

Abstract
Reaction of 1-adamantyl azide with iron(I) diketiminate precursors gives metastable but isolable imidoiron(III) complexes LFe=NAd (L = bulky β-diketiminate ligand; Ad = 1-adamantyl). This paper addresses: (1) the spectroscopic and structural characterization of the Fe=N multiple bond in these interesting three-coordinate iron imido complexes, and (2) the mechanism through which the imido complexes form. The iron(III) imido complexes have been examined by 1H NMR and EPR spectroscopies and temperature-dependent magnetic susceptibility (SQUID), and structurally characterized by crystallography and/or X-ray absorption (EXAFS) measurements. These data show that the imido complexes have quartet ground states and short (1.68 ± 0.01 Å) iron-nitrogen bonds. The formation of the imido complexes proceeds through unobserved iron–RN3 intermediates, which are indicated by QM/MM computations to be best described as iron(II) with an RN3 radical anion. The radical character on the organoazide bends its NNN linkage to enable easy N2 loss and imido complex formation. The product distribution between imidoiron(III) products and hexazene-bridged diiron(II) products is solvent-dependent, and the solvent dependence can be explained by coordination of certain solvents to the iron(I) precursor prior to interaction with the organoazide.
Keywords: imido complexes, iron, QM/MM calculations, EXAFS, organoazide complexes
Introduction
Imido (RN2−) ligands form strong bonds with the transition metals in groups 3-7, particularly those in high formal oxidation states. As a result, imidos often act as unreactive spectator ligands in early metal complexes, for example in the molybdenum olefin metathesis catalysts of Schrock and coworkers.1 This strong interaction is a result of π-donation from the two filled nitrogen p orbitals into empty metal d orbitals, which results in a formal bond order of up to three.2 In late transition metals (groups 8 – 11), on the other hand, the metal-nitrogen π-interactions are usually destabilized, because in octahedral complexes the antibonding metal dπ and nitrogen pπ orbitals are filled.3 Thus, late transition metals in the most common geometries typically form weaker bonds with imido fragments, and structurally characterized imido complexes of the late transition metals are uncommon. Though isolating them is difficult, understanding these species is potentially beneficial because the weaker metal-nitrogen bond can enable thermodynamically favorable nitrene transfer to organic compounds, and can also lower the activation barriers to stoichiometric and catalytic reactions. Thus, these “electrophilic” late transition metal imido complexes4 are of great interest as intermediates in catalytic nitrene transfer reactions, particularly aminations5 and aziridinations.6
There have been several innovations in stabilizing late transition metals with terminal imido ligands.7 Stone and coworkers reported in the 1960s and 1970s that several IrI, RhI, Ru0, and Os0 compounds react with fluoroalkylazides to give crystalline products, and these were formulated as fluoroalkylimido complexes on the basis of IR, NMR, and elemental analysis data.8 However, these putative imido complexes have never been completely characterized, and the assignments remain in doubt. In the late 1980s and early 1990s, work by Bergman and others generated Os,9 Ir, 10 and Ru 11 half-sandwich complexes with terminal imido ligands, which were characterized using X-ray crystallography. In the last ten years, several research groups have published isolable first-row transition metal complexes with terminal imido ligands including those of FeII,12 FeIII,13 FeIV,14 FeV,15 CoIII,16 NiII,17 and NiIII.18 All of these isolated group 8-10 complexes with terminal imido ligands feature bulky ligands that enforce a coordinatively unsaturated metal center. A low coordination number at the metal has emerged as an important feature, as tetrahedral and trigonal metal centers have π-symmetry orbitals that are not doubly occupied.19 Thus, even metals with a high formal d-electron count can form stabilizing π-interactions with donors such as NR2− by appropriately manipulating the geometry using the supporting ligands.
The recent successes in isolating late-metal complexes with terminal imido ligands should not lead one to underestimate the difficulty in preparing them. A few have been generated by deprotonation17a or hydrogen atom abstraction16e from amido complexes, which requires judicious choice of a reagent that can remove the strongly bonded hydrogen without destroying the complex. However, most of the latemetal imido complexes described above arise from the addition of an organoazide to a low-valent, unsaturated metal precursor. This reaction is exothermic and exergonic by virtue of forming N2 as a byproduct. However, the barrier to breaking the N-N bonds of organoazides is usually high, and the addition of organoazides to late transition metal complexes often leads to an organoazide complex in which the N-N bonds have not been cleaved.17b,20 Understanding the mechanism and selectivity for N2 extrusion from metal-organoazide complexes is a current research challenge.21 Proulx and Bergman invoked a four-centered triazametallacyclobutane intermediate to explain how a coordinated phenylazide ligand extruded N2 to afford an imidotantalum complex.20b Recent DFT-based simulations have also addressed possible mechanisms for N2 loss from organoazide complexes.22 Hillhouse recently described the isolation of a nickel-organoazide complex that loses N2 to form an imidonickel complex upon warming.17b
In this contribution, evidence is presented for the formation of a transient formally iron(I) organoazide complex on the way to an isolable iron(III) imido complex. In a novel feature, the metal-RN3 species has significant radical character on the organoazide ligand. It is shown that the addition of donor ligands controls the selectivity between several potential products obtained from organoazide addition. In addition, the geometric and electronic structure of the first structurally characterized three-coordinate iron(III) imido complex is explored in detail using crystallography, EPR, NMR, magnetic susceptibility, X-ray absorption, and computational chemistry techniques. These combined studies lead to new insight about the mechanism of formation of a late-metal imido complex, as well as its bonding and charge distribution.
Results
Products from Iron(I) and Adamantyl Azide
The diiron dinitrogen complexes LRFeNNFeLR (LR = LMe or LtBu, Figure 1) have been shown to be convenient sources of an evanescent two-coordinate iron(I) fragment “LFe” in reactions with alkynes, alkenes, CO, isocyanides, S8, phosphines, and benzo[c]cinnoline.23b,24 Therefore, it was hypothesized that LMeFeNNFeLMe and LtBuFeNNFeLtBu would serve as useful iron(I) precursors in building imidoiron(III) complexes. However, addition of 2 equiv of 1-adamantyl azide (N3Ad) to a pentane solution of LMeFeNNFeLMe did not produce the imido complex as the major product, but instead led to an unusual “hexazene” complex LMeFe(μ-η2:η2-AdNNNNNNAd-1κ2N1,N4:2κ2N3,N6)FeLMe (1) in 74% yield (Scheme 1).25 This product is conceptually derived from addition of two “LFe” fragments to two molecules of N3Ad or, alternatively, the dimerization of two LFe(N3Ad) complexes. The characterization of 1 was reported recently, and Mössbauer spectroscopy and magnetic studies were used to show that 1 is best described as being composed of two iron(II) centers and a dianionic [Ad2N6]2− ligand.25 Therefore, 1 derives from reductive coupling of two N3Ad groups through their terminal nitrogen atoms. The related compound LMeMg(μ-η2:η2-AdNNNNNNAd)MgLMe was recently reported by Jones, Stasch and coworkers, and it was similarly prepared through reductive coupling of N3Ad by the magnesium(I) precursor LMeMgMgLMe.26
Figure 1.
β-Diketiminate ligands used in this study.
Scheme 1.
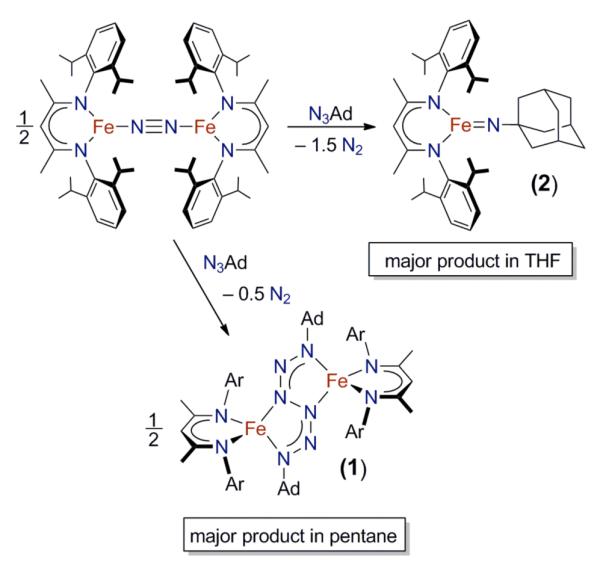
When performing the above reaction in pentane, signals for the desired LMeFeNAd (2) (characterized below) are observed in the 1H NMR spectrum of crude reaction mixtures, but 2 is formed in only 14% conversion. The relative amounts of 1 and 2 are highly dependent on the solvent used in the reaction (Table 1). In each reaction, the hexazene complex 1 is poorly soluble, and thus the yield of 1 is easily determined by filtration of the crude reaction mixture. The yield of imido complex 2 is determined based on 1H NMR integration against an internal standard. The yield of imido 2 is lowest (<30%) in noncoordinating solvents (pentane, diethyl ether, and 2,5-dimethyltetrahydrofuran (Me2THF)), intermediate (40 – 68%) in aromatic solvents (benzene, toluene, and α,α,α-trifluorotoluene (PhCF3)), and highest (80%) in THF. In each of these reactions, the combined yields of hexazene 1 and imido 2 was 79 – 93% based on iron.27
Table 1.
Solvent dependence of the outcome of the reaction between LMeFeNNFeLMe and 2.0 equiv N3Ad. Each reaction performed at [Fe] = 15 mM.
| Solvent | Yield of 1 (%)a |
Yield of 2 (%)b |
Approximate Ratio 2:1c |
|---|---|---|---|
| Pentane | 77 | 14 | 1:6 |
| Et2O | 64 | 25 | 1:3 |
| 2,5-dimethyl-THF | 63 | 28 | 1:2 |
| Toluene | 52 | 41 | 1:1 |
| Benzene | 47 | 43 | 1:1 |
| PhCF3 | 20 | 59 | 3:1 |
| Pentane + 4
equiv 4-tBu-pyridine |
10 | 68 | 7:1 |
| THF | 9 | 80 | 9:1 |
Isolated powder.
Detected in 1H NMR spectrum of crude reaction product.
Rounded to the nearest whole number in the ratio.
Because it gave the highest conversion to 2, THF was chosen for the isolation of pure samples of 2. Addition of a THF solution of N3Ad to a royal purple solution of 0.5 equiv LMeFeNNFeLMe in THF produces vigorous effervescence and an immediate color change to deep yellow. Removal of THF and crystallization of the residue from pentane at low temperature produced a yellow-brown crystalline product in 61% isolated yield. Crystals of 2 are stable for more than 2 weeks in the solid state at −45 °C, and this level of stability enabled spectroscopic and crystallographic characterization. At 25 °C, C6D6 solutions of redissolved crystals of 2 are metastable with t1/2 ~ 48 h. We initially reported the generation of 2 in the presence of 4-tert-butylpyridine (tBupy), but under these conditions t1/2 was only about 0.5 h at 25 °C, and crystallographic characterization was not possible.28
X-ray Crystal Structure of 2
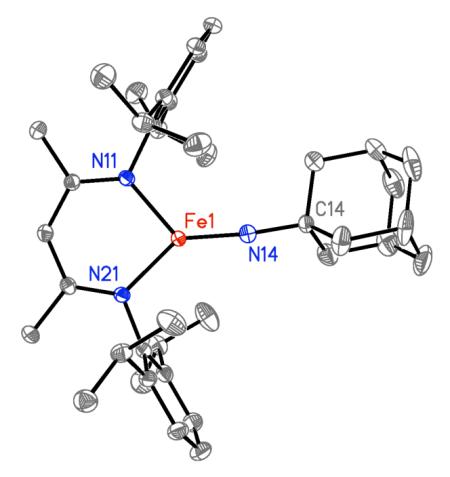
Single crystals of 2 were grown from a saturated pentane solution at −45 °C, and the X-ray crystal structure is shown in Figure 2a. The short Fe=N bond length (1.6699(15) Å) and nearly linear geometry at nitrogen (∠Fe=N–C = 170.40(13)°) highlight the Fe=N multiple bond character. These values are in the range observed in known terminal imidoiron compounds for Fe=N bond lengths (1.61 – 1.73 Å) and Fe=N–C angles (159 – 179°).29 The three-coordinate iron center is planar (sum of N–Fe–N angles is 360.0(1)°), and slightly bent from ideal Y-shaped geometry, as evidenced by the difference in Nimido=Fe–Ndiketiminate angles (126.82(6)° and 139.16(6)°). The Fe–Ndiketiminate bond lengths (1.9285(13), 1.9177(13) Å) are significantly shorter than those in three-coordinate iron(II) diketiminate complexes (average 1.98(2) Å),30 consistent with the assignment of the iron oxidation state in 2 as iron(III).
Figure 2.

Molecular structures of (a) LMeFe=NAd (2), and (b) LMeFe–NHAd (3) using 50% probability thermal ellipsoids. Hydrogen atoms except the amido hydrogen are omitted. Selected bond distances [Å] and bond angles [°] for LMeFe=NAd (2): Fe1-N14, 1.6699(15); Fe1-N11, 1.9285(13); Fe1-N21, 1.9177(13), Fe1-N14-C14, 170.40(13); N11-Fe1-N14, 139.16(6); N21-Fe1-N14, 126.82(6); N11-Fe1-N21, 94.02(6). Selected bond distances [Å] and bond angles [°] for LMeFeNHAd (3): Fe1-N14, 1.860(2); Fe1-N11, 1.974(2); Fe1-N21, 1.986(2), Fe1-N14-C14, 134.7(2); N11-Fe1-N14, 145.86(8); N21-Fe1-N14, 121.04(8); N11-Fe1-N21, 93.09(7).
The structure of the iron(III) imido complex can be compared to the analogous iron(II) amido complex LMeFeNHAd (3) (Figure 2b), which can be generate independently via anion metathesis of [LMeFeCl]2 with 2 equiv LiNHAd in a manner similar to other (β-diketiminato)FeII(amido) complexes.31 The Fe–Namido distance of 1.841(2) Å in 3 is typical of its congeners with NH(p-tolyl), NH(2,6-diisopropylphenyl), and NH(tBu) ligands,31 and is 0.171(3) Å longer than the Fe=N multiple bond in 2. The geometry at iron in the structure of 3 is midway between Y-shaped and T-shaped, with Nimido=Fe–Ndiketiminate angles of 145.88(8)° and 121.03(8)°. Also, the Fe–Ndiketiminate lengths in 3 (1.974(2) and 1.985(2) Å) are ~ 0.05 Å longer than the corresponding bond lengths in 2 because of the lower oxidation state, and are consistent with other three-coordinate iron(II) diketiminate complexes.30
Coordination of Lewis bases to the imido complex 2
The ability of THF to steer the reaction between LMeFeNNFeLMe and N3Ad toward the imidoiron(III) product 2 suggests that Lewis bases are beneficial. Consistent with this idea, performing the reaction in pentane in the presence of at least 4 equiv of tBupy gave a much higher yield of 2 and less than 10% of 1 (Table 1). To investigate the equilibrium between 2 and 2•tBupy (Scheme 2), we added 1 - 20 equivalents of tBupy to a C6D6 solution of 2 that had been previously crystallized. When tBupy was added, the color immediately changed from yellow-brown to dark red-orange. The 1H NMR spectrum of the mixture showed eleven paramagnetic peaks qualitatively similar to pyridine-free 2, as well as three broad peaks that are consistent with tBupy coordination. In contrast, addition of 100 equivalents of THF to a C6D6 solution of 2 did not shift the 1H NMR resonances more than 0.2 ppm or produce a color change, suggesting that THF does not coordinate to 2.
Scheme 2.
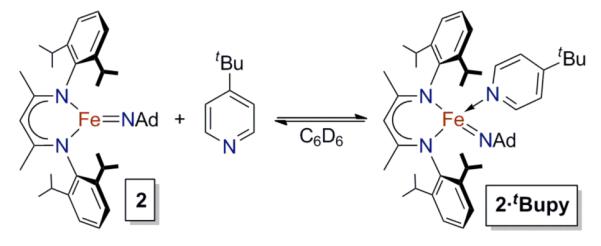
The positions of all peaks in the 1H NMR spectra of mixtures of 2 and tBupy are dependent on the concentration of tBupy (Figure 3), indicating that there is an equilibrium between three-coordinate LMeFeNAd (2) and four-coordinate LMeFe(NAd)(tBupy) (2·tBupy) that is rapid on the 1H NMR time scale. To confirm this hypothesis, the in situ-generated 2·tBupy was treated with a stoichiometric amount of BPh3 to scavenge the Lewis base from solution. This treatment shifted the paramagnetic 1H NMR resonances for 2·tBupy to become identical to those in crystallized 2, and the exchange-broadened tBupy peaks at δ 15.1 and δ 9.3 ppm disappeared (Figure S-2). Five aromatic 1H NMR resonances were observed (δ 6.4 to 8.3 ppm), consistent with the formation of the borane–pyridine adduct tBupy•BPh3.32
Figure 3.
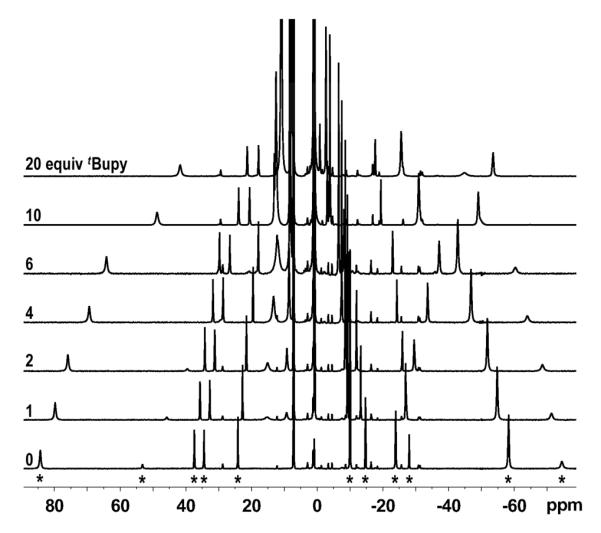
1H NMR spectra of 2 (C6D6, 25 °C) at the specified concentrations of tBupy. The eleven peaks assigned to 2 (marked with *) are indicated in the bottom spectrum. The small peaks with δH independent of [tBupy] are due to trace contamination with the iron-tetrazene complex 5.
The solutions of 2•tBupy were unstable, because the four-coordinate species undergoes intramolecular hydrogen atom abstraction (HAA) from the isopropyl C–H of the diketiminate ligand, with t1/2 on the order of ~0.5 h at 25 °C. This HAA reaction was reported previously,28 and mechanistic details of this reaction shall be explored in detail elsewhere.33 The addition of BPh3 to solutions of 2•tBupy greatly improved the stability of the complex, as t1/2 for decomposition of the mixture increased to ~36 h at 25 °C, similar to that for 2 prepared in THF and without tBupy.
Effect of diketiminate size on the outcome of the reaction
We also investigated the imidoiron complex supported by the bulkier LtBu ligand. This imido complex was obtained by adding 2 equiv of N3Ad to a solution of LtBuFeNNFeLtBu (Scheme 3). At room temperature in pentane, the target imidoiron(III) complex LtBuFeNAd (4) was generated in >80% spectroscopic yield,34 which differs significantly from the low conversion to the LMe analogue 2 (14% yield of imido in pentane). The hexazene complex {LtBuFe}2(Ad2N6) is also formed, but only in <2% crude yield. Larger amounts of hexazene complex (>20% isolated) were only obtained when the reaction was performed at −78 °C.25 Therefore, the use of the bulkier LtBu ligand greatly decreases the formation of the hexazene byproduct.
Scheme 3.
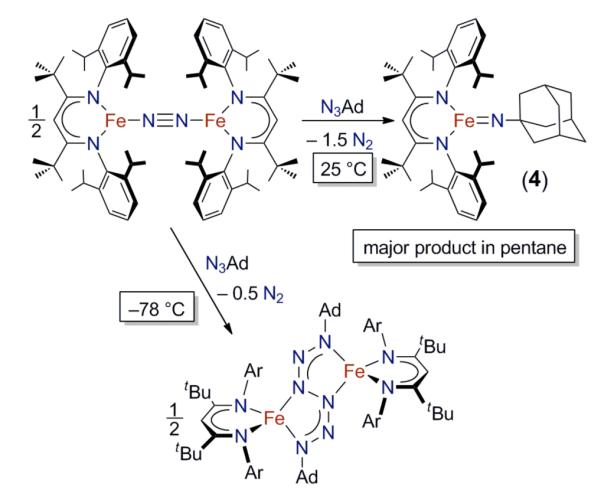
Although we have not been able to obtain single crystals of 4 for crystallography, the assignment of 4 is supported by NMR spectroscopy. As shown in Figure 5, the 1H NMR spectra of 2 and 4 are qualitatively very similar, suggesting they have similar geometries and electronic structures. The most significant difference between the two spectra is the chemical shift of the peaks attributed to the different ligand backbone substituent (Me in 2 at δ −24 ppm; tBu in 4 at δ +20 ppm). The similarity between features in the X-band EPR spectra and EXAFS fits of 2 and 4 will also become evident below.
Figure 5.
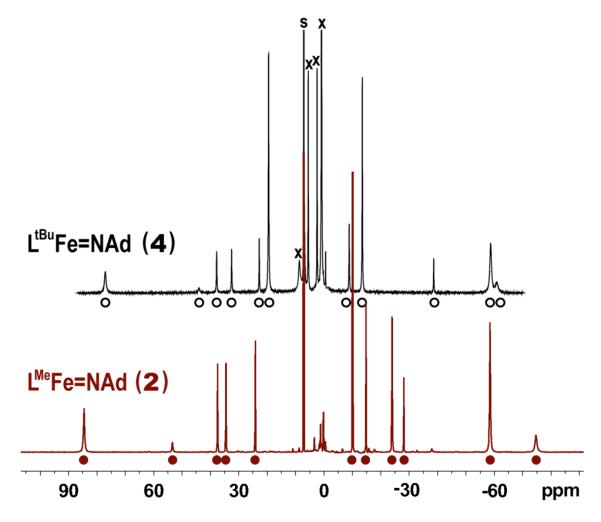
1H NMR spectra for 2 (bottom) and 4 (top) 34 on the same scale. The eleven peaks assigned to each complex are marked with filled circles (2) or open circles (4). Residual solvent signals are marked with “s”. The spectrum of 4 is recorded in the presence of excess 1,4-cyclohexadiene and 4-tert-butylpyridine (marked with “x”) to show that it does not react.
In other ways, compound 4 differs from 2. LtBuFeNAd (4) does not coordinate exogenous donors such as tBupy, and does not react with H· sources such as 1,4-cyclohexadiene or indene that react with 2.28 Despite its greater steric protection, 4 is less stable than 2 in solution, decomposing to a mixture of products with t1/2 of approximately 1 h at 25 °C. Therefore, it has been handled only in solution at low temperatures. However, we have been able to isolate a solid containing LtBuFeNAd in 50 - 70% purity for EXAFS measurements (see below). The nature of the decomposition products is unknown.
EXAFS Characterization of Imido Complexes
To further confirm the identity of the two imido complexes, we obtained Fe K-edge XAS data on imidoiron(III) complexes 2 and 4, as well as on the amidoiron(II) complex 3. XAS data for each compound were obtained in the solid state as dilutions in boron nitride.35 A comparison of the Fe K-edges for 2 and 3 are shown in Figure 6. The rising edge is at higher energy in the imidoiron(III) complex 2 than in the amidoiron(II) complex 3, consistent with the higher oxidation state. There is essentially no change in the pre-edge energies on going from 2 to 3, but the pre-edge intensity is somewhat larger for 2 than 3. A tentative interpretation is that the overall ligand field is similar, but there is greater covalency in the imido complex 2.
Figure 6.
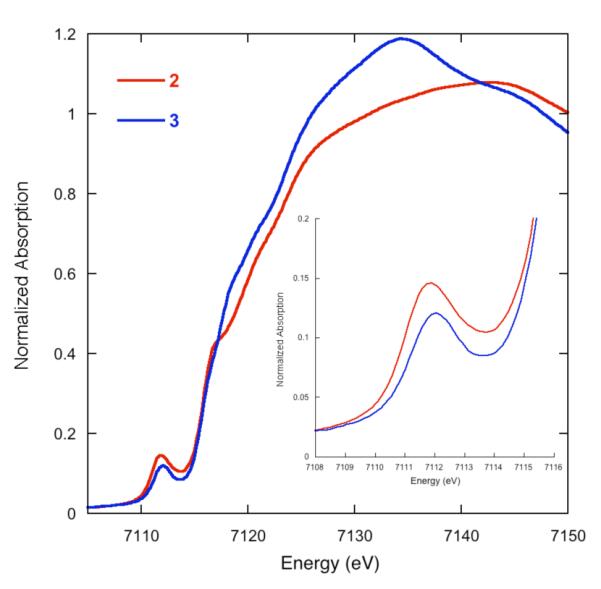
Comparison of the normalized Fe K-edge XAS data for 2 and 3. The inset shows the pre-edge feature at greater magnification.
The EXAFS data for 2, 3 and 4 (together with the corresponding fits) are shown in Figure 7. The non-phase-shift-corrected Fourier-transformed (FT) data are shown in Figure 8. The overall EXAFS beat pattern, as well as the FTs, for imido complexes 2 and 4 are very similar. On the other hand, for the amido complex 3 there is a distinctly different beat pattern and a broader first shell feature in the FT. The data for 2 were successfully modeled with two nitrogen atoms at 1.94 Å, and one nitrogen atom at 1.69 Å (Table 3). These are assigned as Fe-Ndiketiminate and Fe=Nimido bonds since they closely match the corresponding bond lengths from the crystal structure (1.93 and 1.67 Å, respectively). The fit was significantly poorer if the short Fe=N distance was omitted from the model, with the error increasing from 0.079 to 0.316. Although there is no distinct peak in the Fourier-transformed spectrum that corresponds to the short Fe-N scatterer, a similar trend has been noted for other complexes with short Fe-N bonds; the lack of a distinct peak arises from an interference effect that results in a narrower peak in the FT in the presence of a short Fe-N vector (see Figure 8).36 Hence, the EXAFS data corroborate the short Fe=N distance observed in the X-ray crystal structure.37
Figure 7.
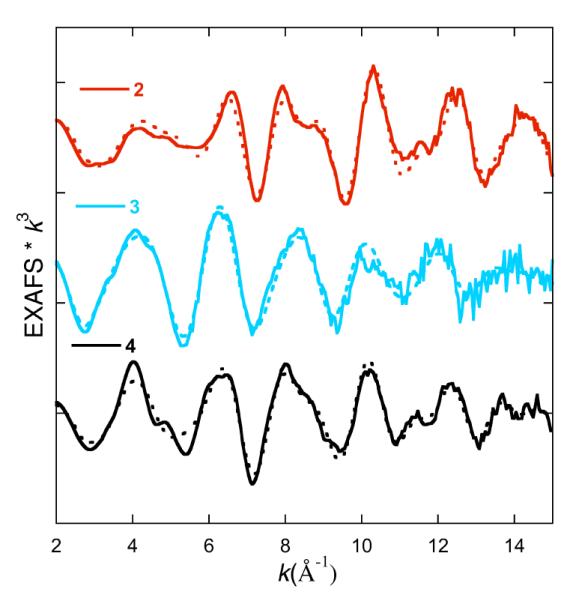
The k3-weighted EXAFS data and the corresponding fits for 2, 3 and 4.
Figure 8.
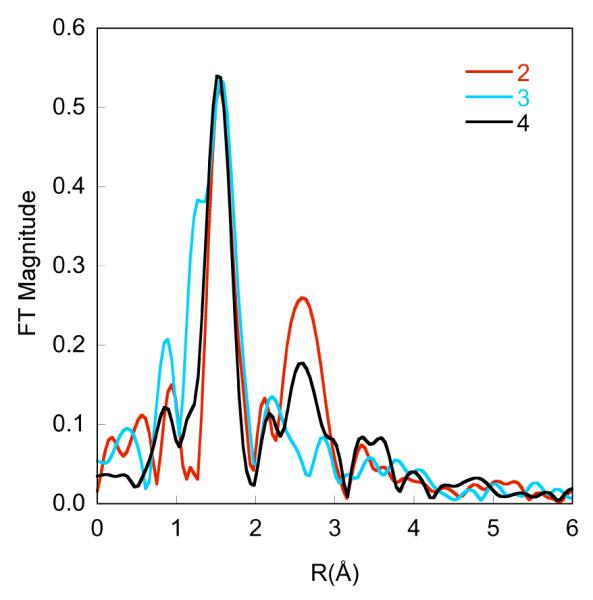
The non-phase shift corrected FT (Fourier transformed) data for 2, 3 and 4.
Table 3.
EXAFS Fit Results
| 2 | 3 | 4 | ||||||
|---|---|---|---|---|---|---|---|---|
| r (Å) | σ2 (Å2) | r (Å) | σ2 (Å2) | r (Å) | σ2 (Å2) | |||
| 2 Fe-N | 1.94 | 0.0039 | 3 Fe-N/O | 2.00 | 0.0067 | 2.5 Fe-N | 1.99 | 0.0062 |
| 1 Fe-N | 1.69 | 0.0052 | 0.5 Fe-N | 1.69 | 0.0045 | |||
| 6 Fe-N-C | 2.89 | 0.0038 | 6 Fe-N-C | 2.97 | 0.0075 | 6 Fe-N-C | 2.91 | 0.0028 |
| ΔE0 (eV) | −8.60 | ΔE0 (eV) | −4.45 | ΔE0 (eV) | −7.71 | |||
| errora | 0.079 | errora | 0.189 | errora | 0.069 | |||
Error is given by Σ[(χobsd - χcalcd)2k6]/Σ[χobsd2 k6].
The EXAFS data for 4 are of particular interest because this compound has not been subjected to crystallographic characterization. In this case, the data fit to 2.5 nitrogen atoms at 1.99 Å and 0.5 nitrogen atoms at 1.69 Å, distances similar to those in the fit for 2. Inclusion of 1 Fe-N at 1.69 Å resulted in an unreasonably large σ2 value (0.032 Å2), suggesting that the imido complex 4 represents only ~1/2 of the species present. Since 4 was not purified through crystallization and is less stable than 2, it is not surprising that the imido complex is less pure. However, the EXAFS data for 2 and 4 are still quite similar, and both show short (<1.7 Å) Fe=N distances, suggesting that they both contain imido ligands.
Magnetism of 2
We previously described the X-band EPR spectra of solution-generated 434 and 2•tBupy.38 The rhombic signal for each of these compounds is similar (geff = 7.0, 1.8, 1.3 for 4; geff = 6.1, 1.9, 1.4 for 2•tBupy) and indicates an S = 3/2 spin system with E/D ~ 0.33. The X-band EPR spectrum of a solution of pyridine-free, crystalline 2 (Figure S-11) was very similar (geff = 6.3, 1.9, 1.5) to that reported previously for 2•tBupy,28 showing that 2 and tBupy interact very weakly, as indicated also by the NMR experiments described above.
Crystalline 2 was also evaluated by magnetic susceptibility measurements. The variable-temperature magnetic susceptibility data (μeff) for solid 2 are shown in Figure 9. The high-temperature limit, corresponding to an effective magnetic moment of ~4.6 μB, exceeds the spin-only value for S = 3/2 (3.87 μB), but it is consistent with the effective magnetic moment measured in C6D6 solution (Evans method, 25 °C) of μeff = 4.4 ± 0.3 μB. The deviation is most probably due to the presence of an unknown paramagnetic contaminant, which is EPR-silent. In previous Mössbauer studies of 2•tBupy, variable amounts of iron(II) impurities were observed,28 and some decomposition is expected because of the metastable nature of 2. By assuming that the impurity is an iron(II) species in 20% abundance with spin S = 2 and roughly the same molar mass, the magnetic data could be successfully fitted to an S = 3/2 model with gav = 2.15 and ∣D∣ = 65(5) cm−1, and E/D = 0.33. The rhombicity parameter of E/D = 0.33 was taken from simulations of the EPR spectrum mentioned above, whereas D and the average g value were refined to fit the magnetic susceptibility data. The refinement gave g = 2.15, which is close to that found for the EPR solution sample (gav = 2.07). The remarkably large value for D was corroborated by variable-temperature measurements at multiple fields (insert of Figure 9). Although the global fit of these traces using the same parameters and including the same S = 2 impurity is not perfect, it shows clearly that the very small nesting of iso-field curves can be reproduced only with ∣D∣ > 50 cm−1 for E/D ≈ 0.3 (or −D > 35 cm−1 for free E/D = 0), because only with such large zero-field splitting are the ms-sublevels of the S = 3/2 manifold sufficiently isolated to exhibit the observed field-independent behavior of the magnetization curves M(μBB/kT).39 In summary, the ability to fit both EPR and magnetic data to a self-consistent quartet model strongly supports a description of 2 having an S = 3/2 ground state. This conclusion is also consistent with the quartet spin state being lowest by 10 kcal/mol in DFT computations.28
Figure 9.
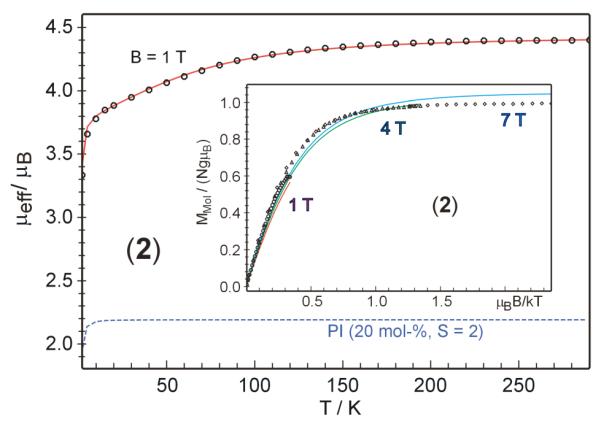
Variable-temperature magnetic susceptibility of crystalline 2 recorded with a field of B = 1 T. The solid red line represents the best fit achieved by adding the values for 20% of an unknown paramagnetic impurity with S = 2, D = 2 cm−1, gav = 2 to the result of a spin Hamiltonian simulation (PI, dotted blue line, assuming same molar mass). The optimized simulation parameters for the target compound with S = 3/2 are gav = 2.15, ∣D∣ = 65 cm−1, and E/D = 0.33. Inset: Multiple-field variable-temperature measurements at B = 1, 4, and 7 T and simulation with the same parameters (red, green and blue traces, respectively).
Computations On A Putative Iron(I)-Adamantylazide Intermediate
Next, we considered the mechanism through which LMeFeNNFeLMe might form 1 and 2. Because LMeFeNNFeLMe reacts with Lewis bases to form monomeric iron(I) adducts LMeFe(Lewis base),23b,24b-d it is reasonable to hypothesize a 1:1 adduct LMeFe(N3Ad) as the first species formed. Other transition metal–organoazide complexes thermally convert to metal–imido complexes.17b,20a-c No intermediate species are observed by 1H NMR spectroscopy during the very rapid reaction of the iron-N2 complex with N3Ad, even at cold temperatures. Therefore, the characteristics and reactions of the putative organoazide species were evaluated using quantum mechanics/molecular mechanics (QM/MM) computations with classical treatment (Universal Force Field) of the 2,6-diisopropylphenyl and methyl substituents of the β-diketiminate ligand and the adamantyl group with the exception of the carbon atom that is directly bonded to nitrogen. The core of the complex was modeled via density functional theory (DFT) at the B3LYP/6-311+G(d) level of theory. All doublet states were considerably higher in energy (>13 kcal/mol) than quartet and sextet states, and are not described here.
Azide Coordination Mode in LMeFe(N3Ad)
Since N3Ad is known to bind metals in three different coordination modes (Figure 10), we first sought to identify the lowest-energy linkage isomer of LMeFe(N3Ad) and elucidate its electronic structure. Isomers of LMeFe(N3Ad) were evaluated with the azide group bonded at the internal nitrogen (LMeFe(N3Ad-κN1)), the terminal nitrogen (LMeFe(N3Ad-κN3)), and side-on (LMeFe(η2-N3Ad)), in both quartet (IS) and sextet (HS) spin states. The triazametallacyclobutene isomer LMeFe(N3Ad-κ2N1 ,N3) was also evaluated, but quartet and sextet models were each found to be considerably (>30 kcal/mol) higher in energy than the other low-energy linkage isomers, and were not considered further.
Figure 10.
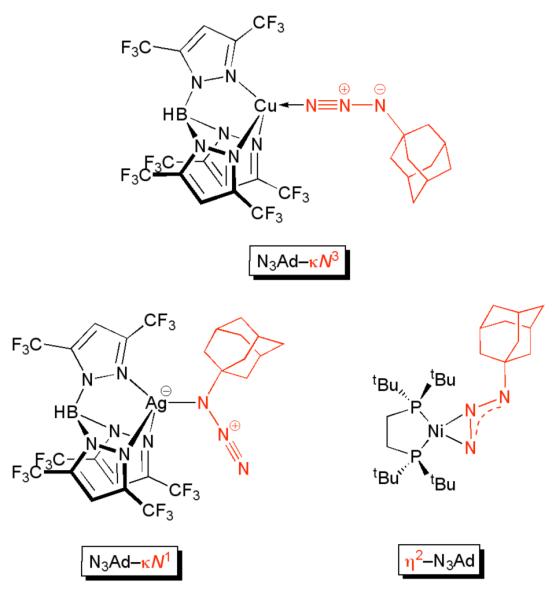
Examples of different coordination modes of N3Ad from literature complexes.17b,40c
The optimized geometries for the lowest spin state of each isomer are shown in Figure 11, and the relative energies of all isomer/spin state combinations are summarized in Table 4. Though sextet LMeFe(N3Ad-κN1) is the lowest energy linkage isomer, the LMeFe(N3Ad-κN3) and LMeFe(η2-N3Ad) linkage isomers are close in energy (lowest spin states +5.0 and +7.1 kcal/mol relative to 6LMeFe(N3Ad-κN3)). In mechanistic discussions below, we shall assume that the different low-energy linkage isomers can interconvert easily.
Figure 11.

QM/MM optimized geometries of three low energy linkage isomers of LMeFe(N3Ad): (a) sextet (κN1), (b) quartet (κN3), and (c) quartet η2. QM atoms are shown as spheres, and MM atoms are shown in wireframe. Hydrogens omitted from figure for clarity.
Table 4.
Calculated Lowest Energy Linkage Isomers of LMeFe(N3Ad)a
Calculated at the B3LYP/6-311+G(d):UFF level of theory.
Relative to the lowest energy linkage isomer in kcal/mol (1 atm, 298.15 K).
A stationary point corresponding to a sextet η2 complex was not found at this level of theory; QM/MM geometry optimizations converted to the sextet κN1 isomer.
Geometry and Electronic Structure of LMeFe(N3Ad)
The calculated Fe-Nazido bond length in 6LMeFe(N3Ad-κN1) is 1.96 Å and the NNN angle is very bent (127°). This is significantly different than in a crystallographically characterized N3R-κN1 complex of copper that has a longer Cu-N distance of 2.079(2) Å and a nearly linear NNN angle of 174.5(2)°.40b Other crystallographically characterized N3R-κN1 complexes also have virtually linear NNN angles (170-178°).40 The bent NNN angle in particular provided an initial structural hint that the adamantyl azide ligand in these complexes might be reduced to the radical anion. Indeed, separate geometry optimization of the radical anion of N3Ad leads to a NNN angle (131°) similar to that observed in 6LMeFe(N3Ad-κN1) (127°). Consistent with this electronic description, the DFT model of LMeFe(N3Ad) has substantial spin density on the organoazide ligand. For the lowest energy sextet-κN1 isomer, substantial spin density is found on the terminal (0.75 e−) and central (0.24 e−) azide nitrogen atoms, along with ~3.8 e− on the iron center and the remaining 0.2 e− on the β-diketiminate nitrogen atoms. For the quartet 4LMeFe(N3Ad-κN3) isomer (ΔGrel = 5.0 kcal/mol, Table 4) there are nearly equal amounts of negative spin density (0.42 e−) on each of the two nitrogen atoms closest to the iron center, with the remainder almost entirely on the iron center (3.59 e−). Finally, for the quartet-η2 isomer the spin density is almost entirely on the iron atom with very little on the organoazide moiety. Figure 12 below catalogues the best Lewis structure descriptions of these states derived from the electronic structure analysis.
Figure 12.
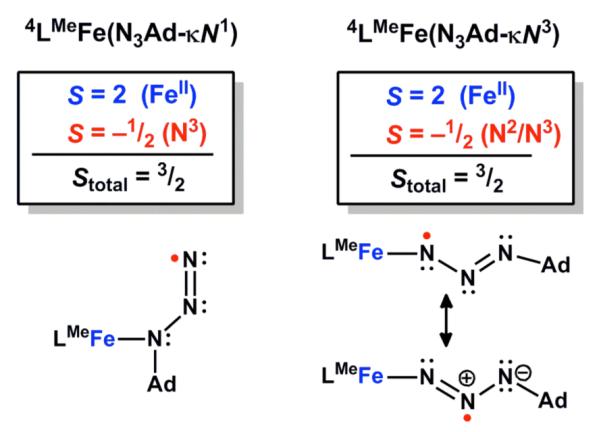
Lewis structures for the two lowest energy isomers of quartet LMeFe(N3Ad) implied by the spin density.
Elimination of N2 from LMeFe(N3Ad)
Two transition states for N2 elimination were examined, and they differed only by spin state (quartet or sextet). These transition states are close in energy (ΔΔG‡ = 1.0 kcal/mol with quartet lower in energy) and are similar in geometry, suggesting facile spin crossover between the quartet and sextet potential energy surfaces. The quartet transition state is depicted in Figure 13, and the sextet transition state is similar. The primary motion from ground state 4LMeFe(N3Ad-κN1) to the transition state is N1-N2 bond lengthening from 1.40 Å to 1.60 Å. The calculated Fe-N1 bond length in the transition state (1.91 Å, Figure 13) is only ~0.05 Å shorter than the corresponding calculated bond length for the ground state, and none of the core bond angles changed by more than ±3°. This similarity of ground state and transition state geometries in conjunction with the exothermicity of N2 loss suggests an “early” transition state. The calculated barrier for N2 loss is ΔG‡ = 12.0 kcal/mol from 6LMeFe(N3Ad-κN1), where the TS is a quartet. The extrusion of N2 is exergonic, with ΔG = −43.2 kcal/mol from 6LMeFe(N3Ad-κN1) to 2 + N2. Thus, as expected from previous results,41 expulsion of the stable N2 and strengthening of the metal-nitrogen linkage provide considerable thermodynamic driving force for imido formation. The overall reaction from 0.5 equiv LMeFeNNFeLMe to form the corresponding imide (2) is exergonic, ΔG = −49.8 kcal/mol.
Figure 13.
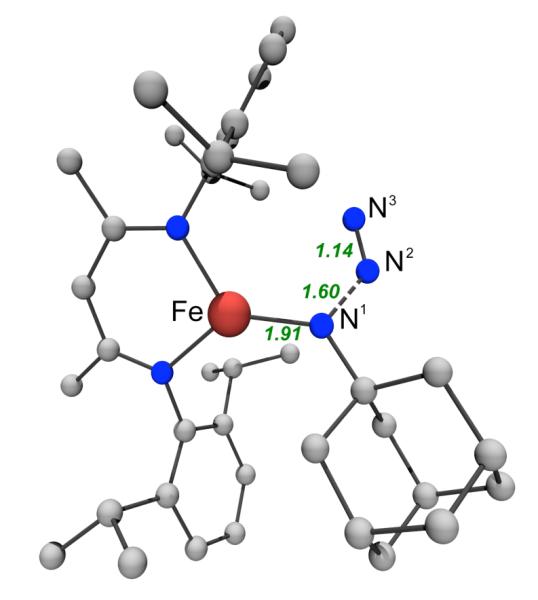
Transition state for N2 loss from quartet LMeFe(N3Ad-κN1). Hydrogens omitted from figure for clarity. Relevant distances and angles in the Fe-N-N-N core in the quartet transition state: Fe-N1, 1.91 Å; Fe-N3, 3.31 Å; N1-N2, 1.60 Å; N2-N3, 1.14 Å; Fe-N1-N2, 118°; N1-N2-N3, 127°. The corresponding sextet transition state is geometrically similar: Fe-N1, 1.94 Å; Fe-N3, 3.19 Å; N1-N2, 1.69 Å; N2-N3, 1.14 Å; Fe-N1-N2, 113°; N1-N2-N3, 123°.
Pyridine coordination to form LMeFe(py)1,2 or LMeFe(N3Ad)(py)
To explore why coordinating solvents such as pyridine greatly improved the yield of imido complex 2 relative to the hexazene complex 1, we next endeavored to understand the influence of pyridine coordination on the geometry and relative reaction barriers for N2 elimination from the Fe-organoazide intermediate. Ligating pyridine to each of the linkage isomers of LMeFe(N3Ad) gave adducts with pyridine coordinated in the apical position of a trigonal pyramid whose base is defined by the nitrogens of the β-diketiminate and the ligating nitrogen of the 1-adamantylazide ligand. For the pyridine-coordinated system, the 4LMeFe(N3Ad-κN3)(py) isomer is lowest in energy (Table S-3 and Figure S-7), which contrasts with “pyridine-free” LMeFe(N3Ad), in which 6LMeFe(N3Ad-κN1) was lowest. Hence, the coordination of pyridine results in a change in preferred AdN3 coordination mode and spin state. Although it is mildly exothermic, the calculated binding of pyridine is calculated to be overall slightly endergonic (ΔGbind = +4.2 kcal/mol) upon including entropy contributions.42 Hence, the calculations suggest that binding of pyridine to form LMeFe(N3Ad)(py) is slightly unfavorable, and that it is expected to be formed only in very small concentrations.
Even though binding pyridine is energetically uphill, we considered the possibility that LMeFe(N3Ad)(py) could eliminate N2 more rapidly than LMeFe(N3Ad), explaining the greater yield of 2 in the presence of pyridine discussed above. The calculated barrier to N2 elimination from 4LMeFe(N3Ad-κN3)(py) (the lowest energy linkage isomer) is calculated to be ΔG‡ = +9.8 kcal/mol.43 Thus, the total barrier for N2 elimination from 6LMeFe(N3Ad-κN1) through a pathway involving pyridine coordination is ΔG‡ = +14 kcal/mol, which is similar to that without pyridine (ΔG‡ = +12 kcal/mol). Therefore, there is no compelling computational evidence that coordination of a donor to LMeFe(N3Ad) facilitates N2 loss. We also evaluated the energetics of LMeFe(py) and LMeFe(py)2, the potential products of AdN3 displacement by pyridine. Both LMeFe(py) and LMeFe(py)2 are 11-12 kcal/mol uphill from LMeFe(N3Ad) (see Figure S-9), suggesting that these species are unlikely to be formed if an organoazide is present.
Diiron Intermediates Leading to Hexazene 1
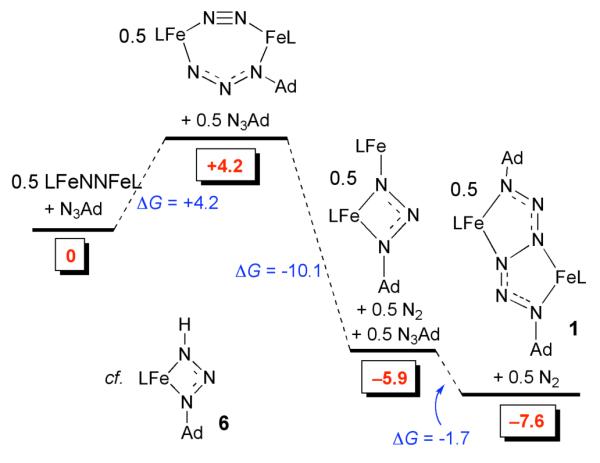
Instead of forming a 1:1 Fe:organoazide intermediate, another possibility is that N3Ad reacts with the iron(I) dimer LMeFeNNFeLMe to give a diiron–organoazide intermediate. The possible intermediates LMeFe(μ-N2-1κ-N1:2κ-N2)(μ-N3Ad-1κ-N1:2κ-N3)FeLMe and LMeFe(μ-N3Ad-1κ2-N1,N3:2κ-N3)FeLMe (shown in Scheme 4) were thus investigated; energies below are calculated as 0.5 equivalent of dimer to maintain consistent energy accounting relative to the monomeric iron complexes in other mechanisms. The formation of LMeFe(μ-N2-1κ-N1:2κ-N2)(μ-N3Ad-1κ-N1:2κ-N3)FeLMe from LMeFeNNFeLMe and N3Ad is mildly endergonic (ΔG = +4.2 kcal/mol), and loss of N2 from this species to give LMeFe(μ-N3Ad-1κ2-N1,N3:2κ-N3)FeLMe is exergonic (ΔG = −10.1 kcal/mol). Consistent with a formulation as a diiron(II) complex and a dianionic AdN 32− ligand, the spin density in LMeFe(μ-N3Ad-1κ2-N1,N3:2κ-N3)FeLMe is primarily on the iron atoms.
Scheme 4.
Though bimetallic complexes with organoazides in the μ-1κ2-N1,N3:2κ-N3 mode are rare, two examples in the literature with crystallographic characterization support the feasibility of dianionic bridging organoazides with the same coordination. The dialuminum complex ((Me3Si)2HC)2Al(μ-N3SiMe3-1κ2-N1,N3:2κ-N3)Al(CH(SiMe3)2)2 has been characterized,44 and assuming the aluminum(III) oxidation state suggests that the bridge is dianionic, as postulated in the calculated diiron intermediate. More recently, the bulky terphenyl group R* (R* = 2,6-bis(2,6-diisopropylphenyl)phenyl) was used to support R*Cr(μ-N3Ad-1κ2-N1,N3:2κ-N3)CrR*, in which the bridging 1-adamantylazide is again dianionic.45 The core angles and distances in the crystal structures of these molecules are similar to those in the computational model of the diiron intermediate (see Figure S-4). The four-membered FeNNN ring of LMeFe(μ-N3Ad-1κ2-N1,N3:2κ-N3)FeLMe also bears a striking resemblance to that in the previously characterized iron(II) triazenido complex LtBuFe(AdNNNH-κ2-N1,N3) (6, see diagram in Scheme 4).24d
During attempts to crystallize 4 (with the bulkier LtBu ligand), we fortuitously obtained a crystal structure (Figure S-3) of LtBuFe(μ-N3Ad)FeLtBu, which has the core structure of the second hypothetical intermediate.46 Although we have not been able to generate this species reproducibly for further characterization, and the crystal was of poor quality, the preliminary observation of a species with this connectivity supports the feasibility of bimolecular organoazide-bridged intermediates in the LMe system.
The computations indicate that the diiron organoazide complex is capable of leading to the final hexazene product (1). Addition of an additional 0.5 N3Ad to 0.5 LMeFe(μ-N3Ad)FeLMe to give 0.5 equiv of 1 is slightly exergonic, ΔG = −1.7 kcal/mol. The overall reaction from 0.5 LMeFeNNFeLMe to 0.5 equiv 1 is exergonic as well, ΔG = −7.6 kcal/mol. The potential energy surface showing the relative energies of these species is shown in Scheme 4.
Discussion
In the synthesis of late transition metal imido complexes, organoazides are an excellent source of the “nitrene” fragment because N2 is the only byproduct, and because organoazides are generally easy to synthesize. In this work, we used the addition of 1-adamantyl azide to an iron(I) synthon to create a well-characterized iron(III) imido complex with a trigonal planar geometry. Similar reactions have been used to generate isolable three-coordinate cobalt(III) and nickel(III) imido complexes of a smaller β-diketiminate ligand, though studies on the mechanism of these interesting reactions have not yet been reported.16b,18 Our computational and synthetic studies suggest that interaction of diketiminate-iron(I) species with organoazide forms an iron-organoazide complex LMeFe(N3Ad).
Comparison of LMeFe(N3Ad) to literature organoazide complexes
Vaddadi et al. have complemented the experimental studies of Hillhouse17b by investigating the mechanism of decomposition of coordinated organoazides via DFT calculations on (dhpe)Ni(N3Me) and (dhpe)Ni(N3Ns) (Ns = p-nitrophenylsulfonyl) model systems, dhpe = H2PCH2CH2PH2.47 Morello and Cundari have extended this computational research to the study of the decomposition of prototypical nitrene transfer reagents used in organic synthesis, such as tosyl azides, Chloramine-T, and iodonium imides.22b For a d10 CuI–scorpionate complex, a κN3 linkage isomer has been structurally characterized by Dias and coworkers (Figure 10a). In this complex, the CuNN and NNN angles are observed to be nearly linear by X-ray crystallography, suggesting a neutral organoazide ligand. Most other examples of κN3 coordination also have essentially linear NNN angles,40 and computations40c support the assignment of a neutral N3R ligand. These late transition metal complexes are distinct from the κN3 organoazides ligated to complexes of earlier transition metals, such as the tantalum(V) and vanadium(V) organoazide complexes reported by the groups of Bergman20a,b and Cummins,20c respectively. In these complexes, the short M–N bond and bent NNN and NNC angles are suggestive of a prevailing diazenylimido (LnM=N–N=N–R) bonding description with a dianionic ligand.
In this work, we describe computations that support the best description of LMeFe(N3Ad) with a form of coordinated organoazide that is different than those described above. In the complex, the organoazide is best described as a one-electron reduced N3Ad•− ligand, which is coordinated through the internal nitrogen atom (κN1). The assignment as a radical anion ligand is based on the bond lengths and N-N-N bond angle that are very similar to those calculated for the free N3Ad•− anion, and by the observation of roughly one electron of spin density on the coordinated organoazide. This assignment implies that the iron is in the iron(II) oxidation state. High-spin iron(II) can magnetically couple with the organoazide radical to give overall quartet or sextet states, and the nearly identical energies calculated for these two states suggests a very small value of the exchange coupling J despite the short distance between the two paramagnetic subsites. Because the species so rapidly reacts to give other products, we were not able to evaluate its properties experimentally except through the distribution of products derived therefrom (see below).
The one-electron reduction of a coordinated ligand by the “LMeFe” fragment has ample precedent. In the tetrazene complex LMeFe(AdNNNNAd-κ2N1,N4) (5), magnetic and spectroscopic studies showed that the tetrazene ligand exists as a radical anion coordinated to iron(II).48 Similarly, a combination of spectroscopic and computational studies showed that LMeFe(alkyne) complexes have one electron localized in a π* orbital of the alkyne fragment.24b In bimetallic examples, the dinitrogen complex LMeFeNNFeLMe is best described as a diiron(II) complex of the N22− anion, and reaction of the iron(I) fragment with acetophenone gives pinacol coupling.23b In an analogy to the radical organoazide ion postulated here, Peters recently reported an iron complex with N3Ad-κN3 coordination and an NNN angle of 147(4)°, for which DFT computations showed a spin density of 0.77 e− on the organoazide, suggesting the assignment of a monoanionic N3Ad ligand.49
Another useful comparison is to complexes (dtbpe)Ni(η2-N3R) (dtbpe = bis(di-tert-butylphosphino)ethane; R = Ad, Mes; see Figure 10c), which were structurally characterized and spontaneously convert into the stable imido complexes (dtbpe)Ni=NR.17b The crystal structures of (dtbpe)Ni(N3R) each show the η2-N3R isomer, in contrast to LMeFe(N3Ad), in which the κN1 isomer has the lowest energy by DFT. It is interesting to note the difference in preferred coordination mode, despite both (dbtpe)Ni(N3R) and LMeFe(N3Ad) utilizing bulky bidentate supporting ligands.
Mechanism of Forming Hexazene Products
The hexazene complex 1 is conceptually derived from reductive coupling of two N3Ad molecules to give an Ad2N62− bridge.25 Given the computational results that indicate LMeFe(N3Ad) has substantial unpaired spin density on the organoazide ligand when coordinated η1, we initially considered that formation of the diiron(II) hexazene complex could result from simple radical dimerization of two molecules of LMeFe(N3Ad·). However, QM/MM computations presented above indicate that there is not a significant thermodynamic driving force for this dimerization (ΔG = −0.6 kcal/mol per Fe), especially in comparison to the formation of the imido product 2 (ΔG = −42.8 kcal/mol) in which the barrier height is calculated to be low (ΔG‡ = 12 kcal/mol). Although the dimerization of LMeFe(N3Ad) molecules may be a contributing mechanism, the mechanism involving diiron intermediates (Scheme 4) is also reasonable, and more easily explains the solvent dependence of the product distribution (see below). The feasibility of bimetallic intermediates of the type {LFe}2(μ-N3Ad) is shown by the fortuitous crystallization of LtBuFe(μ-N3Ad)FeLtBu, although rational synthesis and characterization of this compound has not been achieved at this time.
Why is a coordinating solvent needed during the synthesis of 2?
We now turn our attention to the solvent effect on the outcome of the reaction LMeFeNNFeLMe + 2 N3Ad. The most coordinating solvents (THF, pyridine or arenes50) gave the highest yields of imido product 2 and lowest conversion to hexazene 1 (Table 1). The dominance of solvent coordination (rather than polarity) is most convincingly demonstrated by the difference between the outcomes of reactions performed in THF and 2,5-dimethyl-THF. This pair of solvents has previously been used to distinguish between rate effects of polarity (the solvents have similar polarities) versus coordination (dimethyl-THF is sterically prevented from coordinating).51 In THF, the yield of 2 is 80% (9:1 imido:hexazene), while in the much less coordinating 2,5-dimethyl-THF the yield of 2 is only 28% (1:2 imido:hexazene). Likewise, PhCF3 gives more imido complex (59% yield, 3:1 imido:hexazene) than toluene (41% yield, 1:1 imido:hexazene) because it is a better π-acceptor, and should bind more strongly to iron(I).24c,50
Hypothesis 1: Donor Solvents Compete with Organoazide for Coordination
We first considered an explanation in which donor solvents displace organoazide from LMeFe(N3Ad), decreasing the concentration of this key intermediate. Because conversion of LMeFe(N3Ad) to 1 is bimolecular and to 2 is unimolecular, any decrease in the concentration of LMeFe(N3Ad) from a donor solvent would disfavor coupling to form 1. This is consistent with the production of 2 in donor solvents and 1 in other solvents. To test this idea, we used computations to gauge the influence of pyridine, one of the most effective donors, on potential intermediate species. However, the QM/MM calculations show that pyridine is not capable of displacing organoazide from LMeFe(N3Ad); the exchange is calculated to be uphill by ΔG = +12 kcal/mol. The displacement of organoazide should be even less favorable for weaker donors such as THF. Therefore, the computations cast significant doubt on the verity of this hypothesis.
Hypothesis 2: Lewis Bases Catalyze Imido Formation
In a second potential explanation, we hypothesized that donor solvents catalyze N2 loss to form 2 by binding to the LMeFe(N3Ad) intermediate and transiently forming the more crowded complex LMeFe(N3Ad)(solvent). If this binding were to lower the barrier to N2 elimination, a donor solvent would accelerate imido formation relative to hexazene formation. However, computations indicated that addition of pyridine does not lower the barrier to N2 elimination (ΔG‡ = +12 kcal/mol from LMeFe(N3Ad), and ΔG‡ = +14 kcal/mol from LMeFe(N3Ad)(py)). Alternatively, the rate of dimerization of LMeFe(N3Ad)(solvent) giving hexazene 1 should be slower than the dimerization of LMeFe(N3Ad) since the terminal N is less exposed in the solvent-coordinated complex. However, LMeFe(N3Ad)(py) is computed to be +4.2 kcal/mol higher in energy than LMeFe(N3Ad), and thus the concentration of LMeFe(N3Ad)(py) would not be significant enough to slow down the rate of hexazene formation. Thus, neither displacement of N3Ad by solvent forming LMeFe(solvent) nor coordination of pyridine forming LMeFe(N3Ad)(solvent) adequately explains the observed solvent effect.
Hypothesis 3: Separate Pathways to Hexazene and Imido
Elimination of the two aforementioned explanations leaves the possibility that hexazene and imido products proceed through different mechanisms: one mechanism from the bridging N2 complex LMeFeNNFeLMe that leads to hexazene 1 in the absence of a donor solvent, and another mechanism from the mononuclear LMeFe(solvent) species that leads to imido 2. This explanation is fully consistent with the computational results. First, AdN3 is capable of displacing donor solvents and N2, because it binds strongly. Second, computations show that dinuclear LMeFeNNFeLMe can access an energetically favorable bimetallic reaction with AdN3 that leads directly to hexazene 1. The bimetallic mechanism proceeds without formation of LMeFe(N3Ad) (Scheme 3), and requires no coordinating solvent. Coordination of solvent would partially or completely displace N2 in LMeFeNNFeLMe, eliminating the bimetallic mechanism that gives 1, and opening up the monometallic mechanism through LMeFe(N3Ad) to 2.
Overall Mechanistic Picture of the Reaction of Adamantyl Azide with Low-Valent Iron
The most consistent mechanistic picture relating 1, 2, 2·solvent, and LMeFe(N3Ad) is given in Scheme 5, which starts in the upper left with a mixture of LMeFeNNFeLMe and LMeFe(solvent)n. Adamantyl azide either displaces the N2 ligand in LMeFeNNFeLMe, forming LMeFe(μ-N3Ad)FeL (pathway A), or displaces solvent in LMeFe(solvent)n, forming LMeFe(N3Ad) (pathway B). Neither iron–organoazide intermediate is observed during the reaction. In noncoordinating solvents such as pentane, iron is only present as the bimetallic N2 complex, which primarily proceeds along pathway A to give the diiron organoazide species LMeFe(μ-N3Ad)FeLMe, and leads to the diiron(II) hexazene product 1 upon addition of the second N3Ad molecule. A small amount of 2 is also formed through crossover to pathway B, which is accessed if any of the dimers are cleaved (marked “minor pathway” in Scheme 5). In coordinating solvents such as THF or pyridine, the diiron-N2 complex is in equilibrium with the solvated iron(I) compound LMeFe(solvent)n, and the exergonic addition of N3Ad gives discrete monomeric LMeFe(N3Ad) or LMeFe(N3Ad)(solvent) along pathway B, leading to 2 as the major product. Formation of small amounts of 1 could occur through radical coupling of LMeFe(N3Ad) (marked “minor pathway” in Scheme 5), or from conversion of LMeFe(N3Ad) to LMeFe(μ-N3Ad)FeLMe.
Scheme 5.

Once formed, complex 2 also reversibly coordinates pyridines such as tBupy, forming small amounts of 2•tBupy. This is important, since four-coordinate imido complex 2•tBupy is much more reactive to sources of H· than 2.28,33 The fast reaction of 2·tBupy with 1,4-cyclohexadiene (CHD) to afford the amido species LMeFe(NHAd)(tBupy) (3•tBupy)28 is a convenient test of whether 1 can transiently form 2 in solution. Complex 1 did not form detectible quantities of LMeFe(NHAd)(tBupy) by 1H NMR spectroscopy when heated to 80 °C in the presence of 100 equiv 1,4-cyclohexadiene and 10 equiv tBupy. Therefore, the formation of 1 is irreversible and cannot access the imido complex 2.
The most synthetically useful solvent for the preparation of 2 has proven to be THF, since it is coordinating enough to steer the product distribution mostly away from hexazene, but does not coordinate to the imido product. Although small amounts of tBupy also gave a desirable imido:hexazene ratio (7:1), the presence of pyridine catalyzes the decomposition of 2 by “turning on” hydrogen atom abstraction (HAA) pathways.28,33
Implications For the Larger LtBu System
The mechanistic picture relating 1, 2, and LMeFe(N3Ad) in Scheme 5 is explicit only for the LMe system. Using LtBu as the supporting ligand leads to a good yield of imido complex 4 even in pentane, and thus coordinating solvents are not necessary to avoid hexazene formation at room temperature. This observation can be accommodated within the mechanistic picture in Scheme 5 using different relative rates of the individual steps. We speculate that in the LtBuFe system, the reaction begins along the diiron pathway A (as for the LMe system in pentane), giving LtBuFe(μ-N3Ad)FeLtBu (see above) as a key intermediate. Since the added steric bulk of the LtBu ligand destabilizes the diiron intermediates at the top of Scheme 5, the fragmentation to LtBuFe(N3Ad) (red “minor pathway”) would be more rapid, and the attack of a second azide (giving hexazene) would be slower than in the LMe system. This corresponds to crossover from the bimetallic manifold A to the monoiron manifold B (i.e., the downward “minor pathway” arrow in Scheme 5 becomes the major pathway). Though the inability to characterize intermediates prevents us from further supporting this mechanistic scheme for the LtBu system, it is important that a single mechanistic scheme can rationalize all the trends in the reaction outcomes for both LMe and LtBu complexes.
Properties of Imidoiron(III) Complex 2
Though 2 is only metastable (solids or solutions must be kept at reduced temperature to have lifetimes of more than a few hours), it has been crystallized and characterized through several methods. Crystallographic data show a short iron-nitrogen bond similar to those in other iron(III)-imido complexes (further analysis below). These data are supported by X-ray absorption studies, which also show similar features in the uncrystallized LtBuFeNAd (4). Therefore, the spectroscopic evidence gleaned from solutions of 2•tBupy28 and 434 in previous communications are now supported by compelling structural data.
The greater purity of 2 has enabled the use of additional physical techniques. For example, 1H NMR spectra show that 2 reversibly binds pyridine but not THF. Note that the 1H NMR spectra of 2 are surprisingly narrow for an iron(III) complex. This is the result of the intermediate-spin (S = 3/2) ground state of the iron(III) ion, which has fairly low-lying excited states that enable rapid electronic relaxation and hence slow nuclear relaxation.52 The quartet ground state was inferred from X-band EPR spectra of mixtures containing 2 in previous studies,28 and is now supported by magnetic susceptibility studies on crystalline 2, as well as X-band EPR spectra of purified 2.
Structural Trends in Fe, Co, and Ni Imido Complexes
As a result of the recent successes in stabilizing imido complexes of the late first-row transition metals,12,13,14,15,16,17,18 there now exist enough structurally characterized examples (Table S-6) to begin to evaluate trends.
The first notable feature is that the isolable Fe, Co, and Ni imido complexes always have coordination numbers of three or four.53 This generalization can be rationalized by examining the ligand field splitting, which shows that trigonal-planar and tetrahedral geometries (as well as square-planar if not low-spin) lead to incomplete occupation of the d-orbitals that have the correct symmetry for π-bonding with the imido group.12,13,16,18,19,28 The interaction of empty (or partially empty) d orbitals with the p-orbitals of sp-hybridized nitrogen in the imido ligand leads to π-bonding. In low-spin octahedral and square-planar geometries, the appropriate d orbitals are filled, and there can be no stabilizing π-interactions.3
Further details can be gleaned from a search of the Cambridge Crystallographic Database. Figure 14 shows a scatter plot of M=N bond length versus M=N–C bond angle for all known structurally characterized examples of terminal imido complexes of Fe, Co, and Ni, organized by both metal and coordination geometry.54
Figure 14.

Correlation of M=N bond lengths with M=N–C bond angle in literature Fe, Co, Ni complexes with terminal imido ligands, organized by (a) metal and (b) coordination geometry. For crystal structures with multiple molecules in the asymmetric unit, the average bond parameters are used. The data are tabulated in Table S-6.
Although there is significant scatter in the data, a few points are worth mentioning. First, there is little correlation between M=N bond length and M=N–C bond angle, an observation which has also been noted in early transition metal imido complexes.55 Second, neither the average M=N bond length (Fe, 1.64(3) Å (n = 19; Co, 1.65(2) Å (n = 9); Ni, 1.69(2) Å (n = 4)) nor the M=N–C bond angle (Fe, 172(7)°; Co, 173(6)°; Ni, 168(8)°) are dependent on the identity of the metal. Third, Figure 14b does suggest that the bond metrics are dependent on the coordination geometry of the metal. Thus, tetrahedral imido complexes have a more linear M=N–C angle (176(3)°, n = 21) than trigonal (166(7)°, n = 9) imido complexes (p = 0.0017), and also have a more linear angle than square-planar (162(5)°, n = 2) imido complexes (p = 0.11). Tetrahedral complexes can give two M=N π-bonds (metal-nitrogen triple bond), but partial occupation of M=N π* orbitals in the planar complexes gives less metal-nitrogen π bonding.12,13,16,18,19 It is reasonable that the M=N-C angle would be the metrical parameter that is most responsive to this effect, because computational studies have shown that bending the imido ligand at nitrogen is “soft” (i.e., not energetically costly).2,55
It is interesting to note that the most reactive Fe, Co, and Ni imido complexes feature a trigonal or square-planar ligand field, the same geometry that gives somewhat more acute M=N-C angles. The only catalytic reactions that have been reported using isolable Fe, Co, or Ni imido complexes are organoazide hydrogenation with a square-planar Fe(III) imido complex,13c and formation of carbodiimides and isocyanates by trigonally coordinated iron34 and nickel56 imido complexes. Trigonal copper-imido/copper-nitrene complexes have been implicated as the active intermediates in catalytic C-H amination reactions and catalytic diazene formation reported by Warren and coworkers.57 Therefore, the M=N–C bending seen in planar imido complexes may correlate with an increase in their reactivity. On the other hand, Peters recently showed catalytic diazene formation through a spectroscopically characterized, presumably trigonal-bipyramidal, imidoiron(III) species.49 Also, Gallo recently reported porphyrin-cobalt complexes that catalyze amination.58 Thus, putative higher-coordinate imido species are likely to be reactive as well.
Conclusions
The reaction of adamantyl azide with the iron(I) precursors LRFeNNFeLR (LR = bulky β-diketiminate ligand) gives several unusual results. DFT analysis of the putative metal-organoazide complex LMeFe(N3Ad) shows that LMeFeII(N3Ad·−) is the best valence description. The bending of the NNN unit in the organoazide complex leads to facile N2 loss and formation of a trigonal iron(III) imido complex LMeFe=NAd (2).
Though reactive and unstable, this imidoiron complex has been characterized in great detail. X-ray absorption and diffraction experiments demonstrate that the Fe=N bond is short, in the range of a double bond. Solid-state magnetic susceptibility and solution EPR spectroscopy of 2 show that it has an isolated quartet ground state, consistent with computational studies.28 Comparison to literature iron-imido species suggests that a planar geometry at the metal, as found here, may be responsible for the bending of the M=N–C angle and heightened reactivity.
The reaction to form LMeFeNAd is complicated by formation of the diiron hexazene complex {LMeFe}2(N6Ad2) (1). The ratio of imido and hexazene products is solvent dependent, with coordinating solvents (THF, tBupy) steering the reaction towards imido formation. Computational studies led to a mechanistic scheme in which donor solvents separate the bimetallic complex LMeFeNNFeLMe into monometallic units, which are less likely to undergo a bimetallic reaction pathway toward hexazene. We have shown that the traditional synthesis of imidometal complexes from organoazide and a low-valent metal can be hampered by unexpected side reactions such as organoazide coupling, and that making subtle adjustments to the reaction (THF vs. pentane solvent) can greatly influence the outcome. These insights contribute to the burgeoning field of late transition metal imido complexes, and help to shed light on the formation and properties of these catalytically active species.
Experimental Section
General Considerations
All manipulations were performed under a nitrogen atmosphere in an MBraun glovebox maintained at or below 1 ppm of O2 and H2O. 1-Adamantyl azide was purchased from Aldrich and crystallized twice from pentane prior to use. 1,4-Cyclohexadiene was purchased from Aldrich, vacuum distilled from calcium chloride, and stored over activated 3 Å molecular sieves. 4-tert-Butylpyridine was purchased from Aldrich, vacuum distilled from CaH2, and stored over activated 3 Å molecular sieves. The compounds [LMeFeCl]2,31 LMeFeNNFeLMe,23 and LtBuFe=NAd34 were prepared as previously described. Pentane, diethyl ether, THF and toluene were purified by passage through activated alumina and Q5 columns from Glass Contour Co. (Laguna Beach, CA). Benzene-d6 was dried over flame-activated alumina. Toluene-d8 and THF-d8 were vacuum transferred from purple sodium benzophenone ketyl solutions. THF and THF-d8 were stored over Na metal. Before use, an aliquot of each solvent was tested with a drop of sodium benzophenone ketyl in THF. Celite was dried at 250 °C overnight under vacuum. All glassware was dried overnight in a 150 °C oven. NMR data were collected on either a Bruker Avance 400 or Bruker Avance 500 spectrometer and spectra are referenced to residual C6D5H (δ 7.16 ppm), C4D7HO (δ 3.58 ppm), or C7D7H (δ 2.08 ppm). The NMR probe temperature for the variable-temperature measurements was calibrated using either ethylene glycol or methanol.59 IR data were recorded on a Shimadzu 8400S spectrometer using KBr. UV-Vis spectra were recorded on a Cary 50 spectrometer using screw-cap or Schlenk-adapted cuvettes. Elemental analysis was determined by Columbia Analytical Services, Tucson, AZ. Room temperature solution magnetic susceptibilities were determined by the Evans method.60
LMeFeNAd (2)
A 20-mL scintillation vial was loaded with LMeFeNNFeLMe (298 mg, 0.306 mmol) and THF (15 mL) to give a royal purple solution. A solution of N3Ad (108 mg, 0.611 mmol) in THF (2 mL) was added dropwise causing effervescence and a color change to dark yellow-brown. The reaction was stirred for 15 minutes, and the volatile materials were removed under vacuum. The residue was redissolved in pentane (~8 mL), the solution was filtered through Celite, and the volatile materials were removed under vacuum. The residue was dissolved in a minimum amount of pentane (~4 mL) and cooled to −45 °C, giving crystalline 2 in two crops (231 mg, 61%). 1H NMR (500 MHz, C6D6): δ 84.4 (6H, Ad-Hα), 53.4 (1H, α-CH), 37.4 (3H, Ad-Hβ or Ad-Hγ), 34.5 (3H, Ad-Hβ or Ad-Hγ), 24.2 (3H, Ad-Hβ or Ad-Hγ), −10.1 (12H, CH(CH3)2), −14.8 (4H, m-Ar or CH(CH3)2), −24.0 (6H, Me), −28.1 (2H, p-Ar), −58.4 (12H, CH(CH3)2), −74.6 (4H, m-Ar or CH(CH3)2) ppm. IR: 3056 (w), 2958 (s), 2920 (s), 2900 (s), 2865 (m), 2845 (m), 1522 (s), 1458 (m), 1437 (m), 1384 (s), 1319 (s), 1260 (w), 1177 (w), 1100 (w), 1056 (w), 1027 (w), 936 (w), 797 (m), 757 (m) cm−1. UV/vis (pentane): 323 (19 mM−1cm−1), 410 (4.0 mM−1cm−1), 480 (sh, ~2 mM−1cm−1), 580 (sh, ~0.4 mM−1cm−1) nm. μeff (C6D6, 25 °C): 4.4 ± 0.3 μB.
LMeFeNHAd (3): Method A (from LMeFeNAd (1) and 1,4-cyclohexadiene)
A J-Young NMR tube was loaded with 2 (11.7 mg, 18.8 μmol) and C6D6 (0.6 mL). 1,4-Cyclohexadiene (17.8 μL, 188 μmol) was added, and the reaction was monitored by 1H NMR spectroscopy at 23 °C. After 90 minutes no 2 remained and LMeFeNHAd was formed in 83% yield as measured by integration against an internal integration standard (sealed capillary of Cp2Co in C6D6). Method B (from [LMeFeCl]2 and LiNHAd). [LMeFeCl]2 (1.1 g, 1.08 mmol) and LiNHAd (343 mg, 2.18 mmol) were stirred in pentane (40 mL) for 8 h. The brown mixture was filtered and concentrated under vacuum to 20 mL. The brown pentane solution was cooled to −35 °C and yellow crystals of LMeFeNHAd were isolated in good yield (1.2 g, 90%). 1H NMR δH (C6D6): 125 (1, backbone), 98 (6, Me or Ad-α), 63 (3, Ad-β or Ad-γ), 41 (3, Ad-β or Ad-γ), 31 (3, Ad-β or Ad-γ), 19 (6, Me or Ad-α), −19 (4, m-Ar or CH(CH3)2), −36 (12, CH(CH3)2), −78 (2, p-Ar), −115 (12, CH(CH3)2), -125 (4, m-Ar or CH(CH3)2) ppm. 1H NMR δH (THF-d8): 70 (6, Me or Ad-α), 52 (3, Ad-β or Ad-γ), 31 (3, Ad-β or Ad-γ), 25 (3, Ad-β or Ad-γ), 12 (6, Me or Ad-α), −12 (12, CH(CH3)2), −16 (4, m-Ar or CH(CH3)2), −65 (2, p-Ar), −86 (12, CH(CH3)2), −96 (4, m-Ar or CH(CH3)2) ppm. UV/vis (pentane): 240 (15 mM−1cm−1), 300 (12 mM−1cm−1), 330 (14 mM−1cm−1) nm. μeff (C6D6, 25 °C): 4.8 ± 0.3 μB. Elem Anal Calcd: C, 75.10; H, 9.21; N, 6.74. Found: C, 74.66; H, 7.55; N, 6.28.
LMeFe(AdNNNNAd) (5)
We have previously shown that 5 is formed from LMeFeNNFeLMe and 4 equiv N3Ad in the presence of tert-butylpyridine.48 This compound can also be prepared from isolated 2, or from LMeFeNNFeLMe and N3Ad in THF instead of tert-butylpyridine via the following methods. Method A (From isolated 2): A vial was loaded with 2 (19.2 mg, 30.8 μmol) and pentane (2 mL), giving a dark yellow-brown solution. A solution of N3Ad (5.5 mg, 31 μmol) in pentane (1 mL) was added, and the resulting mixture was stirred for 2 h, during which the color changed to a dark olive-brown and a dark precipitate was evident. The mixture was cooled to −45 °C, and the olive-green precipitate was collected and washed with 3 mL of cold (−45 °C) pentane, affording spectroscopically pure 5 (19.2 mg, 78%). Method B (From LMeFeNNFeLMe in THF): A simpler procedure is given for the preparation of LMeFe(AdNNNNAd) without the use of tert-butylpyridine. A vial was loaded with LMeFeNNFeLMe (61.4 mg, 63.0 μmol) and THF (5 mL) to give a royal purple solution. A solution of N3Ad in THF (1 mL) was added, causing effervescence and a color change to dark yellow-brown. The reaction was stirred for 12 h, and the volatile materials were removed under vacuum. The residue was slurried in cold (−45 °C) pentane, and an olive-green powder was collected on a glass fritted funnel and washed with 5 mL of cold pentane to afford spectroscopically pure 5 (82 mg, 81%). Full characterization of this complex has been reported previously.48
X-ray Absorption Spectroscopy
XAS data were recorded at the Stanford Synchrotron Radiation Laboratory (SSRL) on focused beam line 9-3, under ring conditions of 3 GeV and 60-100 mA. A Si(220) double-crystal monochromator was used for energy selection and a Rh-coated mirror (set to an energy cutoff of 10 keV) was used for harmonic rejection. Internal energy calibration was performed by assigning the first inflection point of the Fe foil spectrum to 7111.2 eV. The solid samples were prepared by dilution in boron nitride, pressed into a pellet and sealed between 38 μm Kapton tape windows in a 1 mm aluminium spacer. The solution samples were prepared by dilution in toluene (~10-15 mM) and loaded into a Delrin XAS sample holder, with a 38 μm Kapton window. All samples were maintained at 10 K during data collection using an Oxford Instruments CF1208 continuous flow liquid helium cryostat. Data were measured in transmission and fluorescence mode (using a Canberra Ge 30-element array detector), respectively. XAS data were measured to k = 15 Å−1. The data were calibrated and averaged using EXAFSPAK.61 Pre-edge subtraction and splining were carried out using PYSPLINE.62 A three-region cubic spline of order 2, 3, 3 was used to model the smooth background above the edge. Normalization of the data was achieved by subtracting the spline and normalizing the post-edge region to 1. The resultant EXAFS was k3-weighted to enhance the impact of high-k data. Theoretical EXAFS signals χ(k) were calculated using FEFF (version 7.0)63 and fit to the data using EXAFSPAK.62 The non-structural parameter E0 was also allowed to vary but was restricted to a common value for every component in a given fit. The structural parameters varied during the refinements were the bond distance (R) and the bond variance (σ2). The σ2 is related to the Debye-Waller factor, which is a measure of thermal vibration and to static disorder of the absorbers/scatterers. Coordination numbers were systematically varied in the course of the analysis, but they were not allowed to vary within a given fit.
Magnetic Susceptibility
Magnetic susceptibility data were measured from powder samples of solid material in the temperature range 2 - 300 K by using a SQUID susceptometer with a field of 1.0 T (MPMS-7, Quantum Design, calibrated with standard palladium reference sample, error <2%). Multiple-field variable-temperature magnetization measurements were done at 1 T, 4 T, and 7 T with the magnetization equidistantly sampled on a 1/T temperature scale. The experimental data were corrected for underlying diamagnetism by use of tabulated Pascal’s constants,64 as well as for temperature-independent paramagnetism. The susceptibility and magnetization data were simulated with our own package julX for exchange coupled systems.65 The simulations are based on the usual spin-Hamilton operator for mononuclear complexes:
| (1) |
where S is the total spin, g is the average electronic g value, and D is the axial zero-field splitting parameter, and and E/D is the rhombicity of the zero-field splitting. Diagonalization of the Hamiltonian was performed with the routine ZHEEV from the LAPACK Library and the magnetic moments were obtained from first order numerical derivative dE/dB of the eigenvalues. Intermolecular interactions were considered by using a Weiss temperature, ΘW, as perturbation of the temperature scale, kT’ = k(T-ΘW) for the calculation. Powder summations were done by using a 16-point Lebedev grid.
Computational Details
All computations employed the Gaussian03 suite of programs.66 Whether quantum (truncated models) or hybrid quantum/classical (full experimental models) simulations, all calculations utilize the B3LYP hybrid density functional67 and the extended Pople basis set, 6-311+G(d), which incorporates diffuse and polarization basis sets on main group elements, and an f-polarization function on iron.
For the QM/MM calculations, the classical region (Universal Force Field) contained the 2,6-diisopropylphenyl and methyl substituents of the β-diketiminate ligand and the adamantyl group with the exception of the carbon that is directly bonded to nitrogen. The remainder of the complex was modeled at the B3LYP/6-311+G(d) level of theory. The ONIOM methodology of Morokuma and coworkers was used for all quantum/classical hybrid simulations.68
All open shell species were modeled within the unrestricted Kohn-Sham formalism. Inspection of total spin expectation values suggested some degree of spin contamination for the quartet states, so results should be viewed with this caveat. All models (full and truncated) were geometry optimized without symmetry constraint using gradient methods. The energy Hessian was evaluated at all stationary points to designate them as either minima or transition states at the pertinent levels of theory. Reported free energies are at 298.15 K and 1 atm and are calculated with unscaled vibrational frequencies.
Synopsis.
Reaction of diketiminate-iron(I) sources with adamantyl azide gives initial adducts that are best described as iron(II)-organoazide radical anion complexes, according to QM/MM computations. These subsequently convert into metastable but isolable three-coordinate imidoiron(III) complexes LFe=NAd (L = β-diketiminate), which have been characterized with spectroscopic, magnetic, and structural techniques. The mechanism of formation of the imidoiron(III) complex and byproducts is examined in detail.
Supplementary Material
Acknowledgements
The authors acknowledge financial support from the Petroleum Research Fund (44942-AC to P.L.H.), the Department of Energy (DE-FG02-03ER15387 to T.R.C.), the National Science Foundation (CHE-0911314 to P.L.H.; Graduate Research Fellowship to R.E.C.), and the Department of Chemistry and Chemical Biology at Cornell University (S.D.). SSRL operations are funded by the Department of Energy, Office of Basic Energy Sciences. The Structural Molecular Biology program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and by the Department of Energy, Office of Biological and Environmental Research. Calculations employed the UNT computational chemistry resource, for which T.R.C. and N.J.D. acknowledge the NSF for support through CRIF grant CHE-0741936. We thank Dr. William Brennessel for assistance with X-ray crystallography.
Footnotes
Supporting Information Available. Spectroscopic and crystallographic details, additional computational results, compiled data from the Cambridge Structural Database, and full list of authors for ref 66.
References and Notes
- 1 (a).Schrock RR. Chem. Rev. 2009;109:3211–3226. doi: 10.1021/cr800502p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schrock RR. Angew. Chem. Int. Ed. 2006;45:3748–3759. doi: 10.1002/anie.200600085. [DOI] [PubMed] [Google Scholar]; (c) Schrock RR. J. Mol. Catal. A: Chem. 2004;213:21–30. [Google Scholar]; (d) Schrock RR, Hoveyda AH. Angew. Chem. Int. Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; (e) Schrock RR. Tetrahedron. 1999;55:8141–8153. [Google Scholar]; (f) Schrock RR. Pure Appl. Chem. 1994;66:1447–1454. [Google Scholar]
- 2 (a).Nugent WA, Mayer JM. Metal-Ligand Multiple Bonds. Wiley; New York: 1988. [Google Scholar]; (b) Wigley DE. Prog. Inorg. Chem. 1994;42:239–482. [Google Scholar]
- 3.Mayer JM. Comments Inorg. Chem. 1988;8:125–135. [Google Scholar]
- 4.Note that some imide transfer reactions have been achieved with early transition metals, for example: Breslow R, Gellman S H. Chem. Commun. 1982:1400–1401. Katsuki T. Synlett. 2003:281–297. Nishikori H, Katsuki T. Tetrahedron Lett. 1996;37:9245–9248. Noda K, Hosoya N, Irie R, Ito Y, Katsuki T. Synlett. 1993:469–471. Lai T-S, Kwong H-L, Che C-M, Peng S-M. Chem. Commun. 1997:2373–2374. Simonato J-P, Pécaut J, Scheidt R, Marchon J-C. Chem. Commun. 1999:989–990. Du Bois J, Tomooka C S, Hong J, Carreira E M. J. Am. Chem. Soc. 1997;119:3179–3180. Yu X-Q, Huang J-S, Zhu N, Che C-M. Org. Lett. 2000;2:2233–2236. doi: 10.1021/ol000107r. Yang J, Weinberg R, Breslow R. Chem. Commun. 2000:531–532. Kohmura Y, Katsuki T. Tetrahedron Lett. 2001;42:3339–3342.
- 5.For recent reviews on catalytic C-H amination reactions, see: Müller P, Fruit C. Chem. Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. Dauban P, Dodd R H. Synlett. 2003;11:1571–1586. Davies HM L, Long M S. Angew. Chem. Int. Ed. 2005;44:3518–3520. doi: 10.1002/anie.200500554. Halfen J A. Curr. Org. Chem. 2005;9:657–669. Denini S, Gallo E, Caselli A, Ragaini F, Fantauzzi S, Piangiolino C. Coord. Chem. Rev. 2006;250:1234–1253. Compain P, Toumieux S. Catalytic intramolecular C-H aminations: a powerful tool for the synthesis of various heterocyclic systems. In: Attanasi O A, Spinelli D, editors. Targets in Heterocyclic Systems. Italian Society of Chemistry; Rome: 2007. Davies HM L, Manning J R. Nature. 2008;451:417–424. doi: 10.1038/nature06485. Von Zezschwitz P. Nachr. Chem. 2008;56:897–901. Collet F, Dodd R H, Dauban P. Chem. Commun. 2009:5061–5074. doi: 10.1039/b905820f. Fantauzzi S, Caselli A, Gallo E. Dalton Trans. 2009:5434–5443. doi: 10.1039/b902929j.
- 6.For recent reviews on aziridines and catalytic aziridination reactions, see: Müller P, Fruit C. Chem. Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. Halfen J A. Curr. Org. Chem. 2005;9:657–669. Tanner D. Angew. Chem. Int. Ed. Engl. 1994;33:599–619. Osborn HM I, Sweeney J. Tetrahedron: Asymmetry. 1997;8:1693–1715. Sweeney J B. Chem. Soc. Rev. 2003;31:247–258. doi: 10.1039/b006015l.
- 7.Bimetallic group 9-11 complexes featuring bridging imido ligands are also well-studied, for example see: Sharp P R, Ge Y W. J. Am. Chem. Soc. 1987;109:3796–3797. Ge Y W, Sharp P R. Organometallics. 1988;7:2234–2236. Ge Y W, Peng F, Sharp P R. J. Am. Chem. Soc. 1990;112:2632–4260. Ge Y W, Sharp P R. J. Am. Chem. Soc. 1990;112:3667–3668. Ramamoorthy V, Sharp P R. Inorg. Chem. 1990;29:3336–3339. Ge Y W, Sharp P R. Inorg. Chem. 1992;31:379–384. Ge Y W, Sharp P R. Inorg. Chem. 1993;32:94–100. Sharp P R, Yi Y, Wu Z, Ramamoorthy V. Spec. Publ. - R. Soc. Chem. 1993;131:198–201. Ge Y-W, Ye Y, Sharp P R. J. Am. Chem. Soc. 1994;116:8384–8385. Ye C, Sharp P R. Inorg. Chem. 1995;34:55–59. Li J J, Li W, James A J, Holbert T, Sharp T P, Sharp P R. Inorg. Chem. 1999;38:1563–1572. Sharp P R. Comments Inorg. Chem. 1999;21:85–114. Sharp P R. Dalton. 2000:2647–2657. Anandhi U, Holbert T, Lueng D, Sharp P R. Inorg. Chem. 2003;42:1282–1295. doi: 10.1021/ic025987f. Singh A, Anandhi U, Cinellu M A, Sharp P R. Dalton Trans. 2008:2314–2327. doi: 10.1039/b715663d. Dobbs D A, Bergman R G. J. Am. Chem. Soc. 1993;115:3836–3837. Dobbs D A, Bergman R G. Organometallics. 1994;13:4594–4605. Danopoulos A A, Wilkinson G, Sweet T K N, Hursthouse M B. J. Chem. Soc., Dalton Trans. 1996:3771–3778. Arita H, Ishiwata K, Kuwata S, Ikariya T. Organometallics. 2008;27:493–496. Ishiwata K, Kuwata S, Ikariya T. J. Am. Chem. Soc. 2009;131:5001–5009. doi: 10.1021/ja900650j. Nichols P J, Fallon G D, Murray K S, West B O. Inorg. Chem. 1988;27:2795–2800. Ohki Y, Takikawa Y, Hatanaka T, Tatsumi K. Organometallics. 2006;25:3111–3113. Takemoto S, Ogura S-I, Yo H, Hosokoshi Y, Kamikawa K, Matsuzaka H. Inorg. Chem. 2006;45:4871–4873. doi: 10.1021/ic060744z. Zart M K, Powell D, Borovik A S. Inorg. Chim. Acta. 2007;360:2397–2402. doi: 10.1016/j.ica.2006.12.020. Lee S W, Trogler W C. Inorg. Chem. 1990;29:1099–1102. Oro L A, Ciriano M A, Tejel C, Bordonaba M, Graiff C, Tiripicchio A. Chem. Eur. J. 2004;10:708–715. doi: 10.1002/chem.200305378. Takemoto S, Morita H, Kamikawa K, Matsuzaka H. Chem. Commun. 2006:1328–1330. doi: 10.1039/b517222e. Allan R E, Beswick M A, Paver M A, Raithby P R, Steiner A, Wright D S. Angew. Chem., Int. Ed. Engl. 1996;35:208–209. Reib P, Fenske D. Z. Anorg. Allg. Chem. 2000;626:2245–2247. Reiss P, Fenske D. Z. Anorg. Allg. Chem. 2000;626:1317–1331. Badiei Y M, Krishnaswamy A, Melzer M M, Warren T H. J. Am. Chem. Soc. 2006;128:15056–15057. doi: 10.1021/ja065299l. Badiei Y M, Dinescu A, Dai X, Palomino R M, Heinemann F W, Cundari T R, Warren T H. Angew. Chem., Int. Ed. 2008;47:9961–9964. doi: 10.1002/anie.200804304. Cundari T R, Dinescu A, Kazi A B. Inorg. Chem. 2008;47:10067–10072. doi: 10.1021/ic801337f.
- 8 (a).Ashley-Smith J, Green M, Mayne N, Stone FGA. Chem. Commun. 1969:409. [Google Scholar]; (b) McGlinchey MJ, Stone FGA. Chem. Commun. 1970:1265. [Google Scholar]
- 9.Michelman RI, Andersen RA, Bergman RG. J. Am. Chem. Soc. 1991;113:5100–5102. [Google Scholar]
- 10 (a).Glueck DS, Hollander FJ, Bergman RG. J. Am. Chem. Soc. 1989;111:2719–2721. [Google Scholar]; (b) Glueck DS, Wu J, Hollander FJ, Bergman RG. J. Am. Chem. Soc. 1991;113:2041–2054. [Google Scholar]
- 11 (a).Danopoulos AA, Wilkinson G, Hussain-Bates B, Hursthouse MB. Polyhedron. 1992;11:2961–2964. [Google Scholar]; (b) Burrell AK, Steedman AJ. Organometallics. 1997;16:1203–1208. [Google Scholar]
- 12.Brown SD, Peters JC. J. Am. Chem. Soc. 2005;127:1913–1923. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]
- 13 (a).Brown SD, Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:322–323. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]; (b) Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]; (c) Bart SC, Lobkovsky E, Bill E, Chirik PJ. J. Am. Chem. Soc. 2006;128:5302–5303. doi: 10.1021/ja057165y. [DOI] [PubMed] [Google Scholar]; (d) Mehn MP, Brown SD, Jenkins DM, Peters JC, Que L. Inorg. Chem. 2006;45:7417. doi: 10.1021/ic060670r. [DOI] [PubMed] [Google Scholar]; (e) Lu CC, Saouma CT, Day MW, Peters JC. J. Am. Chem. Soc. 2007;129:4–5. doi: 10.1021/ja065524z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Scepaniak JJ, Young JA, Bontchev RP, Smith JM. Angew. Chem. Int. Ed. 2009;48:3158–3160. doi: 10.1002/anie.200900381. [DOI] [PubMed] [Google Scholar]
- 14 (a).Verma AK, Nazif TN, Achim C, Lee SC. J. Am. Chem. Soc. 2000;122:11013. [Google Scholar]; (b) Thomas CM, Mankad NP, Peters JC. J. Am. Chem. Soc. 2006;128:4956–4957. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nieto I, Ding F, Bontchev RP, Wang H, Smith JM. J. Am. Chem. Soc. 2008;130:2716–2717. doi: 10.1021/ja0776834. [DOI] [PubMed] [Google Scholar]
- 15.Ni CB, Fettinger JC, Long GJ, Brynda M, Power PP. Chem. Commun. 2008:6045–6047. doi: 10.1039/b810941a. [DOI] [PubMed] [Google Scholar]
- 16 (a).Jenkins DM, Betley TA, Peters JC. J. Am. Chem. Soc. 2002;124:11238–11239. doi: 10.1021/ja026852b. [DOI] [PubMed] [Google Scholar]; (b) Hu X, Meyer K. J. Am. Chem. Soc. 2004;126:16322–16323. doi: 10.1021/ja044271b. [DOI] [PubMed] [Google Scholar]; (c) Dai X, Kapoor P, Warren TH. J. Am. Chem. Soc. 2004;126:4798–4799. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]; (d) Shay DT, Yap GPA, Zakharov LN, Rheingold AL, Theopold KH. Angew. Chem., Int. Ed. 2005;44:1508–1510. doi: 10.1002/anie.200462529. [DOI] [PubMed] [Google Scholar]; 2006. p. 7870. erratum:; (e) Cowley RE, Bontchev RP, Sorrell J, Sarracino O, Feng Y, Wang H, Smith JM. J. Am. Chem. Soc. 2007;129:2424–2425. doi: 10.1021/ja066899n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jones C, Schulten C, Rose RP, Stasch A, Aldridge S, Woodul WD, Murray KS, Moubaraki B, Brynda M, La Macchia G, Gagliardi L. Angew. Chem. Int. Ed. 2009;48:7406–7410. doi: 10.1002/anie.200900780. [DOI] [PubMed] [Google Scholar]
- 17 (a).Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2001;123:4623–4624. doi: 10.1021/ja010358a. [DOI] [PubMed] [Google Scholar]; (b) Waterman R, Hillhouse GL. J. Am. Chem. Soc. 2008;130:12628–12629. doi: 10.1021/ja805530z. [DOI] [PubMed] [Google Scholar]
- 18.Kogut E, Wiencko HL, Zhang L, Cordeau DE, Warren TH. J. Am. Chem. Soc. 2005;127:11248–11249. doi: 10.1021/ja0533186. [DOI] [PubMed] [Google Scholar]
- 19.Holland PL. Acc. Chem. Res. 2008;41:905–914. doi: 10.1021/ar700267b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20 (a).Proulx G, Bergman RG. J. Am. Chem. Soc. 1995;117:6382–6383. [Google Scholar]; (b) Proulx G, Bergman RG. Organometallics. 1996;15:684–692. [Google Scholar]; (c) Fickes MG, Davis WM, Cummins CC. J. Am. Chem. Soc. 1995;117:6384–6385. [Google Scholar]; (d) Guillemot G, Solari E, Floriani C, Rizzoli C. Organometallics. 2001;20:607–615. [Google Scholar]; (e) Dias HVR, Polach SA, Goh S-K, Archibong EF, Marynick DS. Inorg. Chem. 2000;39:3894–3901. doi: 10.1021/ic0004232. [DOI] [PubMed] [Google Scholar]; (f) Hanna TA, Baranger AM, Bergman RG. Angew. Chem. Int. Ed. Engl. 1996;35:653–655. [Google Scholar]; (g) Barz M, Herdtweck E, Thiel WR. Angew. Chem. Int. Ed. 1998;37:2262–2265. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2262::AID-ANIE2262>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (h) Albertin G, Antoniutti S, Baldan D, Castro J, García-Fontán S. Inorg. Chem. 2008;47:742–748. doi: 10.1021/ic701907y. [DOI] [PubMed] [Google Scholar]
- 21.Cenini S, La Monica G. Inorg. Chim. Acta. 1976;18:279–293. [Google Scholar]
- 22 (a).Wu H, Hall MB. J. Am. Chem. Soc. 2008;130:16452–16453. doi: 10.1021/ja805105q. [DOI] [PubMed] [Google Scholar]; (b) Cundari TR, Morello GR. J. Org. Chem. 2009;74:5711–5714. doi: 10.1021/jo900941u. [DOI] [PubMed] [Google Scholar]
- 23 (a).Smith JM, Lachicotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Rodgers KR, Holland PL. J. Am. Chem. Soc. 2001;123:9222–9223. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]; (b) Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J. Am. Chem. Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 24 (a).Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL. J. Am. Chem. Soc. 2004;126:4522–4523. doi: 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]; (b) Stoian SA, Yu Y, Smith JM, Holland PL, Bominaar EL, Münck E. Inorg. Chem. 2005;44:4915–4922. doi: 10.1021/ic050321h. [DOI] [PubMed] [Google Scholar]; (c) Yu Y, Smith JM, Flaschenriem CJ, Holland PL. Inorg. Chem. 2006;45:5724–5751. doi: 10.1021/ic052136+. [DOI] [PubMed] [Google Scholar]; (d) Yu Y, Sadique AR, Smith JM, Dugan TR, Cowley RE, Brennessel WW, Flaschenriem CJ, Bill E, Cundari TR, Holland PL. J. Am. Chem. Soc. 2008;130:6624–6638. doi: 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sadique AR, Brennessel WW, Holland PL. Inorg. Chem. 2008;47:784–786. doi: 10.1021/ic701914m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowley RE, Elhaïk J, Eckert NA, Brennessel WW, Bill E, Holland PL. J. Am. Chem. Soc. 2008;130:6074–6075. doi: 10.1021/ja801375g. [DOI] [PubMed] [Google Scholar]
- 26.Bonyhady SJ, Green SP, Jones C, Nembenna S, Stasch A. Angew. Chem. Int. Ed. 2009;48:2973–2977. doi: 10.1002/anie.200900331. [DOI] [PubMed] [Google Scholar]
- 27.During the synthesis of 2 from LMeFeNNFeLMe and 2 equiv N3Ad, trace amounts (2 – 5%) of the tetrazene complex LMeFe(AdNNNNAd-κ2N1N4) (5) are unavoidably present in crude reaction mixtures, presumably because the cycloaddition reaction of 2 with AdN3 is kinetically competitive with imido formation. Fortunately, 5 is much less soluble in pentane than 2, and extraction of the crude material into pentane is usually sufficient to reduce the amount of 5 contaminant in 2 to <1%, as judged by 1H NMR spectroscopy. Also, note that 5 was originally obtained by reaction of 2·tBupy with 1 equiv of N3Ad (see reference 48). We have noticed that the reaction proceeds qualitatively faster when base-free 2 is used instead of 2·tBupy, consistent with a mechanism that involves rate-limiting tBupy dissociation to form 2, and subsequent cycloaddition of N3Ad to the Fe=N bond. Complex 5 can be also obtained in 81% yield in one step from LMeFeNNFeLMe and 4 equiv N3Ad in THF, without the use of pyridine in the reaction. Details of this simplified synthesis are given in the Experimental Section.
- 28.Eckert NA, Vaddadi S, Stoian S, Lachicotte RJ, Cundari TR, Holland PL. Angew. Chem., Int. Ed. 2006;45:6868–6871. doi: 10.1002/anie.200601927. [DOI] [PubMed] [Google Scholar]
- 29.The average Fe=N bond length is 1.65(3) Å, and the average Fe=N–C angle is 173(5)° calculated from the 15 imidoiron compounds with terminal imido ligands in the Cambridge Structural Database, v. 5.30 (Feb 2009 update). Allen F H. Acta Cryst. 2002;B58:380–388. doi: 10.1107/s0108768102003890.
- 30.The large library of 3- and 4-coordinate iron–diketiminate complexes in the Cambridge Crystallographic Structure Database has allowed us to determine average Fe–Ndiketiminate bond lengths as a function of coordination number and oxidation state; see SI for details.
- 31.Eckert NA, Smith JM, Lachicotte RJ, Holland PL. Inorg. Chem. 2004;43:3306–3321. doi: 10.1021/ic035483x. [DOI] [PubMed] [Google Scholar]
- 32.The same compound “tBupy•BPh3” is obtained from tBupy + BPh3 in the absence of any Fe complex; see SI for details.
- 33.Cowley RE, Eckert NA, Vaddadi S, Cundari TR, Holland PL. manuscript in preparation.
- 34.Cowley RE, Eckert NA, Elhaïk J, Holland PL. Chem. Commun. 2009:1760–1762. doi: 10.1039/b820620a. [DOI] [PubMed] [Google Scholar]
- 35.Frozen solutions gave somewhat different spectra, probably from greater decomposition. See Supporting Information for details.
- 36 (a).Aliaga-Alcalde N, George S. DeBeer, Mienert B, Bill E, Wieghardt K, Neese F. Angew. Chem. Int. Ed. 2005;44:2908–2912. doi: 10.1002/anie.200462368. [DOI] [PubMed] [Google Scholar]; (b) Berry F, Bill E, Bothe E, George S. DeBeer, Mienert B, Neese F, Wieghardt K. Science. 2006;312:1937–1941. doi: 10.1126/science.1128506. [DOI] [PubMed] [Google Scholar]
- 37.It is also important to note that the EXAFS data of the amido complex 3 show no evidence of a short Fe-N bond; the data are best fit by inclusion of three Fe-N interactions at 2.00 Å. Attempts to separate the first shell into shorter 1.8 Å and longer 2.0 Å components, as indicated by the crystal structure, resulted in the two distance components converging to the same value. This suggests that the separation of these components is beyond the resolution of the EXAFS data.
- 38.The EPR signal assigned to 2 in reference 28 was present in ~70% yield based on integration.
- 39.Trautwein AX, Bill E, Bominaar EL, Winkler H. Struct. Bonding. 1991;78:1–95. [Google Scholar]
- 40 (a).Albertin G, Antoniutti S, Baldan D, Castro J, García-Fontán S. Inorg. Chem. 2008;47:742–748. doi: 10.1021/ic701907y. [DOI] [PubMed] [Google Scholar]; (b) Barz M, Herdtweck E, Theil WR. Angew. Chem. Int. Ed. 1998;37:2262–2265. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2262::AID-ANIE2262>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (c) Dias HVR, Polach SA, Goh S-K, Archibong EF, Marynick DS. Inorg. Chem. 2000;39:3894–3901. doi: 10.1021/ic0004232. [DOI] [PubMed] [Google Scholar]
- 41 (a).Cundari TR, Pierpont AW, Vaddadi S. J. Organomet. Chem. 2007;692:4551–4559. [Google Scholar]; (b) Harrold ND, Waterman R, Hillhouse GL, Cundari TR. J. Am. Chem. Soc. 2009;131:12872–12873. doi: 10.1021/ja904370h. [DOI] [PubMed] [Google Scholar]
- 42.Note that this binding energy is not the same as the experimental pyridine binding earlier in the paper; Figure 3 describes the binding of pyridine to the imido complex rather than the organoazide complex.
- 43.A potential energy surface showing the relative energies of LMeFe(N3Ad), LMeFe(N3Ad)(py), and the N2 elimination transition states is given in Figure S-7.
- 44.Uhl W, Gerding R, Pohl S, Saak W. Chem. Ber. 1995;128:81–85. [Google Scholar]
- 45.Ni C, Ellis BD, Long GJ, Power PP. Chem. Commun. 2009:2332–2334. doi: 10.1039/b901494b. [DOI] [PubMed] [Google Scholar]
- 46.This structure resulted from an unsuccessful attempt to isolate 4. Concentrated pentane solutions of LtBuFeNNFeLtBu and N3Ad (1:2) were added together at −45 °C and kept at −45 °C overnight, resulting in the deposition of an amorphous red-brown solid and a very small quantity of well-formed orange crystals of LtBuFe(μ-N3Ad)FeLtBu.
- 47.Cundari TR, Vaddadi S. THEOCHEM. 2006;801:47–53. [Google Scholar]
- 48.Cowley RE, Bill E, Neese F, Brennessel WW, Holland PL. Inorg. Chem. 2009;48:4828–4836. doi: 10.1021/ic900001y. [DOI] [PubMed] [Google Scholar]
- 49.Mankad NP, Müller P, Peters JC. J. Am. Chem. Soc. 2010;132:4083–4085. doi: 10.1021/ja910224c. [DOI] [PubMed] [Google Scholar]
- 50.Arenes are competitive ligands for iron(I) due to the strength of backbonding from Fe to π* orbitals of the arene fragment. Ref. 24b-c show examples of diketiminate-supported iron(I)-arene complexes.
- 51.Wax MJ, Bergman RG. J. Am. Chem. Soc. 1981;103:7028–7030. [Google Scholar]
- 52.La Mar GN, Horrocks WD, Holm RH. NMR of Paramagnetic Molecules. Academic Press; New York: 1973. [Google Scholar]
- 53.Treating the six-electron donors Cp* or arene as three “electron pairs” is necessary to account for the imido complexes in refs 9-11. See also: Glueck D S, Green J C, Michelman R I, Wright I N. Organometallics. 1992;11:4221–4225.
- 54.Included in the analysis are all the examples included up to the Feb 2010 update of the CSD (v. 5.31), the structures reported in reference 16f, and compound 2 in this work. See SI for details.
- 55.Cundari TR, Russo M. J. Chem. Inf. Comput. Sci. 2001;41:281–287. doi: 10.1021/ci0000068. [DOI] [PubMed] [Google Scholar]
- 56.Laskowski CA, Hillhouse GL. Organometallics. 2009;28:6114–6120. [Google Scholar]
- 57(a).Badiei Y M, Dinescu A, Dai X, Palomino R M, Heinemann F W, Cundari T R, Warren T H. Angew. Chem. Int. Ed. 2008;47:9961–9964. doi: 10.1002/anie.200804304. (b) For a theoretical study on these Cu-nitrene complexes, see: Cundari T R, Dinescu A, Kazi A B. Inorg. Chem. 2008;47:10067–10072. doi: 10.1021/ic801337f.
- 58.Fantauzzi S, Caselli A, Gallo E. Dalton Trans. 2009:5434–5443. doi: 10.1039/b902929j. [DOI] [PubMed] [Google Scholar]
- 59 (a).Ammann C, Meier P, Merbach AE. J. Magn. Reson. 1982;46:319–321. [Google Scholar]; (b) Kaplan ML, Bovey FA, Cheng HN. Anal. Chem. 1975;47:1703–1705. [Google Scholar]
- 60 (a).Baker MV, Field LD, Hambley TW. Inorg. Chem. 1988;27:2872–2876. [Google Scholar]; (b) Schubert EM. J. Chem. Educ. 1992;69:62. [Google Scholar]
- 61.George GN. EXAFSPAK. Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center, Stanford University; Stanford, CA: [Google Scholar]
- 62.Tenderholt A. PySpline. Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center, Stanford University; Stanford, CA: [Google Scholar]
- 63 (a).de Leon J. Mustre, Rehr JJ, Zabinsky SI, Albers RC. Phys. Rev. B. 1991;44:4146–4156. doi: 10.1103/physrevb.44.4146. [DOI] [PubMed] [Google Scholar]; (b) Rehr JJ, de Leon J. Mustre, Zabinsky SI, Albers RC. J. Am. Chem. Soc. 1991;113:5135–5140. [Google Scholar]
- 64 (a).O’Connor CJ. Prog. Inorg. Chem. 1982;29:203–283. [Google Scholar]; (b) Weast RC, Astle MJ. CRC Handbook of Chemistry and Physics. CRC Press Inc.; Boca Raton, Florida: 1979. [Google Scholar]
- 65. Available from E.B.: http://ewww.mpi-muelheim.mpg.de/bac/logins/bill/julX_en.php.
- 66.Frisch MJ, et al. Gaussian 03, Revision D.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 67 (a).Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 68.Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K. J. Phys. Chem. 1996;100:19357–19363. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.