

Abstract

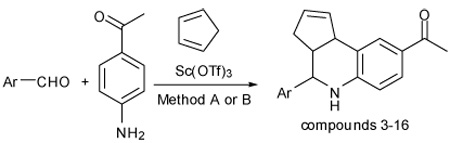
The GPR30 agonist probe G-1 and structural analogs were efficiently synthesized using multicomponent or stepwise Sc(III)-catalyzed aza-Diels Alder cyclization. Optimization of solvent and reaction temperature provided enhanced endo-diastereoselectivity.
Introduction
The discovery of the G protein-coupled estrogen receptor GPR30, (IUPHAR designation: GPER), has revealed a new pathway for non-genomic estrogen signaling. This 7-transmembrane G-protein coupled receptor is expressed in tissues throughout the body and implicated in several biologically important signaling pathways that affect normal and pathogenic states including cancer, reproduction, neuroendocrine and cardiovascular systems.1 Distinguishing the different biological roles of GPR30 from the classical nuclear estrogen receptors (ERα/β) is complicated by similarities in ligand specificity for 17β -estradiol (E2) and xenoestrogens, with overlapping cellular responses mediated by distinct signaling pathways. The development of GPR30-selective chemical probes would provide valuable molecular tools to distinguish these complex systems in vitro and in vivo.

Recently, a combined virtual and biomolecular screen identified the first GPR30-selective agonist G-1 (1), a substituted tetrahydro-3H- cyclopenta[c]quinoline.2 This compound has been used to investigate GPR30-mediated biological effects in a wide variety of different cell types and in vivo models.3 An evaluation of chemical probes developed through the National Institutes of Health Molecular Libraries and Imaging initiative rated G-1 with high confidence for use in vitro and in vivo.4 Therefore, we were interested in developing a high yield, diastereoselective preparative scale synthesis of G-1 to provide sufficient quantities for expanded biological studies, and for construction of related analogs to investigate structure-activity relationships of GPR30-mediated signaling.
The tetrahydroquinoline scaffold is accessible using the three-component aza-Diels Alder (Povarov) reaction that can be catalyzed by a variety of Bronsted and Lewis acids.5 We sought conditions that would provide rapid reaction times, maximum product yields, and increased diastereoselectivity favoring the endo- or syn-product in comparison with the original synthesis that achieved ~70% yield employing trifluoracetic acid.2 Following the precedent for lanthanide catalysis,6 we found Sc(OTf)3 effected the cyclization of 6-bromopiperonal, p-aminoacetophenone and cyclopentadiene in acetonitrile (MeCN) to produce G-1 in near quantitative yield (Table 1). This procedure was successfully performed on multi-gram scale. The endo diastereomer was obtained as the major product; the syn orientation of protons H-3a and H-4 was assigned by the characteristic scalar coupling constant J (3a, 4) = 3.1 Hz, compared with significantly higher coupling constant for the exo diastereomer J (3a, 4) = 9.4 Hz. Recrystallization from acetonitrile did not further affect the diastereomer ratio.
Table 1.
Optimization of G-1 Synthesis
| Entry | Methoda,b | Solvent | Time(h) | Temp(°C) | Yield(%) | endo/exo |
|---|---|---|---|---|---|---|
| 1 | A, B | MeCN | 2 | rt | 98 | 94:6 |
| 2 | A | MeCN | 3 | 0 | 40 | 94:6 |
| 3 | B | MeCN | 3 | 0 | 98 | 97:3 |
| 4 | A, B | DCM | 2 | rt | 94 | 94:6 |
| 5 | A | DCM | 4 | 0 | 80 | 95:5 |
| 6 | B | DCM | 4 | 0 | 95 | 98:2 |
A:Multicomponent cyclization.
B:Stepwise imineformation, cyclization
Crystals of G-1 suitable for X-ray diffraction were grown by slow evaporation from acetonitrile/methanol (1:1). The ORTEP structure of G-1 confirms the structural assignment based on 1H-NMR exhibiting the endo orientation of cyclopentene ring with the 4-piperonal moiety (Figure 1). Superimposing the X-ray and computed structures of G-1 revealed only a slight difference in the conformation of the acetyl substituent. In the calculated conformation this group aligns with the plane defined by the tetrahydroquinoline ring system, but was rotated at a 15° angle in the crystal structure.
Figure 1.

X-ray ORTEP Rendition of GPR-30 Agonist G-1. X-ray structure of G-1 is depicted in light grey and the conformation obtained from energy minimization is showed in blue.
In order to assess the reaction parameters with regards to product yields and optimize the endo-diastereoselectivity, we evaluated the cyclization in different solvents, and using a stepwise procedure employing the isolated imine (Schiff base). Imines are typically prepared using dehydrating agents or azeotropic removal of water, and recent examples include ionic liquids and microwave heating.7 A simple procedure involving heating the neat aniline and aldehyde components at 170 °C for 5 minutes provided complete conversion to the desired imine, and subsequent Sc(III)-catalyzed cyclization with cyclopentadiene yielded the desired product (method B).
The yields and diastereoselectivities were identical for both methods at ambient temperature, but reducing the cyclization temperature to 0 °C and using dichloromethane (DCM) as solvent achieved the highest endo-selectivity (entry 6, Table 1).
We prepared a series of tetrahydroquinoline analogs from a variety of substituted aldehydes using the multicomponent procedure (Table 2). These conditions were generally effective, but gave relatively low yields for the 4-methoxy- and 4-hydroxybenzaldehyde substrates (entries 3 and 5). The 3-substituted analogs gave higher relative yields (entries 4 and 6). We hypothesized that resonance inhibited formation of the Schiff base intermediates due to the strong electron-donating substituents at the 4-position. Consistent with these expectations, the stepwise procedure for Sc(III)-catalyzed imine cyclization increased product yields. Both methods gave similar results for the catechol analog that is a competitive ligand for the Lewis acid, and the intermediate imine exhibited low solubility under the reaction conditions (entry 7). Increased amounts of the exo-products were observed using 3-nitrobenzaldehyde, 3-formylthiophene and formyl-cyclohexene (entries 12–14), reflecting a trend for decreasing endo-diastereoselectivity associated with more electrophilic imine intermediates.
Table 2.
Synthesis of tetrahydroquinoline analogs
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Aldehyde | Methoda,b | Time(h) | Product | Yield(% )c | endo/exod |
| 1 |  |
A | 2.0 | 3 | 88 | 93:7 |
| 2 | 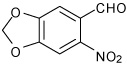 |
A | 3.0 | 4 | 95 | 95:5 |
| 3 | 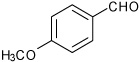 |
A | 5.0 | 5 | 75 | 91:9 |
| B | 2.5 | 5 | 90 | 92:8 | ||
| 4 | 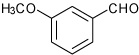 |
A | 2.0 | 6 | 95 | 92:8 |
| 5 | 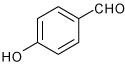 |
A | 5.0 | 7 | 70 | 95:5 |
| B | 15.0 | 7 | 80 | 96:4 | ||
| 6 | 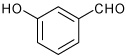 |
A | 1.5 | 8 | 95 | 96:4 |
| 7 | 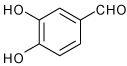 |
A | 5.0 | 9 | 70 | 92:8 |
| B | 15.0 | 9 | 72 | 92:8 | ||
| 8 |  |
A | 2.0 | 10 | 77 | 93:7 |
| 9 | 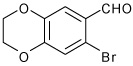 |
A | 3.0 | 11 | 84 | 92:8 |
| 10 | 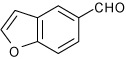 |
A | 3.0 | 12 | 92 | 92:8 |
| 11 | 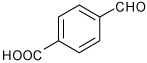 |
A | 2.5 | 13 | 92 | 92:8 |
| 12 |  |
A | 1.0 | 14 | 98 | 89:11 |
| 13 | 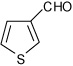 |
A | 1.5 | 15 | 94 | 80:20 |
| 14 | 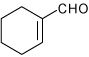 |
A | 1.0 | 16 | 90 | 63:37 |
A: Multicomponent procedure.
B: Stepwise imineformation, cyclization.
Isolated yields.
Endo/exo ratio determined by 1H NMR and HPLC.
The analogous Sc(OTf)3 mediated multi-component cyclization employing 2,3-dihydrofuran (2 eq) as the hetero-dienophile in MeCN at ambient temperature produced the isostructural furo[3,2-c]quinoline derivative 17 in moderate yield with low diastereoselectivity (Table 3).8 The syn orientation of protons H-3a and H-4 in 17-endo are evident from 1H NMR scalar coupling constants of 3 Hz at 5.1 ppm, whereas protons in the anti orientation correspond to J (3a, 4) = 10.5 Hz at 4.5 ppm. The low endo/exo ratio is consistent with other examples of Povarov cyclizations employing 2,3-dihydrofuran as an aza-dienophile, and accentuates the different stereochemical outcome in comparison with cyclopentadiene.6h,8a
Table 3.
Synthesis of furo[2, 3-c]quinoline analogs
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Methoda,b | Solvent | Time (h) |
Temp (°C) |
Yield (%) |
endo/exog |
| 1 | A | MeCNd | 5 | rt | 55 | 40:60 |
| 2 | A | MeCNe | 2 | rt | 15 | 50:50 |
| 3 | B | MeCNc | 2 | rt | 68 | 44:56 |
| 4 | B | MeCNc | 5 | 0 | 50 | 44:56 |
| 5 | B | MeOHc | 4 | rt | 6 | 27:73 |
| 6 | B | DCMc | 5 | 0 | 65 | 72:28 |
| 7 | B | Dioxane c | 3 | 0 | 52 | 75:25 |
| 8 | B | Toluene c | 5 | 0 | 45 | 77:23 |
| 9 | B | DCM/Toluenec | 5 | 0 | 54 | 82:18 |
| 10 | B | DCM/Dioxanec | 3 | 0 | 48 | 80:20 |
| 11 | B | DCMf | 3 | 0 | 50 | 84:16 |
A: Multicomponent procedure.
B: Stepwise imineformation, cyclization.
0.12 mmol of imine/mL of solvent and 2 eq. of 2,3-dihydrofuran.
0.12 mmol of aldehyde and amine/mL and 3 eq. of 2,3-dihydrofuran.
0.12 mmol of aldehyde and amine/mL and 10 eq. of 2,3-dihydrofuran.
0.5 mmol of imine/mL and 2 eq. of 2,3-dihydrofuran.
Endo/exo ratio determined by 1H NMR and HPLC.
We required the isostructural endo-isomer of 17 for comparison of biological activity with G-1 and attempted to optimize the diastereoselectivity of this reaction. Increasing the amount of 2,3-dihydrofuran from 2 to 10 equiv was detrimental to the yield of 17 (entry 2). Under these conditions 2,3-dihydrofuran undergoes ring opening to highly reactive 4-hydroxybutanal that results in formation of the undesired side product 18 from domino coupling.9 The exo-diastereomer was slightly increased using the stepwise procedure (entry 3). Methanol has been successfully used as a solvent for sulfamic acid-catalyzed Povarov cyclizations employing 2,3-dihydrofuran. However, under these conditions the Sc(III)-catalyzed procedure gave low yields and increased amounts of exo-product, accompanied by the undesired byproduct 19 that incorporates methanol through a 4-component coupling process (entry 5).8d,10 These results suggest the reaction with 2,3-dihydrofuran proceeds with a high degree of charge development under these conditions. The desired 17-endo product was obtained as the major isomer (> 70%) employing the stepwise reaction in dichloromethane, 1,4-dioxane and toluene at 0 °C. Further improvement in the endo/exo ratio were achieved using mixed solvents containing toluene or dioxane, and by increasing the concentration of the reaction mixture (entries 9–11). The preferential precipitation of the exo isomer enabled efficient separation of the diastereomers by recrystallization from acetonitrile/water.
Mechanistic studies suggest that Povarov cyclizations can proceed through concerted [4+2] aza-Diels Alder or stepwise pathways.5l The isolation of nucleophilic imine-addition product 19 indicates the stepwise process is favored for the electron rich alkene 2,3-dihydrofuran in polar alcohol solvent.10 Solvent polarity affects charge development and synchronicity of bond formation in the transition state and plays a significant role in determining the stereochemical outcome. Complexation of the Sc(III) catalyst induces polarization of the Schiff base intermediate, thereby altering the orbital energies and possibly enhancing the contributions of secondary interactions with the alkene component in non-polar solvents. The solvent effects observed in this study correlate increased endo-selectivity for the Sc(III)-catalyzed reactions of 2,3-dihydrofuran with decreased dielectric constant of the solvent and reducing the cycloaddition temperature. These results are consistent with a kinetic model in which the endo orientation is favored by π-π contact dispersion interactions, and preference for the transition state with minimum charge development as the main factors governing the observed stereoselectivity.

Conclusions
We have developed an efficient, diastereoselective synthesis of the GPR30 agonist G-1 and related analogs. The endo-structure of G-1 originally assigned on the basis of 1H NMR was verified by single crystal X-ray diffraction. A simple stepwise procedure involving formation of the imine by melting was employed in cases where the multicomponent approach was problematic, and provided increased product yields and endo-diastereoselectivity. The observed solvent effects provide guidance for ongoing efforts to develop related catalytic enantioselective cyclization procedures. The biological activities of these synthetic analogs 3–17 are currently under investigation.
Experimental
All reactions were performed in an efficient fume hood. Solvents and reagents were purchased from commercial sources and were used without further purification. Air sensitive reagents were stored in a glove box and handled according to accepted procedures. Chromatographic separations were performed using medium pressure flash chromatography and ethyl acetate/hexanes as eluent. Deuterated solvents were used without further purification. NMR spectra were acquired at ambient temperatures (18 ± 2 °C) unless otherwise noted. The 1H NMR spectra in CDCl3 were referenced to TMS unless otherwise noted. The 13C {1H} NMR spectra were recorded at 75 or 100 MHz and referenced relative to the 13C {1H} peaks of the solvent. Spectra are reported as (ppm), (multiplicity, coupling constants (Hz), and number of protons). HPLC–MS was performed using a C18 (5 µm, 3.0 × 150 mm) column.
Method A representative multicomponent procedure: (1-[4-(6-Bromo-benzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone) (G-1)
A catalytic amount of Sc(OTf)3 (0.492 g, 1.0 mmol) in anhydrous acetonitrile (2.0 cm3) was added to the mixture of 6-bromopiperonal (2.30 g, 10.0 mmol), p-aminoacetophenone (1.30 g, 10.0 mmol) and cyclopentadiene (3.30 g, 50.0 mmol) in acetonitrile (25 cm3). The reaction mixture was stirred at ambient temperature (~23 °C) for 2.0 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to provide G-1 (4.03 g, 98%, endo:exo = 94:6) as a white solid. mp: 103–105 °C; FT-IR (KBr, cm−1) 3324, 2895, 1659, 1596, 1474; δH(300 MHz; CDCl3; Me4Si) 7.70 (1 H, d, J 1.6, Ar), 7.61 (1 H, dd, J 8.3, 1.6, Ar) 7.09 (1 H, s, Ar), 7.03 (1 H, s, Ar), 6.61 (1 H, d, J 8.3, Ar), 6.00 (1 H, d, J 1.3, OCH2O), 5.99 (1 H, d, J 1.3, OCH2O), 5.96-5.92 (1 H, m, CH=CH), 5.68-5.66 (1 H, m, CH=CH), 4.98 (1 H, d, J 3.1, ArCHNH), 4.12 (1 H, d, J 8.3, ArCHCH=CH), 4.02 (1 H, br s, NH), 3.23-3.15 (1 H, m, CH2CH=CH(1H)), 2.57-2.46 (4 H, m, CH2CH=CH(1H) and COCH3), 1.86-1.77 (1 H, m, CH2CHCH).; δC(75 MHz; CDCl3; Me4Si) 196.5, 149.9, 147.6, 147.5, 133.8, 133.6, 130.4, 130.0, 128.7, 127.6, 125.1, 115.2, 113.1, 112.9, 107.7, 101.8, 56.2, 45.4, 42.0, 31.4, 26.1; HPLC-MS: Elution with 60–90% CH3CN in H2O (gradient 1.5 % min−1), exhibited a peak at 12.42 min. ESI-MS m/z (ES+) calcd for C21H18BrNO3 (M+H)+ 412.05; found 412.18.
Method B general procedure for stepwise cyclization
The aniline (1 mmol) and aldehyde (1 mmol) were dissolved in methanol (2 cm3) then concentrated in vacuo. The resulting viscous oil was heated at 170–180 °C under an argon atmosphere for 5 min. The volatiles were removed to provide the corresponding imine. The imine was dissolved in the specified solvent (4 cm3), then cyclopentadiene (0.330 g, 5.0 mmol) and a solution of scandium triflate (0.049 g, 0.1 mmol) in the solvent (0.5 mL) were added and the reaction was stirred at the specified temperature for 2–15 h. The product formation was monitored by thin layer chromatography using ethyl acetate/hexanes eluent. The volatiles were removed in vacuo. The crude product was purified as specified. The endo:exo ratio of the product was determined by integration of the 1H NMR spectra and HPLC chromatograms.
E-1-(4-((6-bromobenzo[d][1,3]dioxol-5-yl)methyleneamino) phenyl)ethanone (2)
Following Method B p-aminoacetophenone (0.135 g, 1 mmol) and 6-bromopiperonal (0.229 g, 1 mmol) were combined to provide the imine 2 (0.346 g, 100%) as a pale yellow solid. mp: 155–158 °C; νmax(KBr)/cm−1 1675, 1580, 1483, 1261, 1041; δH (300 MHz; CDCl3; Me4Si) 8.70 (1 H, s), 8.00 (2 H, d, J 7.8), 7.70 (1 H, s), 7.22 (2 H, d, J 7.8), 7.05 (1 H, s) 6.06 (2 H, s), 2.61 (3 H, s); δC (75 MHz; CDCl3; Me4Si) 197.1, 159.9, 155.9, 151.4, 147.9, 134.6, 129.7, 128.1, 121.0, 119.3, 112.8, 107.8, 102.3, 26.5.
1-((3aS,4R,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5, 9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethanone (G-1)
Following Method B catalytic amount of Sc(OTf)3 (0.049 g, 0.1 mmol) in DCM (0.2 cm3) was added to the mixture of imine, 2 (0.346 g, 1 mmol) and cyclopentadiene (0.330 g, 5.0 mmol) in DCM (4 cm3). The reaction mixture was stirred at 0 °C for 4.0 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to provide G-1 (0.390 g, 95%, endo:exo = 98:02) as a white solid.
1-(4-Benzo[1,3]dioxol-5-yl-3a,4,5,9b-tetrahydro-3H-cyclopen ta[c]quinolin-8-yl)-ethanone (3)
Following Method A piperonal (0.075 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were combined in anhydrous acetonitrile (2 cm3) and stirred for 2 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 3 (0.146 g, 88%, endo:exo = 93:7) as a colorless solid. mp: 66–68 °C; νmax(KBr)/cm−1 3324, 2887, 1651, 1596, 1228; δH(300 MHz; CDCl3; Me4Si) 7.69 (1 H, d, J 2.0), 7.59 (1 H, dd, J 8.5 and 2.0), 6.89-6.78 (3 H, m), 6.58 (1 H, d, J 8.5), 5.95 (2 H, s), 5.94-5.90 (1 H, m), 5.67-5.63 (1 H, m), 4.60 (1 H, d, J 3.2), 4.24 (1 H, br s), 4.08 (1 H, d, J 9.0), 2.97-2.91 (1 H, m), 2.54-2.52 (4 H, m), 1.90-1.81 (1 H, m); δC(75 MHz; CDCl3; Me4Si) 196.4, 150.0, 147.7, 146.7, 135.7, 133.7, 130.4, 129.9, 128.3, 127.7, 124.8, 119.3, 114.8, 108.2, 106.7, 101.0, 57.0, 45.9, 45.5, 31.3, 25.9; HRMS (EI-MS): calcd [M+H]+ for C21H19NO3 334.1443, found 334.1438.
1-[4-(6-Nitro-benzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta [c]quinolin-8-yl]-ethanone (4)
Following Method A 6-nitropiperonal (0.098 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol), cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were combined in anhydrous acetonitrile (2 cm3) and stirred for 3 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 4 (0.180 g, 95%, endo:exo = 95:5) as a colorless solid. mp: 118–120 °C; νmax(KBr)/cm−1 3318, 2906, 1659, 1597, 1248; δH(300 MHz; CDCl3; Me4Si) 7.68 (1 H, d, J 2.0), 7.60 (1 H, dd, J 8.4 and 2.0), 7.48 (1 H, s), 7.34 (1 H, s), 6.61 (1 H, d, J 8.4), 6.14 (1 H, d, J 1.1), 6.13 (1 H, d, J 1.1), 5.99-5.95 (1 H, m), 5.75-5.65 (1 H, m), 5.26 (1 H, d, J 2.9), 4.14 (1 H, d, J 8.6), 4.07 (1 H, br s), 3.34-3.25 (1 H, m), 2.64-2.52 (1 H, m), 2.49 (3 H, s), 1.91-1.82 (1 H, m); δC (75 MHz; CDCl3; Me4Si) 196.5, 152.0, 149.5, 147.01, 142.6, 134.0, 133.8, 130.2, 130.1, 128.9, 127.5, 125.0, 115.3, 107.1, 105.7, 103.0, 52.7, 45.5, 43.2, 31.3, 20.0. HRMS (EI-MS): calcd [M+H]+ for C21H18N2O5 379.1294, found 379.1298.
1-[4-(4-Methoxy-phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta [c]quinolin-8-yl]-ethanone (5)
Following Method B the imine prepared from p-aminoacetophenone (0.067 g, 0.5 mmol) and 4-methoxybenzaldehyde (0.127 g, 0.5 mmol) was dissolved in dichloromethane (2 cm3) at ambient temperature, then cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were added and the mixture stirred for 2.5 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 5 (0.144 g, 90%, endo:exo = 92:8) as a colorless solid. mp: 58–60 °C; νmax(KBr)/cm−1 3338, 2928, 1658, 1596, 1284; δH(300 MHz; CDCl3; Me4Si) 7.69 (1 H, s), 7.61 (1 H, dd, J 8.4 and 2.0), 7.31 (2 H, d, J 8.8), 6.91 (2 H, d, J 8.8), 6.93 (1 H, d, J 8.4), 5.93-5.90 (1 H, m), 5.67-5.64 (1 H, m), 4.66 (1 H, d, J 3.1), 4.23 (1 H, s), 4.10 (1 H, d, J 8.4), 3.81 (3 H, s), 3.00-2.95 (1 H, m), 2.59-2.47 (4 H, m), 1.86-1.80 (1 H, m); δC(75 MHz; CDCl3; Me4Si) 196.4, 159.0, 150.3, 133.9, 133.8, 13.5, 130.08, 128.4, 127.7, 127.6, 125.0, 114.8, 114.0, 56.9, 55.3, 46.0, 45.7, 31.5, 26.0; HRMS (EI-MS): calcd [M+H]+ for C21H21NO2 320.1651, found 320.1660.
1-[4-(3-Methoxy-phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta [c]quinolin-8-yl]-ethanone (6)
Following Method A m-methoxybenzaldehyde (0.073 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 2 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 6 (0.165 g, 95%, endo:exo = 92:8) as a colorless solid. mp: 56–58 °C; IR (KBr, cm−1) 3337, 2931, 1656, 1596, 1285; δH(300 MHz; CDCl3; Me4Si) 7.71 (1 H, d, J 2.0), 7.62 (1 H, dd, J 8.4 and 2.0), 7.31 (1 H, m), 7.02-6.96 (2 H, m), 6.85 (1 H, dd, J 8.4 and 2.0), 6.60 (1 H, d, J 8.4), 5.93-5.89 (1 H, m), 5.68-5.65 (1 H, m), 4.69 (1 H, d, J 3.3), 4.23 (1 H, br s), 4.12 (1 H, d, J 8.6), 3.83 (3 H, s), 3.06-2.98 (1 H, m), 2.60-2.54 (1 H, m), 2.50 (3 H, s), 1.88-1.81 (1 H, m). δC(100 MHz; CDCl3,)1096.6, 159.6, 150.1, 143.4, 133.6, 130.4, 129.9 129.4, 128.1, 127.6, 124.8, 118.5, 114.8, 112.4, 112.0, 57.1, 55.1, 45.6, 45.4, 31.4, 25.8; HRMS (EI-MS): calcd [M+H]+ for C21H21NO2 320.1651, found 320.1655.
1-[4-(4-Hydroxy-phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta [c]quinolin-8-yl]-ethanone (7)
Following Method B, the imine prepared from p-aminoacetophenone (0.067 g, 0.5 mmol) and 4-hydroxybenzaldehyde (0.061 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) was dissolved in dichloromethane (2 cm3) at ambient temperature and the mixture stirred for 15 h. The volatiles were removed in vacuo. The residue was purified by recrystallization from chloroform (5 cm3) to give the product 7 (0.125 g, 80%, endo:exo = 96:4) as a colorless solid. mp: 208–210 °C; νmax(KBr)/cm−1 3360, 2917, 1650, 1593, 1287; δH (300 MHz; CD3COCD3) 8.31 (1 H, d, J 2.0), 7.69 (1 H, m), 7.58 (1 H, d, J 8.4), 7.26 (2 H, d, J 8.4), 6.83 (2 H, d, J 8.4), 6.8 (1 H, s), 5.97-5.94 (1 H, m), 5.61-5.60 (1 H, m), 5.54 (1 H, s), 4.63 (1 H, d, J 3.3), 4.09 (1 H, d, J 8.6), 2.99-2.94 (1 H, m), 2.53-2.47 (1 H, m), 2.42 (3 H, s), 1.77-1.73 (1 H, m); δC (100 MHz; CD3COCD3) 195.7, 157.4, 152.0, 135.3, 133.9, 130.8, 130.6, 128.7, 128.4, 128.0, 125.3, 115.9, 115.7, 57.3, 47.1, 46.5, 32.2, 26.0. HRMS (EI-MS): calcd [M+H] + for C20H19NO2 306.1494, found 306.1506.
1-[4-(3-Hydroxy-phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta [c]quinolin-8-yl] ethanone (8)
Following Method A m-hydroxybenzaldehyde (0.061 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol), cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.0246 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 1.5 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 8 (0.150 g, 98%, endo/exo 96:4) as a colorless solid. mp: 210–212 °C; νmax(KBr)/cm−1 3346, 2936, 1640, 1599, 1296; δH(400 MHz, d6-DMSO,) 9.38 (1 H, s), 7.59 (1 H, d, J 2.0), 7.51 (1 H, dd, J 8.4 and 2.0), 7.15 (1 H, m), 6.84-6.83 (2 H, m), 6.75 (1 H, d, J 8.4), 6.66 (1 H, dd, J 8.4 and 1.6), 6.51 (1 H, s) 5.97-5.92 (1 H, m), 5.61-5.56 (1 H, m), 4.55 (1 H, d, J 2.6), 4.04 (1 H, d, J 8.6), 2.93-2.88 (1 H, m), 2.42-2.30 (4 H, m), 1.70-1.61 (1 H, m); δC (100 MHz; d6-DMSO) 195.4, 157.2, 151.1, 143.5, 134.7 129.8, 129.6, 129.1, 127.0, 126.6, 123.6, 117.2, 114.7, 113.9, 113.3, 55.8, 45.2, 44.9, 31.4, 25.9; HRMS (EI-MS): calcd [M+H]+ for C20H19NO2 306.1494, found 306.1495.
1-(4-(3,4-dihydroxyphenyl)-3a,4,5,9b-tetrahydro-3H-cyclopen ta[c]quinolin-8-yl)ethanone (9)
Following Method A 3,4-dihydroxy-benzaldehyde (0.069 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 5 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 9 (0.112 g, 70%, endo/exo = 92:8) as a colorless solid. mp: 95–97 °C; νmax(KBr)/cm−1 3485, 2932, 1658, 1589, 1282; δH(300 MHz; CDCl3; Me4Si) 7.68 (1 H, s), 7.60 (1 H, dd, J 8.5 and 2.0), 6.90-6.87 (2 H, m), 6.76 (1 H, dd, J 8.4 and 2.0), 6.49 (1 H, d, J 8.5), 5.85-5.83 (1 H, m), 5.60-5.57 (1 H, m), 4.50 (1 H, d, J 3.2), 4.22 (1 H, br s), 4.01 (1 H, d, J 8.9), 2.90-2.80 (1 H, m), 2.51-2.31 (4 H, m), 1.84-1.76 (1 H, m); δC(75 MHz; CDCl3; Me4Si) 198.4, 150.9, 144.0, 143.4, 134.1, 133.6, 130.7, 130.5, 128.1, 127.5, 125.0, 118.4, 115.2, 114.9, 113.2, 56.7, 45.8, 45.5, 31.4, 26.0; HRMS (EI-MS): calcd [M+H]+ for C20H19NO3 322.1443, found 322.1448.
1-[4-(2-Bromo-4,5-dihydroxy-phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c] quinolin-8-yl]-ethanone (10)
Following Method A 2-bromo-4,5-dihydroxy-benzaldehyde (0.217 g, 1 mmol), p-aminoacetophenone (0.135 g, 1 mmol) and cyclopentadiene (0.330 g, 5 mmol) and Sc(OTf)3 (0.049 g, 0.1 mmol) were dissolved in acetonitrile (4 cm3) and the mixture stirred for 2 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 10 (0.308 g, 77%, endo/exo = 93:7) as a colorless solid. mp: 209–211 °C; νmax(KBr)/cm−1 3470, 2934, 1660, 1596, 1280; δH(300 MHz, d6-DMSO) 9.29 (1 H br s) 7.60 (1 H, s), 7.52 (1 H, dd, J 8.5 and 2.0), 7.00 (1 H, s) 6.94 (1 H, s), 6.74 (1 H, d, J 8.5), 6.44 (1 H, br s), 5.98-5.93 (1 H, m), 5.65-5.60 (1 H, m), 4.69 (1 H, d, J 3.3), 4.02 (1 H d, J 8.5), 3.03-2.93 (1 H, m), 2.46-2.32 (4 H, m), 1.69-1.61 (1 H, m); δC(75 MHz, d6-DMSO) 195.4, 151.1, 145.4, 145.0, 134.6, 130.3, 129.8, 129.6, 127.0, 126.8, 123.6, 118.9, 115.4, 114.8, 109.5, 54.7, 44.7, 42.0, 31.3, 26.0; HRMS (EI-MS): calcd [M+H]+ for C20H18BrNO3 400.0548, found 400.0555.
1-[4-(7-Bromo-2,3-dihydro-benzo[1,4]dioxin-6-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta [c] quinolin-8-yl]-ethanone (11)
Following Method A 7-bromo-2,3-dihydro-benzo[1,4]dioxine-6-carbaldehyde (0.121 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 3 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 11 (0.180 g, 84%, endo/exo = 92:8) as a colorless solid. mp: 96–99 °C; νmax(KBr)/cm−1 3325, 28.95, 1659, 1598, 1228; δH(300 MHz; CDCl3; Me4Si) 7.69 (1 H, d, J 1.9), 7.61 (1 H, dd, J 8.3 and 1.9), 7.10 (1 H, s), 7.08 (1 H, s), 6.60 (1 H, d, J 8.3), 5.94-5.90 (1 H, m), 5.67-5.63 (1 H, m), 4.91 (1 H, d, J 3.2), 4.26 (4 H, s), 4.12 (1 H, d, J 8.8), 4.03 (1 H br s), 3.28-3.18 (1 H, m), 2.56-2.46 (4 H, m), 1.85-1.76 (1 H, m); δC(75 MHz; CDCl3; Me4Si) 196.5, 150.1, 143.3, 143.1, 133.8, 133.2, 130.4, 130.0, 128.6, 127.6, 125.2, 121.2, 116.2, 115.1, 112.8, 64.31, 55.77, 45.4, 42.0, 31.4, 26.0; HRMS (EI-MS): calcd [M+H]+ for C22H20BrNO3 426.0705, found 426.0690.
1-(4-Benzofuran-5-yl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c] quinolin-8-yl)ethanone (12)
Following Method A 1-benzofuran-5-carbaldehyde (0.165 g, 1.13 mmol), p-aminoacetophenone (0.152 g, 1.13 mmol) and cyclopentadiene (0.330 g, 5.0 mmol) and Sc(OTf)3 (0.055 g, 0.113 mmol) were dissolved in acetonitrile (4.5 cm3) and the mixture stirred for 3 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 12 (0.342 g, 92%, endo/exo = 92:8) as a colorless solid. mp: 68–70 °C; νmax(KBr)/cm−1 3335, 2927, 1659, 1596, 1286; δH(300 MHz; CDCl3; Me4Si) 7.71 (1 H, d, J 2.0), 7.67-7.63 (2 H, m), 7.61 (1 H, dd, J 8.4 and 2.0), 7.49 (1 H, d, J 8.4), 7.32 (1 H, dd, J 8.6 and 1.7), 6.77-6.76 (1 H, m), 6.61 (1 H, d, J 8.4), 5.94-5.90 (1 H, m), 5.67-5.64 (1 H, m), 4.80 (1 H, d, J 3.1), 4.33 (1 H, br s), 4.13 (1 H, d, J 8.4), 3.1-2.96 (1 H, m), 2.65-2.56 (1 H, m), 2.49 (3 H, s), 1.85-1.75 (1 H, m); δC(75 MHz; CDCl3; Me4Si) 196.5, 154.2, 150.2, 145.5, 136.4, 133.7, 130.5, 130.0, 128.3, 127.7, 127.6, 124.9, 122.8, 118.6, 144.8, 111.3, 106.5, 57.0, 46.1, 45.6, 31.4, 26.0; HRMS (EI-MS): calcd [M+H]+ for C22H19NO2 330.1494, found 330.1503.
4-(8-Acetyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-4-yl)-benzoic acid (13)
Following Method A 4-formylbenzoic acid (0.075 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 2.5 h. The product precipitated from the reaction mixture and was isolated by filtration and washed with acetonitrile to provide the product 13 (0.157 g, 95%, endo/exo = 92:8) as a colorless solid. mp: 252–255 °C; νmax(KBr)/cm−1 3365, 2951, 1642, 1710, 1288; δH (300 MHz, CD3OD) 8.04 (2 H, d, J 8.2), 7.69 (1 H, d, J 2.1), 7.62 (1 H, dd, J 8.6 and 2.1), 7.57 (2 H, d, J 8.2), 6.74 (1 H, d, J 8.6), 5.95-5.93 (1 H, m), 5.62-5.60 (1 H, m), 4.78 (1 H, d, J 3.3), 4.18 (1 H, d, J 8.8), 3.07-2.98 (1 H, m), 2.51-2.44 (4 H, m), 1.72-1.67 (1 H, m). δC(75 MHz, d6-DMSO) 195.5, 167.2, 150.8, 147.18, 134.7, 131.6, 129.8, 129.5, 129.3, 127.1, 126.9, 126.7, 123.6, 114.9, 55.7, 44.9, 44.8, 31.2, 26.0. HRMS (EI-MS): calcd [M-H]− for C21H19NO3 332.1292, found 332.1287.
1-(4-(3-nitrophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c] quinolin-8-yl)ethanone (14)
Following Method A m-nitrobenzaldehyde (0.075 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 1 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (15:85) to isolate the product 14 (0.164 g, 98%, endo/exo = 89:11) as a yellow solid. mp: 175–176 °C; νmax(KBr)/cm−1 3310, 2914, 1658, 1576, 1367; δH(300 MHz; CDCl3; Me4Si) 8.31 (1 H, d, J 1.8), 8.18 (1 H, ddd, J 8.2, 2.4 and 1.0), 7.78-7.72 (2 H, m), 7.66-7.55 (2 H, m), 6.67 (1 H, d, J 8.5), 5.96-5.92 (1 H, m), 5.67-5.65 (1 H, m), 4.85 (1 H, d, J 3.5), 4.26 (1 H, br s), 4.16 (1 H, d, J 8.4), 3.10-3.03 (1 H, m), 2.60-2.51 (4 H, m), 1.82-1.73 (1 H, m). δC(75 MHz; CDCl3; Me4Si) 196.5, 149.3, 148.5, 144.2, 133.7, 132.5, 130.1, 129.9, 129.6, 129.0, 127.8, 124.8, 122.6, 121.2, 115.3, 56.8, 45.48, 45.45, 31.3, 26.1. HPLC-MS: Elution with 60–90% CH3CN (gradient 1.5% min−1) in H2O, exhibited a single peak at 7.05 min. ESI-MS m/z [ES+] calcd for C20H18N2O3 [M+H]+ 335.13; found 335.13.
1-(4-Thiophen-3-yl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c] quinolin-8-yl)-ethanone (15)
Following Method A thiophene-3-carbaldehyde (0.056 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.0246 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 1.5 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 15 (0.138 g, 94%, endo/exo = 80:20) as colorless solid. mp: 144–147 °C. νmax(KBr)/cm−1 3338, 2912, 1647, 1597, 1292; δH(300 MHz; CDCl3; Me4Si) 7.68 (1 H, d, J 2.0), 7.59 (1 H, dd, J 8.5 and 2.0), 7.32 (1 H, m), 7.23-7.22 (1 H, m), 7.09 (1 H, dd, J 5.0 and 2.5), 6.39 (1 H, d, J 8.5), 5.90-5.85 (1 H, m), 5.67-5.64 (1 H, m), 4.78 (1 H, d, J 3.5), 4.34 (1 H, s), 4.10 (1 H, d, J 9.0), 3.08-2.98 (1 H, m), 2.61-2.50 (1 H, m), 2.47 (3H, s), 1.97-1.87 (1 H, m); δC(75 MHz; CDCl3; Me4Si) 196.5, 149.9, 143.1, 133.8, 130.4, 129.9, 128.4, 127.7, 126.1, 126.0, 125.1, 120.3, 114.8, 53.9, 45.4, 44.9, 31.9, 26.0; HRMS (EI-MS): calcd [M+H]+ for C18H17NOS 296.1109, found 296.1115.
1-(4-Cyclohex-1-enyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c] quinolin-8-yl)-ethanone (16)
Following Method A cyclohex-1-enecarbaldehyde (0.055 g, 0.5 mmol), p-aminoacetophenone (0.067 g, 0.5 mmol) and cyclopentadiene (0.165 g, 2.5 mmol) and Sc(OTf)3 (0.024 g, 0.05 mmol) were dissolved in acetonitrile (2 cm3) and the mixture stirred for 1 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 16 (0.132 g, 90%, endo/exo = 63:37) as a colorless solid. mp: 173–175 °C; νmax(KBr)/cm−1 3377, 2934, 1647, 1599, 1274; δH(300 MHz; CDCl3; Me4Si) 7.64 (1 H, d, J 2.0), 7.57 (1 H, dd, J 8.5 and 2.0), 6.53 (1 H, dd, J 8.5 and 2.0), 5.89-5.87 (1 H, m), 5.73-5.70 (3 H, m), 4.28 (1 H, br s), 3.95 (1 H, d, J 8.8), 3.23-3.14 (1 H, m), 3.02-2.88 (1 H m), 2.50-2.42 (4 H, m), 2.22-1.81 (6 H, m), 1.68-1.63 (1 H, m), 1.38-1.28 (1 H, m). δC(75 MHz; CDCl3; Me4Si) 196.3, 150.0, 149.9, 134.1, 130.2, 129.8, 127.7, 127.6, 127.2, 126.7, 125.5, 125.1, 125.0, 124.9, 114.34, 114.30, 57.4, 57.0, 45.6, 40.7, 40.5, 36.0, 35.9, 30.8, 30.7, 28.8, 27.8, 25.9, 25.6, 24.8, 24.5; HRMS (EI-MS): calcd [M+H]+ for C20H23NO 294.1858, found 294.1858.
1-[4-(6-Bromo-benzo[1,3]dioxol-5-yl)-2,3,3a,4,5,9b-hexahydro-furo[3,2-c]quinolin-8-yl]-ethanone (17-exo)
Following Method B the imine prepared from p-aminoacetophenone (0.067 g, 0.5 mmol) and 6-bromopiperonal (0.173 g, 0.5 mmol) was dissolved in acetonitrile (4 cm3) at ambient temperature, then 2,3-dihydrofuran (0.070 g, 1.0 mmol) and Sc(OTf)3 (0.0246 g, 0.05 mmol) were added. The mixture was stirred for 2 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate/hexanes (10:90) to isolate the product 17 (0.142 g, 68%, endo/exo = 44:56) as a colorless solid. The diastereomerically pure compounds were obtained by recrystallization from acetonitrile/water (2:1) which resulted in selective precipitation of 17-exo: mp: 77–80 °C; νmax(KBr)/cm−1 3321, 2879, 1659, 1608, 1256; δH(300 MHz; CDCl3; Me4Si) 7.99 (1 H, d, J 2.0), 7.78 (1 H, dd, J 8.5 and 2.0), 7.03 (1 H, s), 6.99 (1 H, s), 6.62 (1 H, d, J 8.5), 6.01-6.00 (2 H, m), 4.62 (1 H, d, J 4.7), 4.54 (1 H, s), 4.51 (1 H, d, J 10.5), 4.14-4.06 (1 H, m), 3.97-3.89 (1 H, m), 2.52 (3 H, s), 2.43-2.36 (1 H, m), 2.13-2.06 (1 H, m), 1.87-1.80 (1 H, m). δC(75 MHz; CDCl3; Me4Si)196.3, 149.1, 148.1, 148.0, 133.01, 132.9, 129.6, 127.8, 118.3, 115.1, 114.2, 112.5, 108.4, 102.0, 75.6, 65.5, 54.4, 43.0, 28.2, 26.1; HRMS (EI-MS):calcd [M+H]+ for C20H18BrNO4 416.0497, found 416.0490.
1-[4-(6-Bromo-benzo[1,3]dioxol-5-yl)-2,3,3a,4,5,9b-hexahydro -furo[3,2-c]quinolin-8-yl]-ethanone (17-endo) 17-endo
mp: 99–101 °C; νmax(KBr)/cm−1 3325, 2881, 1660, 1609, 1254; δH(300 MHz; CDCl3; Me4Si) 7.97 (1 H, d, J 2.0), 7.75 (1 H, dd, J 8.5 and 2.0), 7.17 (1 H, s), 7.04 (1 H, s), 6.61 (1 H, d, J 8.5), 6.02 (1 H, d, J 1.3), 6.0 (1 H, d, J 1.3), 5.27 (1 H, d, J 7.8), 5.10 (1 H, d, J 3.0), 4.14 (1 H, br s), 3.85-3.76 (2 H, m), 3.00-2.92 (1 H, m), 2.53 (3 H, s), 2.10-2.02 (1 H, m), 1.56-1.50 (1H, m). δC(75 MHz; CDCl3; Me4Si)196.5, 148.5, 147.8, 147.7, 132.8, 131.7, 128.8, 128.0 121.5, 114.7, 113.0, 112.9, 107.6, 101.9, 74.9, 66.8, 55.4, 41.5, 26.1, 24.6. HRMS (EI-MS): calcd [M+H]+ for C20H18BrNO3 416.0497, found 416.0478.
Supplementary Material
Scheme 1.

Improved Synthesis of G-1
Acknowledgment
This work was supported by NIH grants R01 CA127731, U54MH074425, U54MH084690, the University of New Mexico Cancer Research and Treatment Center NIH P30 CA118100, the New Mexico Cowboys for Cancer Research Foundation, Oxnard Foundation and the Stranahan Foundation.
Footnotes
Supporting Information Available: Crystallographic data for G-1 (CIF); experimental procedures; copies of 1H and 13C NMR spectra; HPLC-MS, HRMS and other characterization data for new compounds. This material is available free of charge via the Internet at www.rsc.org
References
- 1.For recent reviews on GPR30 see Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Annu. Rev. Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. ; (b) Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Neurosignal. 2008;16:140–153. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]; (c) Filardo EJ, Quinn JA, Sabo E. Steroids. 2008;73:870–873. doi: 10.1016/j.steroids.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 2.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Nat. Chem. Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 3.For selected examples employing G-1 as a chemical probe see: Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Ando S, Maggiolini M. Cancer Res. 2007;67:1859–1866. doi: 10.1158/0008-5472.CAN-06-2909. ; (b) Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ. J. Endocrinol. 2007;193:311–321. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]; (c) Pang Y, Dong J, Thomas P. Endocrinology. 2008;149:3410–3426. doi: 10.1210/en.2007-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Albanito L, Sisci D, Aquila S, Brunelli E, Vivacqua A, Madeo A, Lappano R, Pandey DP, Picard D, Mauro L, Ando S, Maggiolini M. Endocrinology. 2008;149:3799–3808. doi: 10.1210/en.2008-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Teng J, Wang ZY, Prossnitz ER, Bjorling DE. Endocrinology. 2008;149:4024–4034. doi: 10.1210/en.2007-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sirianni R, Chimento A, Ruggiero C, De LucaA, Lappano R, Ando S, Maggiolini M, Pezzi V. Endocrinology. 2008;149:5043–5051. doi: 10.1210/en.2007-1593. [DOI] [PubMed] [Google Scholar]; (g) Wang C, Dehghani B, Magrisso IJ, Rick EA, Bonhomme E, Cody DB, Elenich LA, Subramanian S, Murphy SJ, Kelly MJ, Rosenbaum JS, Vandenbark AA, Offner H. Mol Endocrinol. 2008;22:636–648. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Madak-Erdogana Z, Kieserb KJ, Kimb SH, Kommd BA, Katzenellenbogen JA, Katzenellenbogen BS. Mol Endocrinology. 2008;22:2116–2127. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Alyea RA, Laurence SE, Kim SH, Katzenellenbogen BS, Katzenellenbogen JA, Watson CS. Journal of Neurochem. 2008;106:1525–1533. doi: 10.1111/j.1471-4159.2008.05491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kamanga-Sollo E, White ME, Chung KY, Johnson BJ, Dayton WR. Domest. Anim. Endocrinol. 2008;35:254–262. doi: 10.1016/j.domaniend.2008.06.001. [DOI] [PubMed] [Google Scholar]; (k) Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, Fritzemeier KH. Endocrinology. 2008;149:4846–4856. doi: 10.1210/en.2008-0269. [DOI] [PubMed] [Google Scholar]; (l) Kuhn J, Dina OA, Goswami C, Suckow V, Levine JD, Hucho T. Eur. J. Neurosci. 2008;27:1700–1709. doi: 10.1111/j.1460-9568.2008.06131.x. [DOI] [PubMed] [Google Scholar]; (m) Dun SL, Brailoiu GC, Gao X, Brailoiu E, Arterburn JB, Prossnitz ER, Oprea TI, Dun NJ. J. Neuroscience Research. 2009;87:1610–1619. doi: 10.1002/jnr.21980. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, Meyer MR, Amann K, Ammann E, Perez-Dominguez EA, Genoni M, Clegg DJ, Dun NJ, Resta TC, Prossnitz ER, Barton M. Circ. Res. 2009;104:288–291. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Pang Y, Thomas P. Gen. Comp. Endocrinol. 2009;161:58–61. doi: 10.1016/j.ygcen.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Noel SD, Keen KL, Baumann DI, Filardo EJ, Terasawa E. Mol Endocrinology. 2009;23:349–359. doi: 10.1210/me.2008-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Terasawa E, Noel SD, Keen KL. Journal of Neuroendocrinology. 2008;21:316–321. doi: 10.1111/j.1365-2826.2009.01839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Romano N, Lee K, Abraham IM, Jasoni CL, Herbison AE. Endocrinology. 2008;149:5335–5344. doi: 10.1210/en.2008-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H. J. Immuno. 2009;182:3294–3303. doi: 10.4049/jimmunol.0803205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Xu H, Qin S, Carrasco GA, Dai Y, Filardo EJ, Prossnitz ER, Battaglia G, Doncarlos LL, Muma NA. Neuroscience. 2008;158:1599–1607. doi: 10.1016/j.neuroscience.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (u) Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak T, Bologa C, Leitao A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI, Prossnitz ER. Nat. Chem. Biol. 2009;5:421–427. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]; (v) Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Endocrinology. 2009;150:3753–3758. doi: 10.1210/en.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oprea TI, Bologa CG, Boyer S, Curpan RF, Glen RC, Hopkins AL, Lipinski CA, Marshall GR, Martin YC, Ostopovici-Halip L, Rishton G, Shoichet BK, Ursu O, Vaz RJ, Waller CL, Waldmann H, Sklar LA. Nat. Chem. Biol. 2009;5:441–447. doi: 10.1038/nchembio0709-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Povarov LS. Russ. Chem. Rev. 1967;36:656–670. [Google Scholar]; (b) Grieco PA, Bahsa A. Tetrahedron Lett. 1988;29:5855–5858. [Google Scholar]; (c) Babu G, Perumal PT. Tetrahedron Lett. 1997;38:5025–5026. [Google Scholar]; (d) Babu G, Perumal PT. Tetrahedron. 1998;54:1627–1638. [Google Scholar]; (e) Nagarajan R, Perumal PT. Synthetic comm. 2001;31:1733–1736. [Google Scholar]; (f) Nagarajan R, Chitra S, Perumal PT. Tetrahedron. 2001;57:3419–3423. [Google Scholar]; (g) Nagarajan R, Chitra S, Perumal PT. Tetrahedron. 2001;57:3419–3423. [Google Scholar]; (h) Powell DA, Batey RA. Org. Lett. 2002;4:2913–2916. doi: 10.1021/ol026293d. [DOI] [PubMed] [Google Scholar]; (i) Kumar RS, Nagarajan R, Perumal PT. Synthesis. 2004;6:949–959. [Google Scholar]; (j) Kamal A, Prasad BR, Ramana AV, Babu H, Reddy KS. Tetrahedron Lett. 2004;45:3507–3509. [Google Scholar]; (k) Nagaiah K, Sreenu D, Rao RS, Vashshta G, Yadav JS. Tetrahedron Lett. 2006;47:4409–4413. [Google Scholar]; For a recent review see: Kouznetsov VV. Tetrahedron. 2009;65:2721–2750.
- 6.(a) Kobayashi S, Ishitani H, Nagayama S. Synthesis. 1995:1195–1202. [Google Scholar]; (b) Kobayashi S, Araki M, Ishitani H, Nagayama S, Hachiya I. Synlett. 1995:233–234. [Google Scholar]; (c) Shindo T, Taniguchi Y, Takaki K, Fujiwara Y. Synthesis. 1995:801–804. [Google Scholar]; (d) Kobayashi S, Ishitani H, Nagayama S. Chem. Lett. 1995:423–424. [Google Scholar]; (e) Collin J, Jaber N, Lannou MI. Tetrahedron Lett. 2001;42:7405–7407. [Google Scholar]; (f) Powell DA, Batey RA. Tetrahedron Lett. 2003;44:7569–7573. [Google Scholar]; (g) Twin H, Batey RA. Org. Lett. 2004;6:4913–4916. doi: 10.1021/ol0479848. [DOI] [PubMed] [Google Scholar]; (h) Kudale AA, Kendall J, Miller DO, Collins JL, Bodwell GJ. J. Org. Chem. 2008;73:8437–8447. doi: 10.1021/jo801411p. [DOI] [PubMed] [Google Scholar]
- 7.(a) Keinicke L, Fristrup F, Norrby P-O, Madsen R. J. Am. Chem. Soc. 2005;127:15756–15761. doi: 10.1021/ja054706a. [DOI] [PubMed] [Google Scholar]; (b) Sundararajan G, Prabagaran N, Varghese B. Org. Lett. 2001;3:1973–1976. doi: 10.1021/ol0159221. [DOI] [PubMed] [Google Scholar]; (c) Varma RS, Dahiya R, Kumar S. Tetrahedron Lett. 1997;38:2039–2042. [Google Scholar]; (d) Akbari J, Heydari A. Tetrahedron Lett. 2009;50:4236–4238. [Google Scholar]
- 8.(a) Ma Y, Qian C, Xie M, Sun J. J. Org. Chem. 1999;64:6462–6467. [Google Scholar]; (b) Das B, Reddy MR, Reddy VS, Ramu R. Chem. Lett. 2004;33:1526–1527. [Google Scholar]; (c) Mahesh M, Reddy CV, Reddy KS, Raju PVK, Reddy VVN. Synth. Commun. 2004;34:4089–4104. [Google Scholar]; (d) Nagarajan R, Magesh CJ, Perumal PT. Synthesis. 2004;1:69–74. [Google Scholar]
- 9.Zhang J, Li C-J. J. Org. Chem. 2002;67:3969–3971. doi: 10.1021/jo020131d. [DOI] [PubMed] [Google Scholar]
- 10.Jimenez O, de la Rosa Lavilla G. Angew. Chem. Int. Ed. 2005;44:6521–6525. doi: 10.1002/anie.200501548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.