

Abstract
Morphine, a highly potent analgesic agent, is frequently prescribed for moderate to severe cancer pain. In this study, morphine was administered at a clinically relevant analgesic dose to assess tumor cell-induced angiogenesis and subcutaneous tumor growth in nude mice using mouse Lewis lung carcinoma cells (LLCs). Implantation of mice with a continuous slow-release morphine pellet achieved morphine plasma levels within 250–400 ng/ml (measured using a radioimmunoassay, Coat-A-Count Serum Morphine) and was sufficient to significantly reduce tumor cell-induced angiogenesis and tumor growth when compared with placebo treatment. Morphometric analysis for blood vessel formation further confirmed that morphine significantly reduced blood vessel density (P < 0.003), vessel branching (P < 0.05), and vessel length (P < 0.002) when compared with placebo treatment. Morphine’s effect was abolished in mice coadministered the classical opioid receptor antagonist, naltrexone, and in mu-opioid receptor knockout mice, supporting the involvement of the classical opioid receptors in vivo. Morphine’s inhibitory effect is mediated through the suppression of the hypoxia-induced mitochondrial p38 mitogen-activated protein kinase (MAPK) pathway. Our results suggest that in vitro morphine treatment of LLCs inhibits the hypoxia-induced nuclear translocation of hypoxia-inducible transcription factor 1α to reduce vascular endothelial growth factor transcription and secretion, in a manner similar to pharmacological blockade with the p38 MAPK-specific inhibitor, SB203585. These studies indicate that morphine, in addition to its analgesic function, may be exploited for its antiangiogenic potential.
Pain management is a serious problem in patients with cancer. Morphine is considered the “gold standard” for relieving pain and is currently one of the most effective drugs available clinically for the management of moderate to severe pain associated with cancer.1 Management of cancer pain may be important not only to the quality of the patients’ lives but also to the cancer treatment itself. In the United States, 30 to 40% of newly diagnosed cancer patients and 90% of patients with advanced cancer report moderate to severe pain, which progresses in relation to tumor size, the degree of metastasis, the type of tumor, and its location.2 Effective morphine doses and routes of administration vary between cancer patients, because the dose is titrated against either analgesia or opioid-related side effects.3 In human patients the route of administration, duration, and dosage can independently affect the blood plasma levels required to achieve analgesia (11.0–1400 ng). The daily doses reported to produce analgesia range from 25 to 2000 mg morphine, with an average of between 100 and 250 mg in tolerant patients.1 Dose titration is frequent with both instant-release and modified-release morphine products. Even though adverse effects occur, only 4% of patients discontinue treatment because of intolerable adverse effects.1,2,3,4,5
The effect of morphine on tumor growth is still contradictory. Independent investigations using MCF-7 breast cancer cells show that morphine can either decrease6 or increase7 tumor growth in mice. The discrepancies in results may be due to the differences in either i) the morphine dose administered (analgesic6 versus subanalgesic7) or ii) the mode of administration (systemic6 versus localized7). Successful tumor growth depends on many aspects—primarily tumor cell proliferation and blood vessel formation or angiogenesis. As the solid tumor grows, tumor cells move further away from their vascular supply, and low oxygen tensions, or hypoxia, stimulate tumor cells to secrete proangiogenic factors.8 Vascular endothelial growth factor (VEGF) is one such potent proangiogenic factor secreted by hypoxic tumor cells within the developing tumor mass, initiating endothelial cell sprouting, migration, proliferation, and tube formation.9,10 It was previously shown that morphine inhibits hypoxia-induced VEGF secretion in rat cardiomyocytes and human umbilical vein endothelial cells.11 In addition, natural opioid ligands, such as β-endorphins, have also been shown to inhibit blood vessel proliferation and to produce shorter vessels when compared with placebo controls using a chicken chorion-allantoic membrane model.12,13
Adaptive responses to hypoxia are under the control of hypoxia-inducible transcription factors (HIF). HIF-1 activates genes such as VEGF and VEGF-receptor transcription through the binding of hypoxic response elements (HRE) within gene promoter regions.14 HIF-1 controls the expression of many genes involved in angiogenesis, glucose transport, and metabolism. HIF-1 is made up of one α-subunit and one β-subunit. The regulatory α-subunit is constitutively transcribed and protein stability is tightly controlled through posttranslational modifications, leading to rapid degradation under normal oxygen tensions. The β-subunit is constitutively active and localized to the nuclei irrespective of the oxygenation state.14 Prolyl hydroxylase domain-containing proteins are considered the key enzymes in the oxygen-dependent regulation of HIF-1. The activity of HIF prolyl hydroxylases depends on oxygen, iron, ascorbate, and 2-oxoglutarate for optimal hydroxylating capabilities.15 Within normal oxygen tensions, or normoxia, HIF prolyl hydroxylases are capable of hydroxylating the proline 402/564 residues of HIF-1α, which are quickly ubiquitinated by the E3 ligase, von Hippel Landau, and targeted for proteosomal degradation. Hypoxia or treatment with iron chelating agents such cobalt chloride and desferroxamine (DFO) can induce HIF-1α stability by reducing HIF prolyl hydroxylase activity, leading to cytoplasmic accumulation, HIF-1β dimerization, and nuclear translocation for gene transcription.15
HIF-1α protein stability or transcription varies with either the stimulus or cell-type being examined. Under hypoxia, activation of the several mitogen-activated protein kinases (MAPKs) enhances HIF-1α activity over normoxia.16,17,18 Although deletion studies have yet to identify site-specific residues, which when phosphorylated are capable of altering the hypoxic induction of HIF-1α, in vitro assays have successfully demonstrated the capability of MAPK to directly phosphorylate HIF-1α.17 The phosphorylation of HIF-1α is essential in transcriptional activation pathways and/or may prevent the docking of HIF prolyl hydroxylases during hypoxia, further stabilizing HIF-1.19,20 P38 MAPKα knockout mice or mice lacking upstream components of the p38 signaling pathway are embryonic lethal, and die in mid-gestation with defects in placental and embryonic vasculature.21 In addition, the hypoxia-induced HIF-1α nuclear translocation and protein stability is completely abolished in p38 embryonic null fibroblasts.22 However, p38 embryonic null cells treated with anoxia, a complete lack of oxygen, or DFO retain their ability to induce HIF-1α stability and nuclear localization for gene transcription,22 suggesting that hypoxia signaling pathways that regulate HIF-1 are distinct from those involving other known HIF-1 stabilization agents.
To date, very few studies have investigated the effects of opioids on tumor cell-induced angiogenesis—an essential process mediating tumor growth. Based on previous studies showing morphine’s inhibition on hypoxia-induced VEGF expression11 and blood vessel proliferation,12,13 we hypothesized that morphine will also inhibit tumor cell-induced angiogenesis to suppress tumor growth and progression in mice. Naltrexone, a classical opioid receptor antagonist, has been suggested as an effective cancer therapeutic,23 and it is therefore important to address whether morphine commonly prescribed to cancer patients for pain relief can cause further detriment to these patients by increasing tumor growth through effects involving angiogenesis. In the present studies we show that long-term systemic morphine treatment at a physiologically relevant analgesic dose affects tumor cell-induced angiogenesis and tumor growth using mouse Lewis Lung Carcinoma cells. We further investigated the mechanism by which morphine inhibits hypoxia-driven transcriptional activation of VEGF in tumor cells in vitro.
Materials and Methods
Cell Culture, Hypoxia, and Reagents
Lewis lung carcinoma cells (LLCs) were obtained from ATCC (Manassas, VA) and cultured in RPMI 1640 supplemented with 10% FBS + 1% Penicillin and Streptomycin (Sigma Aldrich, St. Louis, MO). MA148 epithelial ovarian cancer cells were from the laboratory of Dr. S Ramakrishnan. Cells were grown on tissue culture-treated dishes for protein and RNA isolation. Subconfluent cells were treated with 1.0 μmol/L morphine sulfate (Sigma) in normoxic conditions (normoxia, 21% oxygen), defined here as normal room air in a 5% CO2, 37°C cell culture incubator. To achieve hypoxia (∼2% oxygen), cell culture dishes were placed in a modular chamber (Billups Rothenberg, Inc., Del Mar, CA) and flushed with a mix of 0% O2, 5%CO2, 95%N2 at 10L/min for 15 minutes. Chambers remained tightly sealed and placed in a 5% CO2, 37°C incubator. This method achieves pO2 levels less than 35mmHg as determined from cell culture medium analyzed using a blood gas analyzer, Rapid Lab 248 (Chiron Diagnostics Tarrytown, NY); pO2 levels of culture supernatant from cells grown in room air or normal oxygen was 150–160 mm Hg. Rat monoclonal antibody to mouse CD31-PE was purchased from BD Pharmingen (Franklin Lakes, NJ); rabbit anti-mouse HIF-1α and hydroxy-HIF antibodies from Novus Biologicals (Littleton, CO); 20 anti-mouse-IgG-FITC from eBiosciences (San Diego, CA); p38 total and phospho antibodies from Cell Signaling Technology (Danvers, MA), Phospho Plus, p38 MAPK Kinase (Thr 180/Tyr 182), β-actin and α-tubulin from Santa Cruz Biotechnology (Santa Cruz, CA). The p38 MAPK inhibitor SB203585 was purchased from Sigma-Aldrich.
Morphine Treatment
In Vivo
Mice received morphine and placebo through pellet implantation method as described19 with a few modifications. Briefly, 75 mg morphine pellets, placebo, and/or 30 mg Naltrexone pellets (National Institutes of Health [NIH]/National Institute on Drug Abuse [NIDA], Bethesda, MD) were inserted in a small pocket created by a small incision on the animal’s dorsal side; incisions were resealed using surgical wound clips (Stoelting, 9 mm Stainless Steel, Wooddale, IL). Pellets are able to deliver morphine at steady state levels (200–400 ng/ml). Around day 10 to 11 morphine pellets become encapsulated and supplemental doses of morphine were given in the tumor volume assay to prevent tolerance associated with morphine usage. From days 7 to 14 animals received a total of 20 mg/kg/day morphine sulfate intraperitoneally and the morphine dose was further escalated to 30 mg/kg/day from day 15 to 21.
In Vitro
Serum-deprived cells were pretreated with 1.0 μmol/L morphine sulfate for 24 hours before hypoxia treatment.
Enzyme Linked Immunosorbent Assay
The concentration of mouse or human VEGF-A protein expression was determined using either a mouse or human VEGF Duo-set enzyme linked immunosorbent assay (ELISA) kit per manufacturer’s instructions using diluted culture supernatants after 48 hours hypoxia, also using the modular chamber (R&D Systems, Minneapolis, MN).
Polymerase Chain Reaction (PCR)
Tumor cell culture medium was removed and cells were washed in ice-cold PBS. Cell pellets were lysed using RLT buffer (Qiagen, Valencia, CA) that contained 5 mmol/L β-mercaptoethanol (Sigma-Aldrich). Total cellular RNA was isolated using the RNeasy Minikit per manufacturer’s instructions (Qiagen). Total RNA was DNase I treated (Invitrogen, Carlsbad, CA) and quantitated using the A260:A280 ratio. Reverse transcription and real-time PCR was performed as described11 on an ABI Prism 7500 instrument using a SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) for mouse VEGF and ribosomal 18s. All assays were performed in triplicate and normalized to ribosomal 18s mRNA in the same cDNA set. Data are expressed as fold change over control (untreated cells) using the ddCt method as described.24 The results from one representative experiment are shown. Mu-opiate receptors (MOR) expression was determined using the primers described.11
Tumor-Induced Matrigel Plug Assay
Animals were implanted with morphine, placebo pellets, or morphine + naltrexone pellets on day 0 (n = 10 per group). At the same time, 1.0 × 106 LLCs were resuspended in 500 μl of a 3:1 matrigel (Sigma):HBSS mix and injected subcutaneously into the right flank of athymic mice. LLCs are an approved cell line for drug testing by the NIH and derived from spontaneous lung tumors of C57BL/6 mice. After 7 days animals were sacrificed; matrigel plugs were surgically removed and photographed using a dissection scope (Biomedical Imaging and Processing Lab, University of Minnesota). Matrigel plugs were fixed in 10% neutral buffered formalin overnight. Histological analysis was performed at the University of Minnesota, Fairview Clinical Pathology and Lab Services. In subsequent experiments, matrigel plugs were snap-frozen in liquid nitrogen and 8- to 10-micron-thick cryostat sections were used for CD31 staining and blood vessel density measurements (morphometric analysis).
VEGF-Induced Matrigel Plug Assay
Mu-opioid receptor knockout (MORKO) mice developed on a C57BL/6 genetic background and wild-type C57BL/6 mice were first implanted with 75 mg morphine pellets on day 0 (n = 12). Next 500 μl of a 3:1 matrigel (Sigma):HBSS mix containing 100 ng/ml VEGF were injected subcutaneously into the right flank. After 7 days animals were sacrificed; matrigel plugs were surgically removed and fixed in 10% neutral buffered formalin overnight. Histological analysis was performed at the University of Minnesota, Fairview Clinical Pathology and Lab Services. In subsequent experiments, matrigel plugs snap-frozen plug sections were used for CD31 staining and vessel density measurements (morphometric analysis) as described below.
COAT-A-COUNT Serum Morphine in Vitro Radioimmunoassay
On the day of termination of experiments, blood was drawn from experimental mice through cardiac puncture and serum was separated and stored at −80°C. The concentration of morphine in experimental mice serum was determined by Radio-Immuno-Absorbent using equal volumes of serum samples as directed by manufacturer (Diagnostic Products Corporation, Los Angeles, CA). The serum concentration of morphine was expressed in ng/ml.
Morphometric Analysis of Blood Vessel Density
Frozen sections were thawed and fixed using ice-cold acetone. Nonspecific binding was blocked with 1% bovine serum albumin (BSA) in PBS. For CD31-staining, at least five sections per matrigel plug or LLC tumor (n = 10) per treatment group (placebo versus morphine versus morphine + naltrexone) were used. Rat anti-mouse-CD31-PE antibody was used at a 1:50 dilution to determine blood vessel density as previously described.25 Tumor sections were examined in a blinded fashion for the presence of CD31-stained endothelial cells in at least 10 randomly chosen fields under ×20 magnification and within vessel dense areas. Fluorescent digital images were first linearized, binarized, and then skeletonized using the Reindeer Plug-In Functions for Adobe Photoshop before calculating in arbitrary units the relative number of blood vessels (vessel density), the branch-points (vessel branching), and total blood vessel length.
Tumor Growth Assay
Athymic mice 7 to 9 weeks of age (Jackson Labs, Bar Harbor, ME) were housed in a SPF facility, and all animal experiments were performed according to approved protocols by the Institutional Animal Care and Use Committee at the University of Minnesota. LLC suspensions were prepared in HBSS admixed in matrigel. Cells (2.0 × 106) in matrigel/HBSS were injected subcutaneously into the animals’ right flank. Mice received morphine and placebo through the pellet implantation method. Tumor size was measured using calipers by recording the greatest length and width. The tumor volume was calculated using the formula 0.5 × (width)2 × (length). On day 21, animals were sacrificed and tumors were surgically removed and photographed using a digital camera (BIPL, University of Minnesota). Tumor wet weights were recorded, and a part of the tumor was then fixed in 10% neutral buffered formalin for 24 hours and processed for histology. The rest of the tumor samples were snap frozen in liquid nitrogen, and 5-μm-thick cryostat sections were used for CD31 morphometric analysis for vessel density and HIF-1α expression.
TUNEL Staining
To determine the presence of apoptotic cells, terminal deoxynucleotidyl transferase (TdT) mediated dUTP nick end label (TUNEL) staining was performed on cryostat cut sections of frozen 21-day tumor tissues. Sections were prewarmed using a slide warmer at 65°C/30 minutes and deparaffinized in xylenes (2× at 5 minutes) then rehydrated in ETOH (95%, 85, 70%, H2O) for 5 minutes each. Slides were next treated with 10 μg/ml Proteinase at 37°C for 15 minutes in 10 mmol/L Tris (pH 7.5 at 37°C). Slides were then washed in Tris-buffered saline (TBS) (3× at room temperature for 5 minutes) followed by distilled water (2×/room temperature for 5 minutes). Sections were then equilibrated in terminal deoxynucleotidyl transferase (TdT) buffer 10 minutes at 37°C then labeled using terminal deoxynucleotidyl transferase (TdT) buffer + Enzyme-FITC labeled for 1 hour/37°C in a humidified chamber. Slides were washed before visualization in the presence of the nuclear stain, DAPI (4′, 6-diamidino-2-phenylindole). Images were then stained with CD31-PE in 1% BSA at room temperature for 1 hour. Fluorescent images were captured on an Olympus BX60 Upright Microscope (BIPL, University of Minnesota), analyzed and merged using Adobe Photoshop.
Immunohistochemistry for HIF-1α
Sections were prepared from frozen tumor samples and fixed in ice-cold acetone followed by 3× wash for 5 minutes in 1×PBS. Sections were incubated in 0.2% Triton X-100 in PBS and then blocked in 5% BSA in PBS at room temperature for 1 hour. The sections were incubated with primary HIF-1 antibodies at room temperature overnight at a dilution of 1:200. After washing with PBS, sections were incubated with a 20 IgG FITC-conjugated secondary antibody (1:1000) in blocking buffer at room temperature for 1 hour. Sections were then washed with PBS, air dried, and mounted with a DAPI-containing mounting medium and observed under fluorescent microscopy (BIPL, University of Minnesota).
MTT Cell Viability Assay
LLCs were plated in 96-well plates in 1% fetal bovine serum containing growth media. Cells were pretreated with increasing concentrations of morphine for 24 to 72 hours under serum-free conditions. After 72 hours incubation, 20 mg/ml MTT was added to each well and allowed to incubate for another 4 hours in a 37°C incubator. Cells were solubilized with 100% DMSO, and absorbance at 450 nm was measured using a plate reader. Untreated controls were compared with morphine treatment by determining the percent change in cell viability using the formula:
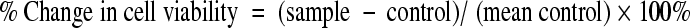 |
Protein Extraction and Western Blot Analysis
Whole cell lysates and cytoplasmic and nuclear protein extracts were prepared on ice for HIF-1α, p38, and hydroxy-HIF Western blot analysis, using a denaturing lysis buffer containing protease inhibitors (Sigma). Bradford protein quantification (BIORAD, Hercules, CA) was used to determine protein concentration and aliquots (60–100 μg) were loaded onto a 12% SDS–PAGE gel then transferred to a nitrocellulose membrane (Pierce, Sigma–Aldrich). The membrane was rinsed in TBS-T solution (0.1% Tween 20 in TBS, pH 7.5) and incubated in a starter block (Pierce, Thermo Scientific, Rockford, IL) at room temperature for 1 hour. Membranes were washed 3× at 5 minutes each with TBS-T followed by 1 hour incubation with HIF-1α, β-actin, α-tubulin, hydroxy-HIF-1α, or overnight incubation with p38 MAPK-phosphophorylation antibodies (1:500) or total antibody (1:1000) anti-rabbit primary antibodies at 4°C overnight. HIF-1α membranes were washed 3× in TBS-T, followed by 1 hour incubation with 5% donkey serum (Sigma Aldrich, St. Louis). Membranes were then washed 3× TBS-T, followed by the appropriate anti-rabbit donkey secondary-HRP antibody (GE Health Care UK Limited) at room temperature for 1 hour. Membranes were washed with 3× in TBS-T and incubated with a secondary HRP-linked antibody provided with the kit. The immunoreactive bands were visualized using chemiluminescent reagents as recommended by Amersham Pharmacia Biotechnology. BIO-Rad software was used for densitometric analysis (BIPL, University of Minnesota).
Electromobility Shift Assay
Nuclear extracts were prepared from LLCs pretreated with 1.0 μmol/L morphine sulfate and exposed to normoxia or hypoxia for comparison with untreated controls using the Nu-CLEAR EXTRACTION KIT (Sigma catalog no. N-XTRACT). 10 ng of nuclear extract was determined using Bradford Reagents and incubated with 0.5 ng 32P-labeled double-stranded oligonucleotide probes (labeled with ATP using a polynucleotide kinase (50,000 cpm/ng) for the hypoxia response element-specific HIF-1a-binding sequence (5′-TCAGTACGTGACCACACTCACCTC-3′) and (3′-AGACATGCACTGGTGTGAGTGGAG-5′) (Promega, Madison, WI) at room temperature in a total volume of 20 μl using the appropriate buffers for 20 minutes. DNA-protein complexes were resolved by electrophoresis on a 4% polyacrylamide gel at 200 V in TBE buffer. The gels were dried overnight and HIF-1α binding to the probe was confirmed after autoradiography with an intensifying screen. To verify the specificity of the HRE–HIF-1α protein interaction, proteins from treated cells were preincubated in the presence and absence of anti-HIF-1 antibody. Briefly, DNA was added to the HRE DNA oligo-protein-buffer mix and protein-antibody protein-buffer mix was incubated 20 minutes at room temperature in the dark. HRE oligos were then added to the protein-antibody-buffer mix and incubated for further 15 minutes at room temperature. DNA-protein complexes were resolved on a 4% native acrylamide gel in the dark. After electrophoresis, the glass plates containing the gel were imaged.
HIF-1α Duo-Set Kit
To further examine HIF-1α stability, nuclear and cytoplasmic proteins were isolated from experimental cells and prepared as recommended by the manufacturer. Aliquots were used to determine protein concentration using the Bradford Assay. Biotin-labeled ds oligonucleotides were added to proteins and incubated for 30 minutes at room temperature along with a control (without nuclear extracts). To determine specificity of binding through competition, 3× the amount of unlabeled ds oligonucleotides were incubated in the presence of biotin-labeled ds oligonucleotides. After incubation with 5% BSA blocking buffer to reduce nonspecific binding, 10 μg of nuclear or 40 μg of cytoplasmic proteins in the appropriate buffer was added and incubated at room temperature for 2 hours. Plates were aspirated and washed in 1× PBS. The recommended concentration (1:200 dilution) of Streptavidin-HRP was added to each well and incubated at room temperature for 20 minutes in the dark. Plates were washed, developing solutions added, and optical density determined using a microplate reader at 450 nm. Data are represented as the relative HIF-1α levels after subtraction of optical density from values of biotin-labeled and unlabeled ds oligonucleotides, adjusting for nonspecific binding.
Statistics
All numerical data are expressed as a mean ± SE. To determine statistical differences between treatment groups, we performed a Student’s t-test; a value of P < 0.05 was considered as statistically significant.
Results
Morphine Inhibits Hypoxia-Induced VEGF Secretion in Cancer Cells
Tumor cells within the developing tumor experience varying levels of low oxygen (hypoxia) and a reduced nutrient supply as the tumor cells move further away from a vascular supply. VEGF is a potent proangiogenic factor secreted from hypoxic and nutrient-deprived tumor cells within the developing solid tumor. VEGF promotes new blood vessel formation or angiogenesis to sustain tumor growth.9,10 We have previously shown that morphine inhibits hypoxia-induced VEGF secretion.11 In the present study, to investigate the effect of morphine on hypoxia-induced VEGF secretion from tumor cells, we used the mouse LLCs and human ovarian cancer cells (MA148). ELISA was used to determine VEGF secretion from diluted cell-free supernatants of tumor cells cultured under room air in a 5% CO2, 37°C incubator and compared with cells cultured in a hypoxia chamber as described in the methods. As shown in Figure 1A, mouse LLCs exposed to hypoxia for 48 hours increased VEGF secretion fivefold over cells cultured under normoxia. LLC hypoxia-induced VEGF secretion was significantly reduced with morphine pretreatment (30%; P < 0.05). Similar to LLCs, hypoxia increased human VEGF protein secretion from MA148 ovarian cancer cells sixfold over cells cultured under normoxic conditions (Figure 1B; P < 0.017, 3000pg/ml vs. 500pg/ml VEGF-A). MA148 hypoxia-induced VEGF secretion was also significantly reduced with morphine pretreatment (50%; P < 0.04). These results suggest that similar to human umbilical vein endothelial cells and rat cardiomyocytes, morphine also inhibits the hypoxia-induced VEGF secretion from hypoxic tumor cells to potentially reduce tumor angiogenesis and growth.
Figure 1.

Morphine treatment affects hypoxia-induced VEGF-A secretion from tumor cells in vitro. Graph shows VEGF-A levels after 48 hours hypoxia (30 mm Hg PO2) using an oxygen-modular chamber and determined using an ELISA and diluted cell culture supernatants derived from mouse Lewis lung carcinoma cells (A) and human ovarian cancer cells (MA148, B). *P < 0.05; **P < 0.005.
Morphine Inhibits Tumor Angiogenesis in Vivo
Based on our previous results that morphine reduced hypoxia-induced VEGF secretion from tumor cells in vitro, we next hypothesized that morphine will inhibit tumor cell-induced angiogenesis in vivo. To examine the effect of morphine on tumor cell-induced angiogenesis we conducted a matrigel plug angiogenesis assay. LLCs were admixed in matrigel and implanted subcutaneously into the flanks of athymic mice (n = 10). At the contra-lateral site mice were implanted with either a placebo or morphine slow-release pellet. LLC matrigel plugs were removed 7 days post implantation, photographed, and hematoxylin and eosin stained for tissue architecture and vascular structures. As shown in Figure 2A, matrigel plugs taken from placebo-treated mice were rosy red in appearance, indicative of successful LLC induced angiogenesis. In contrast, plugs taken from morphine-treated mice were very pale with a few blood vessels appearing mostly at the periphery of matrigel plugs (Figure 2B). Hematoxylin and eosin staining revealed defined vascular structures (arrows) within sections of matrigel plugs taken from placebo- (Figure 2C) and morphine-treated mice (Figure 2D). Morphometric analysis using CD31-PE (red) conjugated antibodies on snap-frozen matrigel plug sections used to further quantify tumor cell-induced angiogenesis. CD31 is present on mature endothelial cells and stained sections were analyzed using fluorescent microscopy. Digital fluorescent images were binarized and skeletonized as described in the methods (Figure 2, E and F). Compared with placebo, chronic morphine treatment reduced blood vessel density (total vessels, Figure 2G; P < 0.015), blood vessel branching (Figure 2H; P < 0.012), and blood vessel length (Figure 2I; P < 0.038). These results show that morphine treatment inhibits tumor cell-induced angiogenesis in vivo.
Figure 2.

Morphine inhibits tumor angiogenesis induced by LLCs. Matrigel containing 1 × 106 LLCs were injected subcutaneously in athymic mice receiving either placebo (A) or morphine treatment (B) through pellet implantation, to achieve mean blood levels of 300 ng/ml morphine. Paraffin-embedded sections of LLC matrigel plugs were hematoxylin and eosin stained to reveal tissue architecture and vascular structures (C, placebo; D, morphine see arrows). Angiogenesis was quantified using (E and F) CD31-PE antibodies and morphometric analysis; F: Fluorescent captured images from morphine treatment were linearized, binarized and skeletonized using Reindeer Plug in Function Software for Adobe Photoshop to determine mean blood vessel density (G) (P < 0.02), blood vessel branching (H) (P < 0.01), and blood vessel length (I) (P < 0.04) in comparison with (E) placebo treatment (n = 10).
Morphine Inhibits Tumor Angiogenesis through Classical Opioid Receptors
To assess the involvement of the opioid receptors in morphine’s inhibition on tumor cell-induced angiogenesis in vivo, we next tested the effect of naltrexone, an opioid receptor antagonist, in the presence of morphine on tumor angiogenesis in vivo. Similarly, LLCs were admixed in matrigel and implanted subcutaneously into the flanks of athymic mice—implanted with either a placebo (group 1), a morphine slow-release pellet only (group 2), or both morphine and naltrexone slow-release pellets (group 3). Morphological examination of matrigel plugs from placebo (Figure 3A) and morphine+naltrexone- (Figure 3C) treated animals were highly vascularized, in contrast to matrigel plugs taken from morphine-treated animals (Figure 3B). Similar to previous experiments and unlike with morphine treatment (Figure 3E), blood vessels containing red blood cells were easily distinguishable within hematoxylin-stained sections taken from either placebo (arrows, Figure 3D) or morphine+naltrexone cotreated mice (arrows, Figure 3F). Quantification of angiogenesis using morphometric analysis showed that morphine treatment (Figure 3H) reduced blood vessel density (Figure 3J; P < 0.003), vessel branching (Figure 3K; P < 0.005), and vessel length (Figure 3L; P < 0.002) when compared with placebo (Figure 3G). Cotreatment with naltrexone (Figure 3I) antagonized morphine’s effect in all parameters measured and levels were similar to placebo treatment (Figure 3, J–L).
Figure 3.

Morphine inhibits tumor angiogenesis through the classical opioid receptors. Athymic mice were injected with LLC/matrigel subcutaneously and implanted with placebo (A), morphine (B), and morphine plus naltrexone (an opioid receptor antagonist) (C) pellets for 7 days. Arrows in hematoxylin and eosin sections show the development of vascular structures from (D) placebo, (E) morphine, and (F) morphine plus naltrexone. Angiogenesis was quantified using CD31-PE antibodies and morphometric analysis (G–I). H: Fluorescent captured images from morphine treatment were linearized, binarized, and skeletonized with Reindeer Plug in Function Software for Adobe Photoshop to determine mean blood vessel density (J) (P < 0.003), blood vessel branching (K) (P < 0.005), and blood vessel length (L) (P < 0.002) in comparison with placebo treatment (G) (n = 10). Naltrexone reversed morphine inhibition of LLC angiogenesis, as blood vessel development was similar to (J–L) placebo. Hematoxylin/eosin (M and N) and CD31-PE (red, O and P) stained sections of 100 ng/ml rhVEGF/matrigel plugs injected subcutaneously in wild-type (WT) and MORKO mice preimplanted with morphine pellets for seven days.
The MORs are primarily responsible for the morphine-induced analgesic responses.26,27 To further investigate the involvement of opioid receptors in tumor cell-induced angiogenesis we performed a matrigel plug VEGF-induced angiogenesis assay in wild-type and MOR knockout (KO) mice receiving morphine through pellet implantation. Briefly, this assay was conducted as described, however LLC tumor cells were replaced with 100 ng/ml recombinant VEGF. In addition to a potent proangiogenic factor, VEGF promotes bone marrow-derived myeloid cell recruitment through direct influences on cell migration to actively participate and aide in angiogenesis.28 As shown in Figure 3, hematoxylin and eosin staining showed that morphine treatment (Figure 3M) reduced host cell infiltration in wild-type but not in MORKO mice (Figure 3N). We further confirmed that the host-infiltrating cells were CD31-positive endothelial cells; and sections of plugs from MORKO-morphine-treated mice (Figure 3P, red) showed greater blood vessel formation than those taken from morphine-treated wild-type mice (Figure 3O, red). In these experiments, plasma morphine levels in experimental mice were determined using a COAT-A-Count Serum Morphine Radioimmunoassay to ensure that this method of drug administration achieved morphine levels within those reported for analgesia, between 11.0 and 1400 ng/ml.3 The concentration of morphine from plasma samples taken from morphine-treated wild-type and MORKO were similar, approximately 300 ng/ml (250–400 ng/ml) and within that reported for analgesia. Taken together, our data suggest that morphine’s inhibition of tumor cell-induced angiogenesis involves the classical opioid receptors.
Morphine Reduces Tumor Growth in Mice
Cancer patients capable of tolerating morphine for analgesia are often exposed long-term and require dose escalation to overcome tolerance associated with morphine use.3,4,5 Because morphine inhibited the ability of hypoxic tumor cells to induce angiogenesis, an essential process required for tumor growth, we next sought to determine whether chronic morphine treatment would also affect tumor growth. To do this, LLCs were admixed in physiological buffer HBSS, and 1 × 106 cells were injected subcutaneously in wild-type mice receiving either placebo or morphine through pellet implantation. Around day 11, pellets can become encapsulated and alter morphine delivery, so supplemental escalating doses were given on day 9 (20 mg/kg/day) to 15 (30 mg/kg/day) as described in the Materials and Methods. Our results show that when compared with placebo, morphine treatment, in a manner similar to that in cancer patients, significantly reduces tumor progression (P < 0.007) (Figure 4A). Tumor wet weights at the time of necropsy further validated morphine’s effect on tumor growth. Gross morphology of representative tumors obtained from placebo-treated mice (Figure 4B) had significantly greater wet-weights compared with morphine treatment (Figure 4C) and on average weighed 52% more than tumors taken from morphine-treated mice (Figure 4D; P < 0.004). Pathological examination of tumor tissues showed greater cell density, indicated by the intense hematoxylin staining and more defined blood vessels (see arrows) within placebo (Figure 4E) as compared with morphine (Figure 4F). To determine whether morphine’s inhibition on tumor growth was indeed due to a decrease in angiogenesis, morphometric analysis for blood vessel density of residual tumors showed that morphine treatment (Figure 4H) significantly reduced overall blood vessel density (P < 0.006; Figure 4I) when compared with placebo treatment (Figure 4G). Tumor growth results from an imbalance between tumor cell death and growth. Morphine or periods of prolonged hypoxia can promote tumor cell apoptosis6,29 independently, so we next assessed apoptosis within the tumor-microenvironment using the TUNEL assay for DNA fragmentation as an indication of apoptosis. TUNEL stained sections of tumors from placebo-treated mice were examined using fluorescent microscopy and exhibited fewer TUNEL-positive apoptotic cells (Figure 4J, green) than tumor sections from morphine-treated mice (Figure 4K-L, P < 0.04). These results suggest that morphine inhibits tumor growth through effects on angiogenesis and tumor cell apoptosis.
Figure 4.

Chronic morphine treatment inhibited tumor growth. Female and male athymic-bearing LLC tumors were implanted with morphine or placebo pellets. Tumor size was measured with calipers, and tumor volume calculated using the formula 0.5 × (width)2 × (length). Graph A shows tumor volume over experimental days (**P < 0.005). Also shown, the gross morphology of representative LLC tumors taken from placebo (B) and morphine-treated mice (C), as well as wet-weights recorded at the time of necropsy (D) (P < 0.01). E: Hematoxylin and eosin sections from placebo tumors show increased cell proliferation and vessel formation (see arrows) over morphine treatment (F). Morphometric analysis for angiogenesis was determined using fluorescently labeled PE-conjugated antibodies to CD31 and Adobe Photoshop, Reindeer Plug-in Functions. Graph shows (I) morphine significantly reduced (H) blood vessel formation (mean vessel number, P < 0.006) when compared with placebo (G). Tunnel labeling, as an indication of apoptosis, was performed on cryostat cut frozen tissue sections. K and L: Morphine treatment resulted in significantly greater (P < 0.04) apoptotic tumor cells (green) than placebo (J).
Morphine Increased Tumor Cell Apoptosis by Decreasing Angiogenesis in Vivo
Next, we wanted to determine whether morphine’s inhibition on tumor cell-induced angiogenesis and tumor growth was due to a direct effect on tumor cell apoptosis because the majority of tumor cells but not endothelial cells (CD31+, red) were apoptotic as determined by TUNEL staining of tumor tissues (green) (see Supplemental Figure S1A at http://ajp.amjpathol.org). LLCs do express the MOR (see Supplemental Figure S1B at http://ajp.amjpathol.org), so we tested the effect of long-term morphine treatment on cell viability and proliferation. An MTT assay for cell viability showed that 72 hours of morphine (10 nmol/L-1.0 μmol/L) treatment did not significantly affect cell viability (see Supplemental Figure S1C at http://ajp.amjpathol.org) when compared with untreated cells. Similarly, cell proliferation studies using BrdU-DNA incorporation showed that morphine also had no significant effects of LLC proliferation in vitro (data not shown). To investigate hypoxia-induced apoptosis we treated LLCs with morphine for 72 hours in vitro. LLCs were grown and treated on chamber slides. Slides were prepared for TUNEL staining as an indication of apoptosis. Our in vitro studies show very high concentrations of morphine, above 1.0 mmol/L, was required to increase hypoxia induced tumor cell apoptosis—a concentration above therapeutic range (data not shown). These results led us to conclude that the increased LLC apoptosis observed in vivo was a consequence of a lack of angiogenesis.
Morphine Inhibits HIF-1 Binding to the HRE Found Within Target Genes
The active HIF-1 complex binds to the HRE within the VEGF gene, mediating this hypoxia-induced VEGF transcription and thus secretion.14 Previous examination of tumor tissues showed that HIF-1α expression (green) was associated primarily with tumor cells rather than CD31+ endothelial cells (see Supplemental Figure S1D at http://ajp.amjpathol.org), so we next examined the effect of morphine on HIF-1α–DNA binding in vitro. As shown in Figure 5A, nuclear extracts isolated from hypoxic LLCs produced greater binding to P32-labeled HRE shifted oligos in an electromobility shift assay than nuclear extracts from normoxic cells (Figure 5A, lanes 2 and 1, respectively) or morphine-pretreated hypoxic cells (Figure 5A, compare lane 2 and 3). To confirm binding specificity to the consensus HRE oligonucleotide, LLC hypoxic nuclear extracts were pretreated with HIF-1α antibody before the addition of the labeled HRE oligos. The addition of HIF-1α antibodies binding specifically to the oxygen-dependent degradation domain (ODD) of HIF-1 resulted in a loss of binding to the HRE-oligos, confirming that the interaction was specific to HIF-1α binding (Figure 5A, lanes 4 and 5). These results suggest that morphine inhibits the hypoxia-induced HIF-1 binding to its consensus DNA element, thereby modulating gene transcription and angiogenesis.
Figure 5.

Morphine reduced HIF-1α DNA binding by inhibiting nuclear localization and accumulation. A, lanes 1–3: HIF-1–DNA binding of nuclear extracts from normoxic and hypoxic cells with and without morphine treatment were subjected to an Electromobility Shift Assay. A, lanes 4–5: Pre-incubation with HIF-1α antibodies before addition of HRE-oligos confirmed specificity of the HRE-HIF-1 interaction. LLC nuclear extracts were analyzed using Western blot analysis and anti-HIF-1α antibodies and normalized to PolII (B) and a HIF-1α duo set kit (C) for HIF-1α nuclear levels. C: Graph shows morphine decreased the hypoxia-induced HIF-1α nuclear expression (HIF-1α Duo-set kit) in LLCs, due to a decrease in cytoplasmic accumulation (D).
Morphine Inhibits Hypoxia-Induced HIF-1α Nuclear Localization and Accumulation in Lung Cancer Cells
The ability of HIF-1 to bind to its respective HRE within target genes depends on HIF-1α stability/accumulation and posttranslational modification.19,20 To understand the observed reductions in DNA binding in the presence of morphine, we next assessed HIF-1α expression in LLC protein extracts using Western blot analysis. Our results showed that hypoxia increased HIF-1α nuclear translocation compared with normoxic cells (Figure 5B, lanes 1 and 2, normalized to nuclear PolII); and this induction was reduced on morphine pretreatment (Figure 5B, lanes 2 and 3). We next, validated the effect of morphine on HIF-1α nuclear localization using another method—a HIF-1α duo-set kit and in the presence of a well known hypoxia mimetic, cobalt chloride (CoCl2) that served as a positive control and a known inducer of HIF-1. Similar to the Western blot analysis, hypoxia increased HIF-1α nuclear expression similar to 100 μmol/L CoCl2 (under normoxia); and morphine pretreatment of hypoxic cells prevented HIF-1α nuclear translocation as HIF-1α was reduced to control levels (Figure 5C).
Hypoxia reduces the molecular oxygen availability for optimal prolyl hydroxylase activity. Any reductions in prolyl hydroxylating activity will lead to a reduction in proteosomal degradation and increased stabilization of cytoplasmic HIF-1α,14,15 so we next analyzed whole cell extracts from normoxia and hypoxic LLCs. Western blot analysis using anti-HIF-1α antibodies showed that 16 hours hypoxia was sufficient to stabilize HIF-1α expression as indicated by HIF-1α cytoplasmic accumulation and posttranslational modification as indicated by a second and slower migrating band (Figure 5D, compare lane 4 and 2). Unlike hypoxia, morphine pretreatment reduced the HIF-1α hypoxia-induced cytoplasmic accumulation and posttranslational modification (Figure 5D, lane 6). Taken together, these results suggest that morphine prevented the hypoxia-induced accumulation and posttranslational modification of HIF-1α that is essential for HIF-1 nuclear translocation and DNA binding.
Morphine Effects are Primarily Through the Hypoxia Signaling Pathways in LLCs
In addition to molecular oxygen, iron (Fe2++) is another essential cofactor required for the catalytic activity of HIF-1α prolyl hydroxylase (PHD).15 Iron chelating agents such as DFO and CoCl2 are excellent hypoxia mimetics and are known stabilizers of HIF-1 under normal oxygen tensions. DFO may directly inhibit the hydroxylation of proline residues and thus reduce the ubiquitin-mediated proteosomal degradation. To test the possibility that morphine may activate prolyl hydroxylase activity leading to an increased degradation and thus the observed reductions in HIF-1α accumulation, we treated LLCs with 100 μmol/L DFO or CoCl2 in the absence and presence of morphine. Western blot analysis of whole cell extracts showed that the DFO or CoCl2 robustly induced HIF-1α accumulation, producing broad diffuse bands, in a manner distinct from hypoxia (Figure 6A). In these studies, morphine pretreatment did not significantly alter this dramatic increase in either DFO-induced or CoCl2-induced HIF-1α nuclear translocation. In addition, nuclear extracts from DFO-treated and morphine-pretreated cells were incubated with HIF-1–HRE labeled oligos and subjected to an electromobility shift assay. As expected, DFO-induced HIF-1α nuclear translocation increased HIF-1 DNA binding, however this was unaffected by morphine pretreatment (Figure 6B). The specificity of the DNA-HIF interaction was confirmed using HIF-1α antibodies and was specific to HIF-1 as indicated by the loss in binding (Figure 6B, lanes 2, 3).
Figure 6.

Morphine treatment does not alter HIF-1 in the presence of hypoxia mimetics or after inhibition of prolyl hydroxylation. Whole cell extracts (WCE) from 100 μmol/L DFO- and 100 μmol/L CoCl2-treated LLCs were analyzed using Western blot analysis and (A) HIF-1α antibodies and (C) hydroxylated-HIF-1α antibodies (HIF-OH). B, lanes 1, 2: HIF-1-DNA binding of nuclear extracts from DFO treated LLCs with and without morphine treatment using an Electromobility Shift Assay. B, lanes 2 and 4: Preincubation with HIF-1α antibodies confirming specificity of the HRE-HIF-1 interaction. HIF-1α and HIF-OH expression was determined using Western blot analysis of WCE taken from normoxic cells treated with (D) Ethyl-3, 4-dihydroxybenzoate (EDHB), an inhibitor of prolyl hydroxylase domain containing enzymes and (E) MG132, a proteosomal inhibitor.
To further eliminate the role of HIF PHDs in morphine signaling, additional studies were conducted assessing the hydroxylation status of HIF-1α in LLCs after hypoxia treatment or hypoxia mimetic treatment. Generally, it is expected that HIF-1α-OH is undetectable under normoxia, because it will be quickly ubiquitinated and degraded. Using antibodies specific to the OH-Pro564 residue of HIF-1α, hydroxylated HIF-1α was more detectable under normoxia than after DFO or CoCl2 treatment, and this was unaffected with morphine pretreatment (Figure 6C). In contrast to the hypoxia mimetics, hypoxia did not completely abolish the hydroxylation of hypoxia-induced HIF-1α (Figure 6C). Instead HIF-1α-OH was more detectable under hypoxia than DFO or CoCl2 treatment and morphine pretreatment did not affect the hypoxia-induced HIF-1α-OH status any further (Figure 6C, lane 8). These results suggest that DFO and CoCl2 inhibit the PHDs to reduce HIF-1α-OH and prevent the ubiquitin-mediated proteosomal degradation that occurs under normoxia to stabilize HIF-1 in a manner distinct from hypoxia; additional mechanisms must exist under hypoxia to stabilize HIF-1 independent of its hydroxylation status.
Ethyl-3, 4-dihydroxybenzoate (EDHB), a specific cell-permeable inhibitor, competitively binds to the ascorbate- and ketoglutarate-binding sites of prolyl hydroxylases inhibiting their activity.30,31 To further explore the effect of morphine on PHDs and HIF-1, we investigated the effect of EDHB on HIF-1α accumulation in normoxic LLCs. We speculate if morphine is increasing PHD enzymatic activity to reduce cytoplasmic HIF-1α levels, then morphine pretreated EDBH cells under normoxia should exhibit reduced HIF-1α levels compared with EDHB treatment alone. Protein expression using Western blot analysis showed that EDHB treatment of normoxic LLCs increased HIF-1α accumulation when compared with untreated cells, and morphine pretreatment did not alter HIF-1α levels after EDHB treatment (Figure 6D). MG132, a potent but reversible cell-permeable 26S proteosome inhibitor,32 reduces ubiquitin-mediated protein degradation and accumulates HIF-OH. Under normal conditions, hydroxylated HIF-α is quickly ubiquitinated by an E3 ligase, VHL, mediating its degradation through the 26S proteosome. To further rule out the PHD-OH-proteosomal pathway, we treated LLCs with 100 nmol/L MG132 under normoxia before and after morphine pretreatment. Our results show that 100 nmol/L MG132 increased HIF-OH detection that was unaffected by morphine treatment (Figure 6E). Taken together these results suggest that morphine-mediated inhibition of HIF-1 α accumulation under hypoxic condition is not through a mechanism that involves PHD activity or the hydroxylation status of HIF-1α.
Morphine Treatment Inhibited Hypoxia-Induced p38 MAPK Activation to Suppress HIF-1α Accumulation
During hypoxia, p38 MAPK is activated as an early response to hypoxia33,34 and inhibition of p38 activity, through genetic22 or pharmacological manipulation,35 suppresses HIF-1α accumulation. Depending on the cell type, p38 activation is necessary for HIF-1α phosphorylation as it reduces the ability to bind VHL and thus prevent proteosomal degradation and promote HIF-1 stabilization.35 Thus we next determined whether inhibition of the p38 signaling under hypoxia contributes to the hypoxic induction of HIF-1α. LLCs were incubated in normoxic or hypoxic conditions for 4 hours after morphine pretreatment, and activation of p38 was monitored by the phosphorylation status. Four hours of hypoxia treatment was sufficient to increase phosphorylated p38 (p38-P04) compared with normoxic cells (Figure 7A, lane 2 and 1). Morphine pretreatment reduced hypoxia-induced p38 activity and addition of the p38 MAPK inhibitor, SB203580 (50 μmol/L), completely abolished hypoxia-induced p38 activity. These results suggest that hypoxic induction of p38MAPK activity is an early response to hypoxia.
Figure 7.

Morphine treatment inhibited hypoxia-induced p38 MAPK activation to suppress HIF-1α accumulation. Western blot analysis of whole cell extracts (WCE) was used to assess p38 MAPK activation (A) using total and phospho-p38 (tyr180/182) MAPK specific antibodies (A, lane 4); specificity was determined using the p38 MAPK inhibitor, SB253085 under hypoxia to confirm the presence of hypoxia-induced p38 MAPK activation in LLCs. Western blot analysis using WCE (B) and nuclear extracts (C) from LLCs showed hypoxic inhibition of p38 MAPK with SB253085 reduced HIF-1α posttranslational modification (indicated by the loss of the appearance of a second higher migrating band, B) and reduced the nuclear accumulation (C). D: Semiquantitative real-time RT-PCR showed that inhibition of hypoxia-induced p38 MAPK with SB253085 prevented the hypoxia-induced VEGF mRNA transcription in LLCs.
To confirm the involvement of p38 in hypoxia-induced HIF-1 regulation, we tested the influence of the p38 MAPK inhibitor SB203580 on HIF-1α expression. As expected, analysis of total cellular proteins isolated from LLCs using Western blot showed 6 hours of hypoxia-induced HIF-1α accumulation and posttranslational modification, possibly phosphorylation. Treatment of LLCs with SB203580 alone reduced the hypoxia-induced HIF-1α accumulation and completely abolished the posttranslational modification (Figure 7B, lane 2 and 4) and was further reduced with morphine pretreatment (Figure 7B, lane 3 and 5). These results suggest that morphine is acting through pathways that involve p38 to suppress the hypoxia-induced posttranslational modifications of HIF-1α required for HIF-1 activation.
To assess whether inhibition of p38 MAPK would affect HIF-1α nuclear translocation, we performed Western blot analysis on nuclear extracts from LLCs treated with the p38 MAPK inhibitor SB203580 under hypoxia. Indeed, Figure 7C showed that inhibition of hypoxia-induced p38 activity suppressed the hypoxia-induced HIF-1α nuclear translocation, and this was further reduced with morphine pretreatment. These results suggest that the observed posttranslational modification induced by hypoxia depends on p38 MAPK activity, and morphine suppresses the p38 MAPK signaling required for HIF-1α accumulation, thus affecting nuclear translocation.
Hypoxia upregulates VEGF mRNA expression,29 so finally we investigated the effect of inhibiting hypoxia-induced p38 activity on VEGF mRNA expression in LLCs. Cells were plated in cell culture-treated dishes and incubated under normoxia and hypoxia. Total RNA was isolated and VEGF mRNA expression determined by real-time PCR as described in methods. As shown in Figure 7D, 48 hours of hypoxia treatment increased VEGF mRNA expression when compared with normoxia (P < 0.02). In contrast, both morphine and SB253085 pretreatment individually reduced the hypoxia-induced VEGF mRNA expression (P < 0.05) to normoxic levels. Morphine and SB253085 cotreatment of hypoxic LLCs resulted in a further decrease in VEGF transcription when compared with morphine-pretreated hypoxic cells alone. Taken together, our data suggest that morphine treatment concomitantly suppressed the HIF-1α posttranslational modification/phosphorylation by inhibiting the hypoxia-induced p38 MAPK activity required to stabilize HIF-1α, thereby reducing nuclear accumulation and DNA binding for gene transcription.
Discussion
In these studies, we demonstrate that morphine, a potent analgesic commonly prescribed for cancer pain management, inhibits tumor angiogenesis and tumor growth in a murine LLC tumor model. Mechanistic studies using mouse Lewis lung carcinoma cells in vitro show that morphine suppresses the hypoxia-induced p38 MAPK activation, altering the hypoxia-induced HIF-1α posttranslational modifications required for nuclear translocation and DNA binding for gene transcription.
Naltrexone is an opioid receptor antagonist, at the mu-, delta-, and kappa-binding sites. In these studies naltrexone reversed the inhibition of morphine on tumor angiogenesis. This suggests that the opioid receptors are involved in morphine’s inhibition of angiogenesis. Additional studies performed in MORKO mice indicate that morphine fails to suppress angiogenesis in vivo, further confirming the importance of the MOR in this process. These findings are in contrast to previous studies by Gupta et al,7 where it was reported that morphine potentiated angiogenesis and tumor growth in a hormone-dependent breast tumor model. Furthermore, the observed potentiation was naloxone insensitive, implicating nonopioid receptors in these effects. We speculate that the differences in the findings reported in Gupta’s manuscript and that observed in our studies might be due to differences in the morphine doses used, route of administration, and/or plasma doses achieved at steady state. In our studies, we maintained the morphine dose, which at steady state approximated 250 to 400 ng/ml, within levels observed in cancer patients receiving morphine for analgesia (ranges between 11 and 1440 ng/ml4). The dose and route of administration of morphine are critical factors that need to be taken into consideration in clinical settings, such that tolerance and withdrawal are minimized. Single bolus injections of morphine doses that are subanalgesic can sensitize the opioid receptors leading to drug tolerance and withdrawal. In light of the several recent studies, showing that both psychological and physical stress promotes tumor growth,32,33 it is quite conceivable that the increase in tumor growth seen in the animals reported in the Gupta et al study may be a consequence of withdrawal stress. In contrast, in our current studies morphine was administered as a continuous release model, and dose levels were escalated to prevent both tolerance and withdrawal associated with morphine use.
Additionally, the use of the estrogen-dependent MCF-7 breast cancer cell line could have led to the confounding effects, because estrogen has been shown to internalize and reduce MOR function.34 Thus, it is difficult to determine whether the effect of morphine in the Gupta et al studies7 resulted from a direct effect of morphine, because the morphine effects were not antagonized by naloxone, the classical opioid antagonist. Interestingly, another study reported that morphine treatment, using the same MCF-7 hormone-dependent breast cancer cells in a mouse tumor model, increased tumor cell apoptosis through a p53 pathway to inhibit tumor growth.6 In our studies, morphine doses in the clinically relevant range had no significant effects on LLC cell viability and proliferation in vitro, even though RT-PCR showed that LLCs express the MOR (Supplemental Figure S1, at http://ajp.amjpathol.org). We therefore concluded that morphine’s inhibition on tumor growth is primarily through its effects on tumor cell-induced angiogenesis, and the increased tumor cell apoptosis observed in vivo was a consequence of this reduced angiogenesis.
To understand the effects of morphine on LLC-induced angiogenesis and VEGF, we tested the effects of morphine on two known signaling pathways that modulate HIF-1 activation: i) the PHD, and ii) the hypoxia-induced p38 MAPK pathway. In these studies we used a PHD inhibitor, EDBH, and a hypoxia mimetic, DFO, to examine morphine’s inhibition on HIF-1α. Physiologically, the HIF/HRE pathway is controlled by the continuous synthesis of and oxygen-dependent degradation of the HIF-α subunits through prolyl hydroxylation and subsequent proteosomal degradation.15 When LLCs were treated with EDHB, HIF-1α accumulation was enhanced under normoxia. However, morphine pretreatment did not significantly alter EDBH-induced HIF-1α accumulation under normoxia but still prevented the hypoxia-induced posttranslational modification under hypoxia. Subsequent experiments using the 26S proteosomal inhibitor35 MG132 under normoxic conditions showed that LLCs accumulated hydroxylated-HIF-1α (HIF-OH) when compared with untreated cells. We speculated that if morphine acts to alter the hydroxylation status of HIF-1α and thus its degradation, we would expect to see a further increase in HIF-OH in the presence of morphine and MG132. Because morphine treatment did not further alter MG132-induced accumulation of HIF-OH, we conclude morphine’s suppression of HIF-1 activation is independent of PHD activity. Furthermore, DFO can mimic hypoxia by reducing iron availability for prolyl hydroxlase enzymatic activity15 and increase the stability of HIF-1α protein. In our studies, DFO treatment of LLCs under normoxia increased HIF-1α accumulation and nuclear protein binding to the HRE, but again this was unaltered with morphine pretreatment, further supporting that morphine is acting through an alternate pathway to reduce HIF-1 stability in LLCs.
Additional studies using SB253085, a p38 MAPK inhibitor, attenuated the hypoxia-induced HIF-1α nuclear localization and VEGF transcription in LLCs, similar to morphine treatment alone. We also show that morphine and SB253085 synergistically inhibited hypoxia induced HIF-1α and VEGF transcription, suggesting that morphine’s inhibition on VEGF secretion involves p38 MAPK and not PHD enzymatic activity. Several investigations indicate that the hypoxia signaling pathways that involve p38 are distinct from the DFO. Previous studies on the hypoxic induction of p38 MAPK suggest a strong role for the production of reactive oxygen species (ROS) generated through the mitochondrial complex III in the hypoxic activation of HIF-1.22,36 Our preliminary investigations indicate that morphine does not significantly alter the hypoxia-induced ROS production in LLCs. Morphine treatment alone under normoxia has no effect on ROS formation but can inhibit doxorubicin-induced mitochondrial ROS production in SH-SY5Y neuroblastoma tumor cells,37 a pathway known to mediate the hypoxia-induced p38 MAPK activation and HIF-1α phosphorylation. In vitro studies indicate a direct interaction of p38 with HIF-1α because p38-null embryonic fibroblasts lose their ability to activate HIF-1α under hypoxia and when exposed to either anoxia or DFO retained the ability to activate HIF-1α.22 We hypothesize that morphine through the MOR may be acting downstream of this hypoxia induced-ROS production to alter p38 MAPK activity and thus HIF-1 activation.
Morphine administration in our studies is similar and more representative of chronic high-dose morphine used during clinical applications for cancer pain management. Taken together, our results demonstrate that morphine is a potential inhibitor of tumor growth, through the suppression of tumor cell-induced angiogenesis and hypoxia-induced p38 MAPK activation of HIF-1 (Figure 8). In addition to its analgesic potential, morphine can be exploited for its antiangiogenic potential in cancer pain management; these findings support the use of morphine for cancer pain management.
Figure 8.

A signaling model for the effect of morphine on hypoxia-induced HIF-1α. Based on our current findings we propose that morphine alters the hypoxic activation of the p38 MAPK signaling pathway. Hypoxia increased p38 MAPK activity, resulting in decreased HIF-1α hydroxylation and increased phosphorylation leading to an increased HIF-1α nuclear accumulation. Morphine treatment reduced hypoxia induced MAPK p38 and thus HIF-1 activation.
Acknowledgments
We thank the University of Minnesota Biomedical Imaging and Processing Lab Staff for use and guidance in image capturing. We also thank Fairview University Pathology Services for tissue sectioning and staining.
Footnotes
Address reprint requests to Dr. Sabita Roy, University of Minnesota Medical School, Department of Surgery, MMC 195, 420 Delaware Street, SE, Minneapolis, MN 55455. E-mail: royxx002@umn.edu.
Supported by the National Institutes of Health (NIDA/NIH, F31-DA021005-01 to L.K.; CA114340 to S.R.; and NIDA/NIH grants RO1 DA 12104, RO1 DA 022935, KO2 DA 015349, P50 DA 011806 to S.Ro.).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Wiffen PJ, McQuay HJ. Oral morphine for cancer pain. Cochrane Database of Systematic Reviews. 2007;3:1–51. doi: 10.1002/14651858.CD003868.pub2. [DOI] [PubMed] [Google Scholar]
- Cain DM, Wacnik PW, Eikmeier L, Beitz A, Wilcox G, Simone D. Functional interactions between tumor and peripheral nerve in a model of cancer pain in the mouse. Pain Med. 2001;2:15–23. doi: 10.1046/j.1526-4637.2001.002001015.x. [DOI] [PubMed] [Google Scholar]
- Walsh D, Perin ML. Parenteral morphine prescribing patterns among inpatients with pain from advanced cancer: a prospective survey of intravenous and subcutaneous use. Am J Hosp Palliat Care Med. 2006;23:353–359. doi: 10.1177/1049909106292170. [DOI] [PubMed] [Google Scholar]
- Aherne GW, Piall EM, Twycross RG. Serum morphine concentration after oral administration of diamorphine hydrochloride and morphine sulphate. Br J Clin Pharmacol. 1979;8:577–580. doi: 10.1111/j.1365-2125.1979.tb01047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiseo PJ, Thaler HT, Lapin J, Inturrisi CE, Portenoy RK, Foley KM. Morphine-6-glucuronide concentrations and opioid-related side effects: a survey in cancer patients. Pain. 1995;61:47–54. doi: 10.1016/0304-3959(94)00148-8. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Grösch S, Schmidtko A. G protein-independent G1 cell cycle block and apoptosis with morphine in adenocarcinoma cells: involvement of p53 phosphorylation. Cancer Res. 2003;63:1846–1852. [PubMed] [Google Scholar]
- Gupta K, Kshirsagar S, Chang L, Schwartz R, Law PY, Yee D, Hebbel RP. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002;62:4491–4498. [PubMed] [Google Scholar]
- Folkman J, D'Amore PA. Blood vessel formation: what is its molecular basis? Cell. 1996;87:1153–1155. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocrine Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Brekken RA, Thorpe PE. Vascular endothelial growth factor and vascular targeting of solid tumors. Anticancer Res. 2002;21:4221–4229. [PubMed] [Google Scholar]
- Balsubramanian S, Ramakrishnan S, Charboneau R, Wang J, Roy S. Morphine sulfate inhibits hypoxia-induced vascular endothelial growth factor expression in endothelial cells and cardiac myocytes. J Mol Cell Cardiol. 2001;33:2179–2187. doi: 10.1006/jmcc.2001.1480. [DOI] [PubMed] [Google Scholar]
- Blebea J, Mazo JE, Kihara TK, Vu JH, McLaughlin PJ, Atnip RG, Zagon IS. Opioid growth factor modulates angiogenesis. J Vasc Surg. 2000;32:364–373. doi: 10.1067/mva.2000.107763b. [DOI] [PubMed] [Google Scholar]
- Pasi A, Qu BX, Steiner R, Senn HJ, Bär W, Messiha FS. Angiogenesis: modulation with opioids. Gen Pharmacol. 1991;22:1077–1079. doi: 10.1016/0306-3623(91)90580-y. [DOI] [PubMed] [Google Scholar]
- Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. Elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):1–3. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Minet E, Michel G, Mottet D. ERK activation upon hypoxia: involvement in HIF 1 activation. FEBS Lett. 2000;468:53–55. doi: 10.1016/s0014-5793(00)01181-9. [DOI] [PubMed] [Google Scholar]
- Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1α (HIF-1α) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274:32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, Gutkind JS. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer Res. 2000;60:4873–4880. [PubMed] [Google Scholar]
- Minet E, Michel G, Mottet D. Transduction pathways involved in Hypoxia-Inducible Factor-1 phosphorylation and activation. Free Radic Biol Med. 2001;31:847–855. doi: 10.1016/s0891-5849(01)00657-8. [DOI] [PubMed] [Google Scholar]
- Bilton R, Booker GW. The subtle side to hypoxia inducible factor (HIFα) regulation. Eur J of Biochem. 2003;270:791–798. doi: 10.1046/j.1432-1033.2003.03446.x. [DOI] [PubMed] [Google Scholar]
- Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton PA, Lingen MW, Fekete MJ, Garcia JG, Moss J. Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: role of receptor transactivation. Microvasc Res. 2006;72:3–11. doi: 10.1016/j.mvr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Dussault AA, Pouliot M. Rapid and simple comparison of mRNA levels using real-time PCR. Biol Proceed Online. 2006;8:1–10. doi: 10.1251/bpo114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild R, Ramakrishnan S, Sedgewick J, Griffioen AW. Quantitative assessment of angiogenesis and tumor vessel architecture by computer-assisted digital image analysis: effects of VEGF-toxin conjugate on tumor microvessel density. Microvasc Res. 2000;59:368–376. doi: 10.1006/mvre.1999.2233. [DOI] [PubMed] [Google Scholar]
- Pasternak GW. The pharmacology of mu analgesics: from patients to genes. Neuroscientist. 2001;7:220–231. doi: 10.1177/107385840100700307. [DOI] [PubMed] [Google Scholar]
- Pick CG, Nejat RJ, Pasternak GW. Independent expression of two pharmacologically distinct supraspinal mu analgesic systems in genetically different mouse strains. J Pharmacol Exp Ther. 1993;265:166–171. [PubMed] [Google Scholar]
- Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu Z, Hackett NR, Crystal RG, Moore MA, Hajjar KA, Manova K, Benezra R, Rafii S. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- Barr MP, Bouchier-Hayes DJ, Harmey JJ. Vascular endothelial growth factor is an autocrine survival factor for breast tumour cells under hypoxia. Int J Oncol. 2008;32:41–48. [PubMed] [Google Scholar]
- Majamaa K, Günzler V, Hanauske-Abel HM, Myllylä R, Kivirikko KI. Partial identity of the 2-oxoglutarate and ascorbate binding sites of prolyl 4-hydroxylase. J Biol Chem. 1986;261:7819–7823. [PubMed] [Google Scholar]
- Sasaki T, Majamaa K, Uitto J. Reduction of collagen production in keloid fibroblast cultures by ethyl-3, 4-dihydroxybenzoate. Inhibition of prolyl hydroxylase activity as a mechanism of action. J Biol Chem. 1987;262:9397–9403. [PubMed] [Google Scholar]
- Lee JW, Shahzad MM, Lin YG, Armaiz-Pena G, Mangala LS, Han HD, Kim HS, Nam EJ, Jennings NB, Halder J, Nick AM, Stone RL, Lu C, Lutgendorf SK, Cole SW, Lokshin AE, Sood AK. Surgical stress promotes tumor growth in ovarian carcinoma. Clin Cancer Res. 2009;15:2695–2702. doi: 10.1158/1078-0432.CCR-08-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermes GL, Delgado B, Tretiakova M, Cavigelli SA, Krausz T, Conzen SD, McClintock MK. Social isolation dysregulates endocrine and behavioral stress while increasing malignant burden of spontaneous mammary tumors. Proc Natl Acad Sci U S A. 2009;106:22393–22398. doi: 10.1073/pnas.0910753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micevych PE, Rissman EF, Gustafsson JA. Estrogen receptor-alpha is required for estrogen-induced mu-opioid receptor internalization. J Neurosci Res. 2003;71:802–810. doi: 10.1002/jnr.10526. [DOI] [PubMed] [Google Scholar]
- Kisselev A, Goldberg A. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1324–L1329. doi: 10.1152/ajplung.00326.2001. [DOI] [PubMed] [Google Scholar]
- Lin X, Li Q, Wang YJ, Ju YW, Chi ZQ, Wang MW, Liu JG. Morphine inhibits doxorubicin-induced reactive oxygen species generation and nuclear factor-kappaB transcriptional activation in neuroblastoma SH-SY5Y cells. Biochem J. 2007;406:215–221. doi: 10.1042/BJ20070186. [DOI] [PMC free article] [PubMed] [Google Scholar]