimmune diseases.
The IL-12 family of cytokines play critical roles in the differentiation of Th1 and Th17 cells and the pathogenesis of EAE, MS and other autoimmune diseases.8,15,37 Earlier studies showed that targeted disruption or pharmacological inhibition of IL-12 or IL-23 was sufficient to prevent Th1/Th17 responses and pathogenesis of EAE.10,11,40 We have shown that PPARγ agonists inhibit the Th1 response by blocking IL-12 production and IL-12 signalling in EAE.3 To further understand the mechanisms by which PPARδ agonists inhibit Th1 and Th17 responses in EAE, we examined the expression of IL-12 and IL-23 in EAE. We found that in vivo treatment with L165041 and GW501516 decreased the expression of IL-12 and IL-23 in the CNS and lymphoid organs, suggesting that this could be a mechanism by which PPARδ agonists inhibit Th1/Th17 responses in EAE. This is consistent with an earlier report on the inhibition of IL-12 production by PPARγ agonists in immune cells.41 PPARδ agonists are known to inhibit the nuclear factor (NF)-κB signalling pathway.32 As the NF-κB pathway is essential for IL-12p40 expression, blocking this pathway could be a mechanism by which PPARδ agonists regulate IL-12 and IL-23 production in EAE. Although IL-6 and TGF-β have been commonly used to induce Th17 polarization in culture, their regulation by PPARδ agonists in EAE is not known.
Anti-inflammatory cytokines such as IL-4 and IL-10 are critical regulators of inflammation and demyelination in EAE and MS.20 To further understand the mechanisms by which PPARδ agonists inhibit CNS inflammation and demyelination, we then examined the expression of IL-4 and IL-10 in EAE. We found that in vivo treatment with PPARδ agonists increased the expression of IL-4 and IL-10 in the CNS and lymphoid organs of mice with EAE. In view of the regulation of Th1 and Th17 responses by IL-4 and IL-10,42,43 our findings suggest the involvement of these anti-inflammatory cytokines in the amelioration of EAE by PPARδ agonists. Taken together, our findings suggest that PPARδ agonists have several modes of actions in the inhibition of Th1 and Th17 responses in EAE, including the reduction of IL-12 and IL-23 in antigen-presenting cells, T-cell intrinsic inhibition of Th1 and Th17 differentiation and impaired reactivation of Th1/Th17 cells and expression of IFN-γ and IL-17 in response to neural antigens. The molecular mechanism involved in the regulation of Th1 and Th17 responses in EAE by PPARδ agonists is under investigation.
To determine the specific role played by PPARδ in the regulation of EAE, we treated PPARδ−/− mice with L165041 and GW501516 following induction of EAE. We found no significant inhibition of EAE by PPARδ agonists in PPARδ−/− mice, suggesting the involvement of PPARδ-dependent mechanisms in the regulation of EAE by these compounds. This is consistent with our earlier findings on the exacerbation of EAE in PPARγ-deficient heterozygous mice and those treated with PPARγ antagonists.5,28,44 We have observed some differences between L165041 and GW501516 in the regulation of inflammatory responses in EAE. This could be attributed to the difference in the affinities of L165041 and GW501516 (1000- and 120-fold, respectively) to PPARδ compared with other PPAR isoforms. However, our findings highlight the fact that PPARδ agonists ameliorate EAE in association with the regulation of Th1/Th17 responses. In addition to immune cells, oligodendrocytes are known to express PPARδ45 and PPARδ agonists induce oligodendrocyte differentiation in culture,46 suggesting that PPARδ agonists may also promote remyelination and CNS repair in EAE. As an earlier study failed to detect any significant increase in the expression of myelin basic protein in the CNS of mice treated with PPARδ agonist GW0472 following induction of EAE,35 further studies are needed to fully define the role of PPARδ in the regulation of MS and other autoimmune diseases.
Glossary
Abbreviations:
- AMCS
average mean clinical score
- AUC
area under the curve
- EAE
experimental allergic encephalomyelitis
- MCS
mean clinical score
- MMCS
mean maximum clinical score
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- PLP
proteolipid protein
- PPAR
peroxisome proliferator-activated receptor
- qRT-PCR
quantitative reverse transcription–polymerase chain reaction
- RXR
retinoid X receptor
Disclosures
None.
References
- 1.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 2.Whitacre CC, Reingold SC, O’Looney PA. A gender gap in autoimmunity. Science. 1999;283:1277–8. doi: 10.1126/science.283.5406.1277. [DOI] [PubMed] [Google Scholar]
- 3.Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 4.Muthian G, Bright JJ. Quercetin, a flavonoid phytoestrogen, ameliorates experimental allergic encephalomyelitis by blocking IL-12 signaling through JAK-STAT pathway in T lymphocyte. J Clin Immunol. 2004;24:542–52. doi: 10.1023/B:JOCI.0000040925.55682.a5. [DOI] [PubMed] [Google Scholar]
- 5.Raikwar HP, Muthian G, Rajasingh J, Johnson C, Bright JJ. PPARgamma antagonists exacerbate neural antigen-specific Th1 response and experimental allergic encephalomyelitis. J Neuroimmunol. 2005;167:99–107. doi: 10.1016/j.jneuroim.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 6.Natarajan C, Bright JJ. Curcumin inhibits experimental allergic encephalomyelitis by blocking IL-12 signaling through Janus kinase-STAT pathway in T lymphocytes. J Immunol. 2002;168:6506–13. doi: 10.4049/jimmunol.168.12.6506. [DOI] [PubMed] [Google Scholar]
- 7.Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med. 1995;181:817–21. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19:641–4. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 9.Nicoletti F, Patti F, Cocuzza C, Zaccone P, Nicoletti A, Di Marco R, Reggio A. Elevated serum levels of interleukin-12 in chronic progressive multiple sclerosis. J Neuroimmunol. 1996;70:87–90. doi: 10.1016/s0165-5728(96)00101-4. [DOI] [PubMed] [Google Scholar]
- 10.Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med. 1995;181:381–6. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bright JJ, Musuro BF, Du C, Sriram S. Expression of IL-12 in CNS and lymphoid organs of mice with experimental allergic encephalitis. J Neuroimmunol. 1998;82:22–30. doi: 10.1016/S0165-5728(97)00184-7. [DOI] [PubMed] [Google Scholar]
- 12.Mattner F, Smiroldo S, Galbiati F, et al. Inhibition of Th1 development and treatment of chronic-relapsing experimental allergic encephalomyelitis by a non-hypercalcemic analogue of 1,25-dihydroxyvitamin D(3) Eur J Immunol. 2000;30:498–508. doi: 10.1002/1521-4141(200002)30:2<498::AID-IMMU498>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 13.Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. 2006;83:1299–309. doi: 10.1002/jnr.20826. [DOI] [PubMed] [Google Scholar]
- 14.Smith T, Hewson AK, Kingsley CI, Leonard JP, Cuzner ML. Interleukin-12 induces relapse in experimental allergic encephalomyelitis in the Lewis rat. Am J Pathol. 1997;150:1909–17. [PMC free article] [PubMed] [Google Scholar]
- 15.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 16.Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–98. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- 17.Gyulveszi G, Haak S, Becher B. IL-23-driven encephalo-tropism and Th17 polarization during CNS-inflammation in vivo. Eur J Immunol. 2009;39:1864–9. doi: 10.1002/eji.200939305. [DOI] [PubMed] [Google Scholar]
- 18.Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J Clin Invest. 2003;112:1186–91. doi: 10.1172/JCI19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–42. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Rosa F, Francesconi A, Di Virgilio A, Finocchi L, Santilio I, Barnaba V. Lack of Th2 cytokine increase during spontaneous remission of experimental allergic encephalomyelitis. Eur J Immunol. 1998;28:3893–903. doi: 10.1002/(SICI)1521-4141(199812)28:12<3893::AID-IMMU3893>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 21.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–87. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 22.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–4. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–6. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 24.Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol. 2000;164:1364–71. doi: 10.4049/jimmunol.164.3.1364. [DOI] [PubMed] [Google Scholar]
- 25.Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, Tashiro K, Onoe K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol. 2001;116:40–8. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- 26.Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–15. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- 27.Feinstein DL, Galea E, Gavrilyuk V, et al. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- 28.Natarajan C, Muthian G, Barak Y, Evans RM, Bright JJ. Peroxisome proliferator-activated receptor-gamma-deficient heterozygous mice develop an exacerbated neural antigen-induced Th1 response and experimental allergic encephalomyelitis. J Immunol. 2003;171:5743–50. doi: 10.4049/jimmunol.171.11.5743. [DOI] [PubMed] [Google Scholar]
- 29.Woods JW, Tanen M, Figueroa DJ, Biswas C, Zycband E, Moller DE, Austin CP, Berger JP. Localization of PPARdelta in murine central nervous system: expression in oligodendrocytes and neurons. Brain Res. 2003;975:10–21. doi: 10.1016/s0006-8993(03)02515-0. [DOI] [PubMed] [Google Scholar]
- 30.Peters JM, Lee SS, Li W, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta) Mol Cell Biol. 2000;20:5119–28. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham TL, Mookherjee C, Suckling KE, Palmer CN, Patel L. The PPARdelta agonist GW0742X reduces atherosclerosis in LDLR(−/−) mice. Atherosclerosis. 2005;181:29–37. doi: 10.1016/j.atherosclerosis.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 32.Peters JM, Hollingshead HE, Gonzalez FJ. Role of peroxisome-proliferator-activated receptor beta/delta (PPARbeta/delta) in gastrointestinal tract function and disease. Clin Sci (Lond) 2008;115:107–27. doi: 10.1042/CS20080022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takata Y, Liu J, Yin F, et al. PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci USA. 2008;105:4277–82. doi: 10.1073/pnas.0708647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovett-Racke AE, Hussain RZ, Northrop S, et al. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. J Immunol. 2004;172:5790–8. doi: 10.4049/jimmunol.172.9.5790. [DOI] [PubMed] [Google Scholar]
- 35.Polak PE, Kalinin S, Dello Russo C, et al. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;168:65–75. doi: 10.1016/j.jneuroim.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Mo C, Chearwae W, O’Malley JT, et al. Stat4 isoforms differentially regulate inflammation and demyelination in experimental allergic encephalomyelitis. J Immunol. 2008;181:5681–90. doi: 10.4049/jimmunol.181.8.5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trembleau S, Germann T, Gately MK, Adorini L. The role of IL-12 in the induction of organ-specific autoimmune diseases. Immunol Today. 1995;16:383–6. doi: 10.1016/0167-5699(95)80006-9. [DOI] [PubMed] [Google Scholar]
- 38.Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–6. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klotz L, Burgdorf S, Dani I, et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. 2009;206:2079–89. doi: 10.1084/jem.20082771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bright JJ, Rodriguez M, Sriram S. Differential influence of interleukin-12 in the pathogenesis of autoimmune and virus-induced central nervous system demyelination. J Virol. 1999;73:1637–9. doi: 10.1128/jvi.73.2.1637-1639.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakobsen MA, Petersen RK, Kristiansen K, Lange M, Lillevang ST. Peroxisome proliferator-activated receptor alpha, delta, gamma1 and gamma2 expressions are present in human monocyte-derived dendritic cells and modulate dendritic cell maturation by addition of subtype-specific ligands. Scand J Immunol. 2006;63:330–7. doi: 10.1111/j.1365-3083.2006.01745.x. [DOI] [PubMed] [Google Scholar]
- 42.Adlard K, Tsaknardis L, Beam A, Bebo BF, Jr, Vandenbark AA, Offner H. Immunoregulation of encephalitogenic MBP-NAc1-11-reactive T cells by CD4 + TCR-specific T cells involves IL-4, IL-10 and IFN-gamma. Autoimmunity. 1999;31:237–48. doi: 10.3109/08916939908994069. [DOI] [PubMed] [Google Scholar]
- 43.Broberg EK, Salmi AA, Hukkanen V. IL-4 is the key regulator in herpes simplex virus-based gene therapy of BALB/c experimental autoimmune encephalomyelitis. Neurosci Lett. 2004;364:173–8. doi: 10.1016/j.neulet.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 44.Raikwar HP, Muthian G, Rajasingh J, Johnson CN, Bright JJ. PPARgamma antagonists reverse the inhibition of neural antigen-specific Th1 response and experimental allergic encephalomyelitis by Ciglitazone and 15-deoxy-Delta12,14-prostaglandin J2. J Neuroimmunol. 2006;178:76–86. doi: 10.1016/j.jneuroim.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 45.Granneman J, Skoff R, Yang X. Member of the peroxisome proliferator-activated receptor family of transcription factors is differentially expressed by oligodendrocytes. J Neurosci Res. 1998;51:563–73. doi: 10.1002/(SICI)1097-4547(19980301)51:5<563::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 46.Saluja I, Granneman JG, Skoff RP. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia. 2001;33:191–204. [PubMed] [Google Scholar]