

Abstract
Synuclein γ (SNCG), previously identified as a breast cancer-specific gene, is highly expressed in malignant cancer cells but not in normal epithelium. The molecular targets of SNCG during breast cancer progression have not been fully identified. Here we analyzed the effect of SNCG on stimulation of membrane-initiated estrogen signaling. While SNCG expression enhanced estrogen-induced activation of ERK1/2 and mammalian target of rapamycin, knockdown of endogenous SNCG decreased membrane-initiated estrogen signaling. SNCG functions as a molecular chaperone protein for estrogen receptor (ER)-α36, a membrane-based variant of ER-α. SNCG bound to ER-α36 in the presence and absence of functional molecular chaperone heat shock protein 90. Disruption of heat shock protein 90 with 17-AAG significantly reduced ER-α36 expression and membrane-initiated estrogen signaling. However, expression of SNCG prevented ER-α36 degradation and completely recovered 17-AAG-mediated down-regulation of estrogen signaling. The function of SNCG in ER-α36-mediated estrogen signaling is consistent with its ability to stimulate cell growth in response to estrogen. Expression of SNCG also renders tamoxifen resistance, which is consistent with the clinical observation on the association of ER-α36 expression and tamoxifen resistance. The present study indicates that ER-α36 is a new member of the ER-α family that mediates membrane-initiated estrogen signaling and that SNCG can replace the function of heat shock protein 90, chaperone ER-α36 activity, stimulate ligand-dependent cell growth, and render tamoxifen resistance.
Estrogen signaling is mediated by both genomic nuclear-initiated estrogen signaling by nuclear estrogen receptors (ERs) designated as ERα-66, ERα-46, and ERβ through transcriptional activation of the target genes1,2,3,4 and non-genomic membrane-initiated estrogen signaling (MIES), which is thought to be directed via membrane-based ER. MIES was found to activate different cytoplasmic signaling proteins and other membrane-initiated signaling pathways including the adenylate cyclase,5 the phospholipase C,6 G protein coupled receptor-activated,7 and the mitogen-activated protein kinase MAPK.7,8,9 It was reported in the early 1970s that 17β-estradiol (E2) binds to a cell surface receptor and stimulates a rapid generation of cAMP;10 since then evidence has accumulated to indicate a plasma membrane-based ER that transduces membrane-initiated estrogen signaling appeared.11,12,13 Most recently, we reported the identification of a predominantly cell membrane-based 36-kd novel isoform of ER-α66 and designated it as ER-α36.14,15 ER-α36 is generated from a promoter located in the first intron of the ER-α66 and lacks both ligand-independent AF-1 and ligand-dependent transcriptional AF-2 domains of ER-α66 but retains DNA-binding domain and partial ligand-binding domain. ER-α36 is predominantly on the plasma membrane, and also in cytoplasm where it transduces both estrogen- and tamoxifen-induced activation of MAPK/ERK1/2 signaling and stimulates cell growth.15 Thus, ER-α36 plays an important role in mitogenic estrogen signaling. However, the molecular mechanisms underlying the regulation of ER-α36 function are largely unknown.
We previously identified a breast cancer specific gene BCSG1, also named as synuclein γ (SNCG).16 Synucleins are a family of small proteins consisting of three known members, synuclein α (SNCA), synuclein β (SNCB), and SNCG.17 While synucleins are highly expressed in neuronal cells and are abundant in presynaptic terminals, and SNCA and SNCB have been specifically implicated in neurodegenerative diseases,18,19 SNCG is not involved in neurodegenerative diseases but primarily involved in neoplastic diseases.16,20,21,22,23,24 SNCG is highly expressed in breast carcinomas and predicts poor clinical outcome in breast cancer.24,25 When overexpressed, SNCG stimulates growth of hormone-dependent breast cancer cells both in vitro and in nude mice.26,27 Expression of SNCG in mammary gland in the transgenic mice induces a highly proliferative pregnancy-like phenotype of mammary epithelial cells and the gland hyperplasia.28 Investigations aimed to elucidate the molecular mechanisms underlying the oncogenic functions of this protein reveal that expression of SNCG in cancer cells results in a more malignant phenotype with increased cell motility,20 enhanced transcriptional activity of steroid receptors,26,27,28 and accelerated rate of chromosomal instability.29,30 The contribution of SNCG to breast cancer development and progression may be due to its chaperone activity on both estrogen (E2)-dependent and E2-independent pathways. Previously, we demonstrated that SNCG participates in the heat shock protein 90 (Hsp90)-based multichaperone complex for steroid receptors and stimulates ER-α66 transcriptional activity but does not affect ER-β signaling.26,27 The present study demonstrated SNCG as a tumor specific chaperone, which can replace the chaperoning function of Hsp90 and protect and stimulate ER-α36-mediated MIES.
Materials and Methods
Reagents
Antibodies used for immunoprecipitation and Western blot analyses were as follows: anti-γ-synuclein antibody (goat polyclonal antibody E-20); anti-ER-α antibody (rabbit polyclonal antibody HC-20); a specific peptide anti-ER-α36 antibody,14,15 anti-Hsp90 antibody (rabbit polyclonal antibody sc-7947); normal goat IgG (sc-2028); and anti-actin antibody (goat polyclonal antibody sc-1615). These antibodies were from Santa Cruz Biotechnology. Anti-ERK1/21/2, anti-phospho-ERK1/21/2, anti-S6K, and anti-phospho-S6K were from Cell Signaling Technology (Beverly, MA).
Cell Culture
Proliferating subconfluent human breast cancer cells were harvested and cultured in the steroid-stripped condition (phenol red-free IMEM containing 5% dextran-charcoal-stripped fetal calf serum) for 3 days before addition of indicated dose of E2.
Development of Antibody Specific to ER-α36
We developed affinity-purified rabbit polyclonal anti-ER-α36 antibodies (Abs) as a custom service from Alpha Diagnostic (San Antonio, TX). The Abs were raised against a synthetic peptide antigen corresponding to the unique C-terminal 20 amino acids of human ER-α36. The specificity of the Ab was tested in ER-α36 expression vector transfected HEK293 cells that do not express endogenous ER-α.15
Cell Proliferation Assay
Cell growth was measured using a cell proliferation kit (XTT; Roche Molecular Biochemicals, Germany).
Transfection of SNCG into MCF-7 and MDA-MB-435 Cells
SNCG stably transfected MCFB6 and SNCG-435-3 cells were previously established and described.26,20
Stable Expression of SNCG Antisense mRNA in T47D Cells
Knockdown endogenous SNCG expression in T47D cells was achieved by antisense approach as described previously.26 Briefly, a 285-bp DNA fragment corresponding to the exon 1 region (−169 to +116) of SNCG gene was amplified from the plasmid pBS-SNCG759 and was cloned into the EcoRI site of the expression vector pcDNA3.1.Vectors expressing SNCG antisense mRNA (pcDNA-SNCG-As) or SNCG sense mRNA (pcDNA-SNCG-S) were separately transfected into T47D cells by Effectin reagent. Two stably SNCG antisense transfected clones (AS-3 and AS-1) were selected and characterized.26
Knockdown of Endogenous SNCG in MDA-MB-231 Cells by RNA Interference
To knock down endogenous SNCG, we used SNCG siRNA lentiviral particle gene silencers (42290-v from Santa Cruz). SNCG siRNA consists of a pool of five target-specific 19- to 25-nucleotide siRNAs designed to knock down SNCG gene expression. Subconfluent MDA-MB-231 cells were infected with the lentiviral vectors according to the manufacturer’s protocol.
In Vitro Pulldown Assay using Glutathione-S-Transferase
The complete coding region of SNCG was inserted into pGEX-5X-1 (Amersham Bioscience). For the glutathione-S-transferase (GST) pulldown assay, full-length SNCG fused to GST (GST-SNCG) or GST alone was expressed in Escherichia coli BL21, which was then purified on glutathione-Sepharose 4B beads (Amersham Pharmacia). MCF-7 and ER-α36-transfected MDA-MB-435 cells (5 × 106 cells/dish) were rinsed three times with 1 ml of ice-cold PBS and sonicated in 1 ml of lysis buffer (20 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 50 mmol/L NaF, and 1 mmol/L Na3VO4, containing protease inhibitors). Cell lysates were spun at 12,000 × g for 10 minutes at 4°C to remove debris. Supernatants were incubated for 12 hours at 4°C with GST or GST-SNCG bound to beads. The beads were then washed three times in lysis buffer. Proteins were subjected to Western analysis of ER-α36 with a specific peptide antibody.
Immunoprecipitation (IP)
IP was performed as previously described.27
Soft Agar Colony Formation Assay
The anchorage-independent growth was performed in 12-well plates as previously described.26
Tumor Growth in Athymic Nude Mice
A nude mouse tumorigenic assay was performed as we previously described.27 Briefly, 17β-estradiol pellets (0.72 mg/pellet, 60-day releasing, Innovative Research of America, Toledo, OH) were implanted subcutaneously in athymic nude mice (Frederick Cancer Research and Development Center, Frederick, MD) 1 day before the injection of hormone-dependent MCF-7 and MCFB6 tumor cells. Approximately 3 × 106 cells were injected into a 6-week-old female athymic nude mouse. When tumor xenografts were established, mice bearing tumors were randomly allocated to different treatment groups. Tamoxifen treatment was given subcutaneously 3 days/week. Each animal received two injections, one on each side, in the mammary fat pads between the first and second nipples. Tumor size was determined every 10 days by three-dimensional measurements (in millimeters) using a caliper. Only measurable tumors were used to calculate the mean tumor volume for each tumor cell clone at each time point.
Statistical Analysis
Results were reported as the mean ± SD for typical experiments done in three replicate samples and compared by the Student’s t-test. Results were considered significantly different for P < 0.05. All experiments were done at least twice to ensure reproducibility of the results.
Results
Expression of ER-α36 in Breast Cancer Cells
We first conducted Western blot analysis on ER-α36 expression in ER-positive breast cancer T47D and MCF-7 cells, ER-negative MDA-MB-435, MDA-MB-231, MDA-MB-436 cells, and HT29 colon cancer cells as a control for non-breast cells. Figure 1A demonstrates that while ER-α36 is not expressed in ER-negative MDA-MB-435 cells, it is expressed in both MDA-MB-231 and MDA-MB-436 cells, two well-known ER-α66-negative breast cancer cell lines. ER-α36 is also expressed in ER-positive breast cancer T47D and MCF-7 cells. ER-α36 expression was not detected in HT29 colon cancer cells. These data are consistent with previous publication on ER-α36 expression in both ER-negative and-positive breast cancer cells.15 We transfected ER-negative MDA-MB-435 cells with hER-α36 expression vector. This transfection showed an abundant ER-α36 expression in MDA-MB-435/ER36 cells. To determine whether ER-α36 is subjected to hormonal regulation, expression of ER-α66 and ER-α36 in MCF-7 and T47D cells was analyzed (Figure 1B). While limited amount of ER-α36 was observed when both cell lines were cultured in the absence of estrogen, treatment of the cells with 17β-estradiol (E2) and tamoxifen induced a significant increase in expression of ER-α36. As expected, both E2 and tamoxifen treatment resulted in a down-regulation of ER-α66.
Figure 1.

Expression of ER-α36. A: Expression of ER-α36 in different cells. Western blot analysis of cell extracts with an ERα36-specific Ab generated against the C terminus of the human ER-α36 protein.15 For transfection, MDA-MB-435 cells were transiently transfected with hERα-36 plasmid pCI-ER36 or the control vector pCI-neo and then selected with G418. B: Regulation of ER-α36 expression by E2 and tamoxifen. MCF-7 and T47D cells were cultured in 5% stripped fetal calf serum (phenol red free IMEM) for three days and then treated with 2 nmol/L E2 or 1 μmol/L tamoxifen for two days. Total cell lysates were isolated and subjected to Western analyses with anti-ERα antibody (RB9016, Lab Vision) and specific peptide antibody against ER-α36.
Enhancement of Membrane-Initiated Estrogen Signaling by SNCG
We previously demonstrated that SNCG expression in MCF-7 cells significantly enhanced transactivation of ER-α66.26,27,28 We decided to test whether SNCG also regulates MIES. We examined ERK1/2 activation in SNCG-negative MCF-7 cells and previously established SNCG stably transfected MCFB6 cells26,27 in the absence and presence of E2 or tamoxifen. In contrast to genomic nuclear-initiated estrogen signaling, which requires a longer time treatment; hormone-induced MIES is rapid and occurs several minutes after the treatment. As shown in Figure 2A, treatment of MCF-7 cells with E2 for 15 minutes induced a rapid 2.9-fold phosphorylation of ERK1/2. The magnitude of hormone-induced ERK1/2 activation was significantly enhanced in MCFB6 cells; E2 treatments resulted in a 6.2-fold phosphorylation of ERK1/2 over control cells. The effect of SNCG on E2-stimulated MIES was further demonstrated in the previously established AS-3 cells derived from T47D cells with stable SNCG knockdown, which reduced SNCG expression to 15% of that of control T47D cells.26 Whereas E2 strongly stimulated ERK1/2 activation in control T47D cells that express endogenous SNCG, resulting in a 5.8-fold ERK1/2 phosphorylation, knockdown of SNCG expression significantly reduced E2-stimulated ERK1/2 activation with only a 2.2-fold increase (Figure 2B).
Figure 2.

Stimulation of membrane ER-mediated ERK1/2 and mTOR activation by SNCG. A: MCF-7 and its SNCG stably transfected clone MCFB6 cells are cultured in the stripped condition. Cells were treated with 2 nmol/L E2 for 15 minutes. Total cell lysates were subjected to Western analyses of ERK1/2, phosphorylated ERK1/2, and SNCG. B: T47D and its SNCG knockdown AS-3 cells. Cells were cultured and treated as described for MCF-7 and MCFB6 cells. C: Enhancement of E2-induced mTOR signaling. MCF-7 and SNCG stably transfected MCFB6 cells were cultured in the stripped condition. Cells were treated with or without E2 (2 nmol/L) for 15 minutes. Total protein was subjected to Western analyses of S6K, phosphorylated S6K, and SNCG. Comparing the intensity of bands on a Western blot was determined by densitometry.
A growing body of evidence has suggested that mammalian target of rapamycin (mTOR) acts as a sensor that integrates extracellular and intracellular events, coordinating cell proliferation and survival.31 Rapamycin, an inhibitor of mTOR, possesses antitumor activity against many tumors including breast tumors, and particularly against ER-positive breast cancer cell lines. The sensitivity of these cells to rapamycin has been attributed to activation of the PI3K/Akt/mTOR pathway by nongenomic ER signaling.32,33,34 We tested if SNCG expression affects E2-stimulated mTOR activation (Figure 2C). Treatment of MCF-7 cells with E2 stimulates the phosphorylation of S6K, a downstream effector of mTOR. However, an E2-stimulated S6K phosphorylation in MCFB6 cells stably transfected a SNCG expression vector was significantly enhanced versus control MCF-7 cell. The data suggest that SNCG enhances the E2-inducerd rapid activation of the mTOR signaling. Taken together, the increased E2-stimulated ERK1/2 and mTOR activation in SNCG-transfected MCF-7 cells and the down-regulated ERK1/2 activation in SNCG knockdown T47D cells indicated that SNCG enhances ligand-dependent MIES.
SNCG Stimulates ER-α36-Mediated ERK1/2 Activation
While alternation of SNCG expression in MCF-7 and T47D cells affects MIES, it is not clear which ER-α isoform mediates this effect. Furthermore, the E2-induced activation of non-genomic MIES in MCF-7 and T47D cells could also be mediated by GPR30, a G protein-coupled receptor for estrogen.35 In an effort to dissect the contributions of different receptors—ER-α66, ER-α46, ER-α36, and GPR30—to observed SNCG activity in E2-stimulated ERK1/2 activation, and to determine whether ER-α36 can mediate E2-induced MIES, we studied ERK1/2 activation in ER-α-negative MDA-MB-435 and ER-α36 transfected MDA-MB-435/ER36 cells. As shown in Figure 3A, while treatment MDA-MB-435 cells with E2 did not stimulate activation, enforced expression of ER-α36 in MDA-MB-435 cells rendered ERK1/2 activation in response to E2, resulting in a 3.1-fold increase in ERK1/2 activation. The ER-α36-stimulated stimulation of ERK1/2 activation was ligand-dependent because ER-α36 had no significant effect on the ERK1/2 activation in the absence of E2. We next co-transfected MDA-MB-435 cells with SNCG and ER-α36 and studied the effect of SNCG on ER-α36-mediated ERK1/2 activation by E2. A significant stimulation of E2-induced ERK1/2 activation by SNCG was observed in MDA-MB-435 cells co-transfected with ER-α36 and SNCG expression constructs. While SNCG had no effect on the basal levels of the ERK1/2 activation, SNCG increased ligand-dependent ERK1/2 activation twofold over the control SNCG-negative cells.
Figure 3.

SNCG stimulates ER-α36 signaling. A: Activation of ERK1/2 in ER-α36-transfected MDA-MB-435 cells. Cells were co-transfected with ER-α36 and pCI-SNCG or ER-α36 and the control vector pCI-neo and then selected with G418. Cells were cultured in stripped condition for two days and then treated with or without 2 nmol/L E2 for 15 minutes. Total cell lysates were isolated and subjected to Western analyses with antibodies against phosphorylated ERK1/2, total ERK1/2, and ERα−36. B: Effect of knockdown of endogenous SNCG on ERK1/2 activation on MDA-MB-231 cells. Control siRNA (siCtrl) and SNCG siRNA infected MDA-MB-231 cells were treated with 2 nmol/L E2 for 15 minutes. Phosphorylated ERK1/2, ERK1/2, and SNCG were analyzed by Western blot. Comparison of the intensity of bands on a Western blot was determined by densitometry. C: SNCG stimulated ligand-dependent and ER-α36-mediated cell proliferation. Control MDA-MB-435, ER-α36 transfected MDA/ER36, SNCG transfected MDA/SNCG, and ER-α36/SNCG double transfected MDA/ER36/SNCG cells were cultured in the stripped condition for three days before the hormone treatments. Cells were treated with or without 1 nmol/L E2 for four days. Cell proliferation was measured by using a cell proliferation kit (XTT). The numbers represent means ± SD of three cultures. The open bar represents non-treated cells; closed bar represents E2-treated cells. All values were presented as the percentage of stimulation over the control non-treated cells, which were taken as 100%. The asterisk indicates a statistical comparison of P < 0.001. Statistical comparisons for E2-treated ER-α36/SNCG double transfected cells relative to E2-treated ER-α36 single transfected cells indicate *P < 0.001.
Although there has been a controversy over the past several years about the true origin of the human MDA-MB-435 cell line, which might be derived from M14 melanoma cells, recent evidence suggests that the idea that the MDA-MB-435 cell line indeed represents a poorly differentiated, aggressive breast tumor line, with expression of both epithelial and melanocytic markers, should be reconsidered.36 We also used ER-negative (ER-α66-negative) but ER-α36- and SNCG-positive MDA-MB-231 breast cancer cells to study the effect of SNCG on ER-α36-mediated MIES. The effect of SNCG expression on ER-α36-mediated MIES was demonstrated by inhibiting endogenous SNCG expression in MDA-MB-231 cells (Figure 3B). SNCG siRNA significantly reduced endogenous SNCG expression in MDA-MB-231 cells. Knockdown SNCG significantly reduced E2-stimulated ERK1/2 activation. Treatment of control siRNA-infected cells with E2 induced a 4.3-fold increase in ERK1/2 activation; the E2-induced ERK1/2 activation was greatly reduced to 1.5-fold in the SNCG knockdown cells. These data suggest that membrane-initiated estrogen signaling is mediated, at least in part, by ER-α36, and SNCG elevated ER-α36-mediated signaling.
ER-α36 and SNCG Synergistically Mediates Estrogen-Stimulated Cell Proliferation
To pursue the significance of MIES through ER-α36, we tested whether ER-α36 can mediate E2-stimulated cell proliferation. Data in Figure 3C show that E2 stimulated about twofold proliferation of ER-α36-transfected MDA-MB-435 cells compared with cells treated with vehicle. This ER-α36-mediated and E2-stimulated cell proliferation was also previously observed in HEK 293 cells.15 These results indicated that ER-α36 acts as a functional receptor to mediate mitogenic estrogen signaling. To determine the biological significance of SNCG in the mitogenic estrogen signaling mediated by ER-α36, we analyzed the effect of SNCG expression on E2-stimulated growth in ER-α36 transfected cells. To determine whether SNCG overexpression affects ligand-dependent and ligand-independent cell growth, the cellular proliferation of ER-α36 and SNCG co-transfected MDA-MB-435 cells were compared with that of control and cells transfected with ER-α36 or SNCG expression vector individually. SNCG had no significant effect on the proliferation of SNCG-transfected cells compared with control cells in the absence of E2, which was consistent with the previous observations that SNCG only stimulates hormone-dependent growth of breast cancer cells both in vitro and in nude mice.16,27 Overexpression of SNCG significantly stimulated the ligand-dependent and ER-α36-mediated proliferation. Treatment of MDA435/ERα36 cells with E2 stimulated cell proliferation 2.2-fold over controls. However, E2 treatment of MDA435/ERα36/SNCG cells resulted in a 4.5-fold increase in the proliferation versus controls. These data suggest that SNCG renders cells more responsively to E2-stimulated and ER-α36-mediated cell proliferation.
SNCG Chaperones ER-α36 and Prevents Hsp90 Inhibition Induced Down-Regulation of MIES
We previously demonstrated that SNCG physically binds to unliganded ER-α66 complex and chaperones ER-α66 transcriptional activity.27 Since these studies were performed in the absence of E2 and we demonstrated that E2 treatment stimulates ER-α36 expression (Figure 1B), we are interested in studying if SNCG is physically associated with ER-α36 in the presence of E2. As shown in Figure 4A, in the absence of ligand, IP SNCG efficiently co-precipitated ER-α66 and small amount of ER-α36 (control). In the presence of E2, however, SNCG dissociated from the ER-α66 complex and interacted with ER-α36. These data suggest that SNCG binds to unliganded ER-α66 in the absence of E2; while SNCG dissociates from ER-α66 complex and binds to ER-α36 in the presence of E2. To further confirm this interaction, we then investigated if SNCG interacts with ER-α36 in vitro in the cell free system. We performed a GST pulldown assay using the purified GST-tagged SNCG protein to pull down ER-α36 (Figure 4B). The GST-tagged SNCG was immobilized to GST beads and incubated with lysates of MCF-7 and ER-α36-transfected MDA-MB-435 cells. The eluted proteins were subjected to immunoblot analysis using specific anti-ER-α36 antibody. The results of immunoblotting revealed that ER-α36 was specifically precipitated by immobilized GST-SNCG, indicating that ER-α36 directly interacts with SNCG in vitro.
Figure 4.

Prevention of loss of ER-α36 signaling by SNCG. A: Physical interaction between SNCG and ERα. SNCG stably transfected MCFB6 cells cultured in the stripped condition, were treated with or without 1 nmol/L E2 for 16-hour. Equal amount of protein was subjected to immunoprecipitation with anti-SNCG and followed by Western analysis of ERα. Total represents the total protein from the E2-treated cells before immunoprecipitation. B: GST-SNCG fusion protein was expressed in E. coli, purified, and stained with Coomassie Blue to demonstrate the expression of the fusion proteins (left). Cell extracts from MCF-7 and ER-α36-transfected MDA-MB-435 cells were subsequently incubated either with bead-bound GST as a negative control or GST-SNCG. After the beads were washed, proteins were subjected to Western blot for ER-α36 using the specific anti-ER-α36 peptide antibody (right). C: Preventing Hsp90 inhibition-induced down-regulation of mTOR signaling in MCF-7 cells by SNCG. MCF-7 and MCFB6 cells were treated with or without 1 μmol/L 17-AAG for 15 hours and stimulated with E2 (2 nmol/L) for 15 minutes. Total protein was subjected to Western analyses of phosphorylated S6K, S6K, and SNCG. D: Preventing Hsp90 inhibition-induced down-regulation of ERK1/2 activation in MCF-7 cells by SNCG. MCF-7 and MCFB6 cells were treated with or without 1 μmol/L 17-AAG for 15 hours and stimulated with E2 (2 nmol/L) or tamoxifen (1 μmol/L) for 15 minutes. Total protein was subjected to Western analyses of phosphorylated ERK1/2, ERK1/2, and normalized with actin.
Hsp90 is a molecular chaperone whose association is required for the stability and function of multiple signaling proteins that promote the growth and/or survival of cancer cells. It is well documented that the activation of steroid receptors including ER-α is modulated by an Hsp90-based multiple chaperone complex.37,38 Inhibition of Hsp90 by small molecules, eg, 17-AAG, leads to degradation of Hsp90 client proteins and inactivation of the corresponding signaling pathways.39 The chaperone activity of SNCG on regulation of transcriptional activation of ER-α66 has been demonstrated in breast cancer cells27 as well as in mammary glands.28 Since SNCG enhances ER-α36-mediated MIES and physically interacts with ER-α36 in the presence of estrogen, we reason that SNCG may also chaperone ER-α36-mediated MIES. To determine whether disrupting Hsp90 function with 17-AAG could attenuate MIES and if SNCG expression can restore MIES activity by chaperone ER-α36, we disrupted Hsp90 function by treating the cells with 17-AAG. We demonstrated that under stressful conditions when the chaperone function of Hsp90 was blocked by 17-AAG, the loss of hormone-induced MIES was protected by SNCG. As shown in Figure 4C, while treatment of parental MCF-7 cells with 17-AAG abolished E2-induced mTOR activation as measured by the absence of phosphorylated S6K, expression of SNCG in MCFB6 cells rendered a resistance to the inactivation of mTOR due to Hsp90 disruption. We also studied the chaperone effect of SNCG on protection of ERK1/2 signaling. Both estrogen and tamoxifen treatment stimulated ERK1/2 activation in MCF-7 and MCFB6 cell (Figure 4D). Treatment of MCF-7 cells with 17-AAG resulted in a significant decrease in activated ERK1/2 in both control and hormone-stimulated cells. However, forced expression of SNCG completely restored ERK1/2 activation in MCFB6 cells. These data suggest an important role of SNCG in regulation of ER-α36-mediated MIES, which can replace the chaperone activity of Hsp90 and maintain MIES.
Using MCF-7 as well as MCFB6 cells, we determined the physical interaction among SNCG, Hsp90, and ER-α36 in the presence and absence of 17-AAG (Figure 5A). IP of SNCG in SNCG-positive MCFB6 cells brought down Hsp90 and ER-α36 in the absence of 17-AAG, indicating that SNCG participated in a chaperone complex with Hsp90 and ER-α36 in the absence of Hsp90 inhibitor. As negative controls, control IgG did not pull down SNCG, ER-α36, and Hsp90 in MCFB6 cells; IP of SNCG in SNCG-negative MCF-7 cells did not bring down ER-α36 and Hsp90. Similarly, IP of Hsp90 co-precipitated SNCG and ER-α36 in MCFB6 cells and ER-α36 in MCF-7 cells. As expected, after cells were treated with Hsp90 inhibitor 17-AAG, Hsp90 dissociated from its client protein ER-α36. However, although SNCG dissociated from Hsp90 after the treatment with 17-AAG, it was still able to interact with ER-α36. Our data indicate that although SNCG participates in the chaperone complex with Hsp90, its function on ER-α36 is mediated by Hsp90-independent pathways such as by direct binding to and chaperoning ER-α36. We also investigated the interactions among endogenous SNCG, Hsp90, and ER-α36 in MDA-MB-231 cells (Figure 5B). The same interaction pattern among SNCG, ER-α36, and Hsp90 was observed in MDA-MB-231 cells as that we demonstrated in SNCG-transfected MCF-7 cells. When MDA-MB-231 cells were cultured in the absence of 17-AAG, endogenous SNCG was co-precipitated with Hsp90 and ER-α36. However, after the treatment of 17-AAG, Hsp90 dissociated from ER-α36 and SNCG, but SNCG still bound to ER-α36. Since SNCG is physically associated with ER-α36 at the stressful condition when the function of Hsp90 is disrupted, we reason that SNCG protects its client protein ER-α36 and prevents its degradation due to the loss of Hsp90 function. As demonstrated in Figure 5C, treatment of SNCG-negative MCF-7 cells with 17-AAG resulted in a significant loss of ER-α36; expression of SNCG in MCFB6 cells prevented this Hsp90 disruption-induced degradation of ER-α36. As a negative control, treatment of the cells with antiestrogen ICI 182,780 had no effect on the expression of ER-α36. Similar to MCF-7 cells, while SNCG-positive T47D cells were resistant to Hsp90 disruption; knockdown SNCG expression in AS-3 cells renders the reduction of ER-α36 in response to 17-AAG treatment. Taken together, these data suggest that at the stressful condition when the function of Hsp90 is disrupted, SNCG functions as a chaperone protein, which physically binds to ER-α36 and protects its expression and function.
Figure 5.

SNCG chaperones ER-α36. A and B: Physical interactions among SNCG, ER-α36, and Hsp90 in the presence and absence of 17-AAG. A: MCF-7 and its SNCG-transfected MCFB6 cells. B: MDA-MB-231 cells. Cells were treated with or without 1 μmol/L 17-AAG for 15 hours. Equal amount of protein was subjected to IP with SNCG, control IgG, and Hsp90 followed by Western blot for SNCG, ER-α36, and Hsp90. C: Prevention of ER-α36 degradation by SNCG. MCF-7, MCFB6 cells, T47D, and its SNCG knockdown AS-3 cells were treated either with 17-AAG (1 μmol/L, 12 hours) or ICI 182,780 (10 nmol/L, four hours). ER-α36 expression was analyzed by Western blot and normalized with actin.
SNCG Renders a Resistance to Tamoxifen
Recently, an association between ER-α36 expression and tamoxifen resistance was analyzed in 709 breast cancer patients with a median follow-up of 7.9 years.40 In these studies, ER-α36 expression in ER-α66-positive tumors who received tamoxifen treatment (n = 306) was associated with poorer survival. These studies indicate that ER-α36 is an important predictive marker for tamoxifen resistance in ER-α66-positive breast cancer patients. Since SNCG chaperones ER-α36 expression and function, we reason that SNCG may render tamoxifen resistance. As expected, treatment of MCF-7 cells with tamoxifen inhibited cell growth resulted in 90% fewer cells (Figure 6A). In MCFB6 cells, by contrast, the magnitude of tamoxifen-mediated growth inhibition was significantly attenuated, with only 30% growth inhibition achieved. The effect of SNCG expression on tamoxifen resistance was further demonstrated by inhibiting endogenous SNCG expression in T47D cells. In an anchorage-independent growth (Figure 6B), estrogen stimulated a 4.2-fold colony formation over control in SNCG expressing T47D cells, knocking down endogenous SNCG significantly reduced colony formation in AS-3 cells in response to E2, resulting in only 1.6-fold increase. While tamoxifen alone only slightly decreased colony formation in T47D cells, resulting in a 25% inhibition, a robust 74% reduction of colony formation was observed in AS-3 cells, indicating that SNCG expression renders a cell more resistance to tamoxifen.
Figure 6.
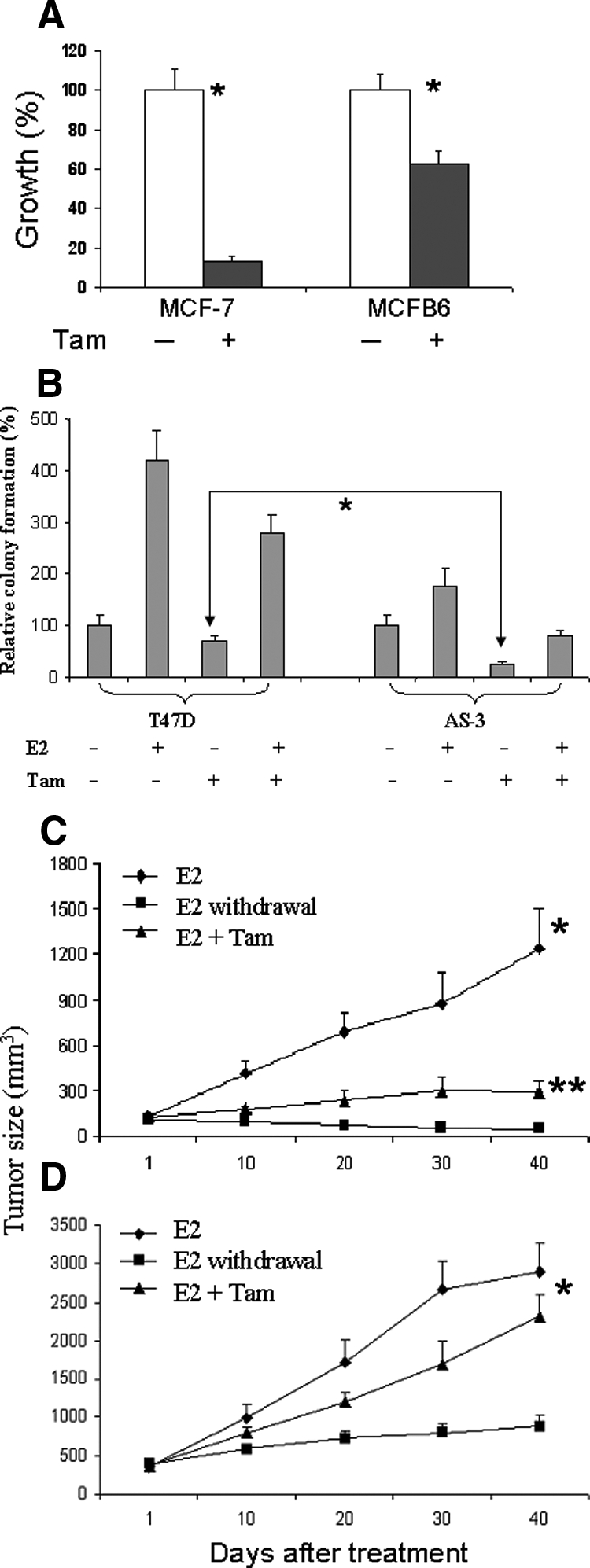
SNCG renders tamoxifen resistance. A: MCF-7 and MCFB6 cells were treated with 10 μmol/L tamoxifen for four days. Media were changed every two days with fresh added tamoxifen. Cell growth was measured using a cell proliferation kit (XTT). All values were presented as the percentage reduction over the control non-treated cells, which was taken as 100%. The numbers represent means ± SD of three cultures. Comparisons for cell growth in control relative to that of tamoxifen treatment indicate *P < 0.001. B: Colony formation of T47D and its SNCG knockdown AS-3 cells. Cells were treated with or without 1 nmol/L E2, 1 μmol/L tamoxifen, or combination of E2 and tamoxifen. The number of colonies was counted after two weeks of plating. Statistical comparison of colony formation for tamoxifen-treated AS-3 cells relative to tamoxifen-treated T47D cells indicates P < 0.01. C and D: Effect of tamoxifen on the growth of MCF-7 and MCFB6 xenografts. MCF-7 (C) and MCFB6 (D) tumor cell injection and estrogen supplement were described in Materials and Methods. There were eight mice in each group and each mouse received two injections, one on each side. Twelve days after injection, xenografts were established and tumors reached to >100 mm3. At this stage (day 1), mice bearing established tumors were randomly allocated to three groups: group 1, continued E2; group 2, E2 withdrawal (removal of the E2 pellet); group 3, E2 plus tamoxifen treatment (500 μg/mouse given subcutaneously, three days/week). Tumor size was determined by three-dimensional measurements (mm) using a caliper. All mice were sacrificed at day 40 after the first treatment. Each point represents the mean of tumors ± SE In MCF-7 xenograft, statistical comparison for tumor size in tamoxifen treated mice relative to control (E2 group) mice indicates *P < 0.001; and comparison of tamoxifen versus E2 withdrawal indicates **P < 0.05. In MCFB6 xenograft, comparisons for tumor size in both control (E2 group) and tamoxifen treated mice relative to that of E2 withdrawal mice indicate *P < 0.001; and comparisons for tamoxifen versus control indicate P > 0.05.
The biological relevance of SNCG-mediated tamoxifen resistance was also investigated in an orthotopic nude mouse model. To establish tumor xenografts, all mice were supplemented with E2 1 day before injection of tumor cells. Mice bearing established tumors were then randomized into three groups: group 1, continued treatment with E2; group 2, E2 withdrawal; and group 3, E2 with tamoxifen. The growth of MCFB6 tumor was stimulated much more by E2 than parental MCF-7 tumor. At 40 days, E2-stimulated MCF-7 tumor was 1.23 cm3 (Figure 6C) compared with that of 2.90 cm3 for MCFB6 tumor (Figure 6D). As expected, the growth MCF-7 xenograft was significantly inhibited by tamoxifen. At 40 days following treatment, tamoxifen inhibited E2-stimulated tumor growth by 77%. Although the tumor growth of MCFB6 cells was also inhibited by tamoxifen, the magnitude of growth inhibition reduced with a slight 19% growth inhibition. E2 withdrawal completely suppressed tumor growth of MCF-7 xenograft, but MCFB6 xenograft continued to grow at very slow path in the absence of E2 supplement. Consistent with in vitro data, these data indicate that SNCG renders tumor tamoxifen resistance.
Discussion
The present study showed that ER-α36 is an important and new player in MIES and that SNCG positively regulates ER-α36-mediated MIES through chaperoning ER-α36 protein. While SNCG overexpression enhanced ligand-dependent MIES, compromising endogenous SNCG expression comprised hormone stimulated MIES. The SNCG-enhanced MIES was demonstrated in four different cell systems including overexpression of SNCG in SNCG-negative MCF-7 cells; knockdown endogenous SNCG expression in SNCG-positive T47D cells; overexpression of SNCG in ER-negative but ER-α36 transfected MDA-MB-435 cells; and knockdown endogenous SNCG expression in ER-α36- and SNCG-positive MDA-MB-231 cells. We provided evidence suggesting that SNCG is a new member of molecular chaperone protein, which protects and stimulates ER-α36 mediated MIES. This evidence includes that SNCG binds to ER-α36 both in vitro in cell-free system and in breast cancer cells; SNCG is able to interact with ER-α36 even in the presence of 17-AAG, in which Hsp90 dissociates from its client protein ER-α; SNCG prevents Hsp90 disruption-induced degradation of ER-α36; SNCG can restore Hsp90 disruption-induced down-regulation of MIES; and SNCG significantly stimulated the mitogenic estrogen signaling mediated by ER-α36. These data suggest a critical role of SNCG in maintaining the stability and function of ER-α36, and thus rendering estrogen membrane-initiated signaling. In fact, it had been demonstrated that expression of SNCG enhances E2-stimulated mammary gland proliferation in transgenic mice28 and the growth of hormone-dependent breast cancer xenografts.27
Previously, we found that ER-α36 strongly attenuates the transactivational activity mediated by both AF1 and AF2 domains of ER-α66,15 indicating that ER-α36 negatively regulated genomic estrogen signaling. Recently, we also found the ER-α66 negatively regulates the promoter activity of the ER-α36 gene.41 Taken together, these results indicated that ER-α isoforms may mutually regulate each other’s activity and suggested that the non-genomic and genomic estrogen signaling have to be coordinated in vivo to maintain normal cell growth. Our finding that SNCG interacts with ER-α66 or ER-α36 depending on the presence of E2 suggested SNCG may function as a coordinator to regulate both genomic and non-genomic estrogen signaling.
We previously developed a specific polyclonal anti-ER-α36 antibody against the 20 amino acids at the C-terminal region that are unique to ER-α36.14,15 Using this antibody, we found that ER-α36 is expressed in both ER-positive and established ER-negative breast cancer cell lines. Expression of ER-α36 was also demonstrated in clinical breast cancer specimens from both ER-positive and ER-negative breast cancer patients.42 These data demonstrated a potential biological and clinical relevance of ER-α36 in breast cancer development and progression. Interestingly, we demonstrated that both E2 and its antagonist tamoxifen stimulate ER-α36-mediated ERK1/2 activation (Figure 4D), which was consistent with the previous observations that estrogen antagonists tamoxifen and ICI 182,780 acted as agonists to activate the ERK1/2 signaling pathway through ER-α36.42 These data indicate that ER-α36 can mediate agonist-like activities of antiestrogens. The antitumor efficacy of tamoxifen has been associated with the inhibition of estrogen binding to the ER-α66 and the subsequent inhibition of receptor transactivation. The mechanism of acquired tamoxifen resistance remains poorly understood. Since tamoxifen treatment up-regulates ER-α36 expression and ER-α36 can mediate agonist-like activities of tamoxifen in stimulation of ERK1/2 pathway, up-regulation of ER-α36 expression and signaling during breast cancer progression may represent an underlying mechanism for tamoxifen resistance. Indeed, we demonstrated that while expression of SNCG renders tamoxifen resistance in the growth of MCF-7 cells; knockdown endogenous SNCG expression in T47D cells makes the cells more responsive to tamoxifen-mediated growth inhibition in colony formation. This in vitro SNCG-mediated tamoxifen resistance is also confirmed in vivo in MCF-7 tumor xenograft model. Since SNCG protects ER-α36 expression and stimulates its signaling, we reason that this chaperoning effect on ER-α36 may serve one of the underlying mechanisms for tamoxifen resistance. It is noteworthy to emphasize that SNCG may serve as a tumor-specific chaperone and regulates many signaling pathways, eg, Akt/mTOR signaling (not published), involved in tumor cell survival, and thus SNCG-induced tamoxifen resistance might be mediated by multiple signaling pathways that regulated by SNCG. Nevertheless, our most recent large scale (n = 709) clinical follow-up studies indicate that expression of ER-α36 in ER-α66-positive breast cancer associates with tamoxifen resistance, while ER-α36 expression is not associated with survival in patients with ER-α66 negative tumors.42
Hsp90 is one of the most abundant cytoplasmic proteins in unstressed cells, where it performs housekeeping functions and controls the activity, intracellular disposition, and proteolytic turnover of a variety of proteins. So far, at least 100 proteins are known to be regulated by Hsp90, many of which, including HER2, Akt, mTOR, and steroid hormone receptors, are known to be important in the development of cancer.37,38,43 We previously demonstrated that SNCG participates in Hsp90-based multiple chaperone complex and regulates ER-α66 transactivation.26,27 One of the critical questions that needs to be addressed is whether SNCG-enhanced and ER-α36-mediated MIES is manifested by Hsp90-based chaperone complex or by its own chaperoning function. Using Hsp90 inhibitor 17-AAG, our data demonstrate that treatment of cells with 17-AAG resulted in a significant down-regulation of E2-induced mTOR activation. However, expression of SNCG completely recovered 17-AAG-mediated loss of mTOR activation. Furthermore, when cells were treated with 17-AAG, while Hsp90 lost it ATPase function and dissociated from its client protein ER-α36, SNCG was still able to physically associate with and chaperone ER-α36. These data suggest that SNCG, which can replace the chaperone function of Hsp90, is an independent chaperone protein and its chaperoning function is not dependent on Hsp90. Thus, SNCG and Hsp90 act cooperatively in ER-α36-mediated MIES.
Unlike the typical chaperone Hsp90, which is essential for a range of indispensable functions in normal tissue, SNCG is not expressed in normal cells but aberrantly expressed in advanced malignant state through epigenetic control by demethylation of CpG sites within SNCG gene,23 suggesting that SNCG is a more tumor-oriented chaperone. Targeting tumor specific chaperones such as SNCG represents a potential alternative to direct Hsp90 inhibition that may offer greater specificity and an improved side effect profile. Clinical follow-up studies indicate that expression of SNCG in breast cancers renders resistance to endocrine and adjuvant therapy.24 The present study demonstrated SNCG as a tumor specific chaperone, which can replace the chaperoning function of Hsp90 and regulate membrane estrogen signaling. The study will potentially lead to a new molecular profile of the tumor for the optimal patient selection for endocrine and Hsp90 disruption and a new strategy of combining SNCG targeting with Hsp90 disruption as a novel advantageous approach for treatment of cancer. Since SNCG positively regulates both genomic and non-genomic estrogen signaling, targeting SNCG for treating hormone-dependent breast cancer warrants further investigation.
Footnotes
Address reprint requests to Dr. Y. Eric Shi, M.D., Ph.D., Department of Radiation Medicine, Long Island Jewish Medical Center, New Hyde Park, NY 11040. E-mail: eshi@lij.edu.
Supported in part by grant 99-028-01-CCE from the American Cancer Society and grants W81XWH-04-1-0569 and W81XWH-07-1-0375 from the United States Army Medical Research and Development Command.
Y.E.S. and Y.C. contributed equally to this work.
References
- Beato M. Gene regulation by steroid hormones. Cell. 1989;56:335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- Norris JD, Fan D, Kerner SA, McDonnell DP. Identification of a third autonomous activation domain within the human estrogen receptor. Mol Endocrinol. 1997;11:747–754. doi: 10.1210/mend.11.6.0008. [DOI] [PubMed] [Google Scholar]
- Flouriot G, Brand H, Denger S, Metivier R, Kos M, Reid G, Sonntag-Buck V, Gannon F. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. EMBO J. 2000;19:4688–46700. doi: 10.1093/emboj/19.17.4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penot G, Le Péron C, Mérot Y, Grimaud-Fanouillère E, Ferrière F, Boujrad N, Kah O, Saligaut C, Ducouret B, Métivier R, Flouriot G. The human estrogen receptor-alpha isoform hERalpha46 antagonizes the proliferative influence of hERalpha66 in MCF7 breast cancer cells. Endocrinology. 2005;146:5474–5484. doi: 10.1210/en.2005-0866. [DOI] [PubMed] [Google Scholar]
- Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci USA. 1994;91:8517–8521. doi: 10.1073/pnas.91.18.8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Mellay V, Lasmoles F, Lieberherr M. Galpha(q/11) and gbetagamma proteins and membrane signaling of calcitriol and estradiol. J Cell Biochem. 1999;75:138–146. doi: 10.1002/(sici)1097-4644(19991001)75:1<138::aid-jcb14>3.3.co;2-9. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999;33:307–319. doi: 10.1210/mend.13.2.0239. [DOI] [PubMed] [Google Scholar]
- Marquez DC, Pietras RJ. Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells. Oncogene. 2001;20:5420–5430. doi: 10.1038/sj.onc.1204729. [DOI] [PubMed] [Google Scholar]
- Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signaling cascade and c-fos immediate early gene transcription. Endocrinology. 1997;138:4030–4033. doi: 10.1210/endo.138.9.5489. [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. 1977;265:69–72. doi: 10.1038/265069a0. [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Driggers PH, Segars JH. Estrogen action and cytoplasmic signaling pathways. Part II: the role of growth factors and phosphorylation in estrogen signaling. Trends Endocrinol Metab. 2002;13:422–427. doi: 10.1016/s1043-2760(02)00634-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12:152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336:1023–1027. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA. 2006;103:9063–9068. doi: 10.1073/pnas.0603339103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Liu YE, Jia T, Wang M, Liu J, Xiao G, Joseph BK, Rosen C, Shi YE. Identification of a breast cancer-specific gene. SNCG, by direct differential complementary DNA sequencing. Cancer Res. 1997;57:759–764. [PubMed] [Google Scholar]
- Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;121:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Jia T, Liu YE, Liu J, Shi YE. Stimulation of breast cancer invasion and metastasis by synuclein gamma. Cancer Res. 1999;59:742–747. [PubMed] [Google Scholar]
- Bruening W, Giasson BI, Klein-Szanto AJ, Lee VM, Trojanowski JQ, Godwin AK. Synucleins are expressed in the majority of breast and ovarian carcinomas and in preneoplastic lesions of the ovary. Cancer. 2000;88:2154–2163. [PubMed] [Google Scholar]
- Wu K, Weng Z, Tao Q, Lin G, Wu X, Qian H, Zhang Y, Ding X, Jiang Y, Shi YE. Stage-specific expression of breast cancer-specific gene gamma synuclein. Cancer Epidemiol Biomarkers Prev. 2003;12:920–925. [PubMed] [Google Scholar]
- Liu H, Liu W, Wu Y, Zhou Y, Xue R, Luo C, Wang L, Zhao W, Jiang JD, Liu J. Loss of epigenetic control of synuclein-gamma gene as a molecular indicator of metastasis in a wide range of human cancers. Cancer Res. 2005;65:7635–7643. doi: 10.1158/0008-5472.CAN-05-1089. [DOI] [PubMed] [Google Scholar]
- Guo J, Shou C, Meng L, Jiang B, Dong B, Yao L, Xie Y, Zhang J, Chen Y, Budman DR, Shi YE. Neuronal protein synuclein gamma predicts poor clinical outcome in breast cancer. Int J Cancer. 2007;121:1296–1305. doi: 10.1002/ijc.22763. [DOI] [PubMed] [Google Scholar]
- Wu K, Quan Z, Weng Z, Li F, Zhang Y, Yao X, Chen Y, Budman D, Goldberg ID, Shi YE. Expression of neuronal protein synuclein gamma gene as a novel marker for breast cancer prognosis. Breast Cancer Res Treatment. 2007;101:259–267. doi: 10.1007/s10549-006-9296-7. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Liu YE, Lu A, Gupta A, Goldberg ID, Liu J, Shi YE. Stimulation of estrogen receptor signaling by gamma synuclein. Cancer Res. 2003;63:3899–3903. [PubMed] [Google Scholar]
- Jiang Y, Liu YE, Goldberg ID, Shi YE. γ synuclein, a novel heat-shock protein-associated chaperone, stimulates ligand-dependent estrogen receptor α signaling and mammary tumorigenesis. Cancer Res. 2004;64:4539–4546. doi: 10.1158/0008-5472.CAN-03-3650. [DOI] [PubMed] [Google Scholar]
- Liu YE, Pu W, Jiang Y, Shi D, Dackour R, Shi YE. Chaperoning of estrogen receptor and induction of mammary gland proliferation by neuronal protein synuclein gamma. Oncogene. 2007;26:2115–2125. doi: 10.1038/sj.onc.1210000. [DOI] [PubMed] [Google Scholar]
- Gupta A, Inaba S, Wong OK, Fang G, Liu J. Breast cancer-specific gene 1 interacts with the mitotic checkpoint kinase BubR1. Oncogene. 2003;22:7593–7599. doi: 10.1038/sj.onc.1206880. [DOI] [PubMed] [Google Scholar]
- Inaba S, Li C, Shi YE, Song DQ, Jiang JD, Liu J. Synuclein gamma inhibits the mitotic checkpoint function and promotes chromosomal instability of breast cancer cells. Breast Cancer Res Treatment. 2005;94:25–35. doi: 10.1007/s10549-005-6938-0. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. mTOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Chang SB, Miron P, Miron A, Iglehart JD. Rapamycin inhibits proliferation of estrogen-receptor-positive breast cancer cells. J Surg Res. 2007;138:37–44. doi: 10.1016/j.jss.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Yin XJ, Wang G, Khan-Dawood FS. Requirements of phosphatidylinositol-3 kinase and mammalian target of rapamycin for estrogen-induced proliferation in uterine leiomyoma- and myometrium-derived cell lines. Am J Obstet Gynecol. 2007;196:176.e1–176.e5. doi: 10.1016/j.ajog.2006.09.037. [DOI] [PubMed] [Google Scholar]
- Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278:32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007;265–266:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AF. MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res. 2009;69:5292–5293. doi: 10.1158/0008-5472.CAN-09-1528. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- Picard D, Khursheed B, Garabedian M, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Tanaka F, Sobue G. Alleviating neurodegeneration by an anticancer agent: an Hsp90 inhibitor (17-AAG). Ann NY Acad Sci. 2006;1086:21–34. doi: 10.1196/annals.1377.012. [DOI] [PubMed] [Google Scholar]
- Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Wang Z, Xie Y. Expression of ER-α36, a novel variant of estrogen receptor α, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27:3423–3429. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Ding L, Coleman M, Wang Z. Estrogen receptor-alpha (ER-alpha) suppresses expression of its variant ER-alpha 36. FEBS Lett. 2009;583:1368–1374. doi: 10.1016/j.febslet.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LMJ, Cao J, Chen P, Gatalica Z, Wang ZY. ER-α36, a novel variant of ER-a, is expressed in ER-α positive and negative human breast carcinomas. Anticancer Res. 2008;28:479–484. [PMC free article] [PubMed] [Google Scholar]
- Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003;63:2139–2144. [PubMed] [Google Scholar]