

Abstract
The carbohydrate moieties on conjugating with 3-(1′-hexyloxyethyl)-3-devinyl pyropeophorbide-a (HPPH) altered the uptake and intracellular localization from mitochondria to lysosomes. In vitro, HPPH-Gal 9 PDT showed increased PDT efficacy over HPPH–PDT as detectable by the oxidative cross-linking of nonphosphorylated STAT3 and cell killing in ABCG2-expressing RIF cells but not in ABCG2-negative Colon26 cells. This increased efficacy in RIF cells could at least partially be attributed to increased cellular accumulation of 9, suggesting a role of the ABCG2 transporter for which HPPH is a substrate. While such differences in the accumulation in HPPH derivatives by tumor tissue in vivo were not detectable, 9 still showed an elevated light dose-dependent activity compared to HPPH in mice bearing RIF as well as Colon26 tumors. Further optimization of the carbohydrate conjugates at variable treatment parameters in vivo is currently underway.
Introduction
Photodynamic therapy (PDTa) is a noninvasive antitumor treatment that requires the combination of photosensitizer (PS), tissue oxygen, and light to produce cytotoxic reactive oxygen species, predominantly singlet oxygen. PDT has potential advantages over surgery and radiotherapy due to its tissue-sparing properties.1 Various PS have different pharmacokinetics, but effective treatment relies in part on greater accumulation of the sensitizer in the tumor extracellular matrix or tumor cells, via undefined binding, relative to normal tissue. Porfimer sodium is the only PS that has received worldwide regulatory approval for the treatment of cancer in humans.2 Porfimer sodium has important limitations in that it (i) consists of a complex mixture of monomers, dimers, and oligomers, (ii) has a short-wavelength absorption (λmax = 630 nm, ε = 5000) that can restrict applications that require deep penetration of excitation light, and (iii) can cause prolonged (4–6 weeks) skin photosensitivity due to its pharmacokinetic and tissue-retention properties. These shortcomings have spurred the development of a plethora of second-and third-generation PS. Of these, chlorin- and bacteriochlor-in-based PS are monomeric compounds, efficient generators of singlet oxygen (Φ = 45–60%), and may be particularly effective in treating large and/or deeply seated tumors due to their long wavelength (650–800 nm) absorption.3 While these characteristics are promising for the improvement of clinical applications, enhanced selectivity toward tumors is still a desirable goal.
Our laboratory has extensively applied synthetic approaches for developing PS with improved photosensitizing efficacy. One avenue has been a classical QSAR approach in which the overall lipophilicity of the molecule is altered by varying the length of the aliphatic chain or by replacing the substituents at the peripheral positions of the tetrapyrrole system.4 A more challenging approach is to design PS conjugates that specifically target cell-surface proteins or receptors overexpressed by tumors.5 In recent years,6 a variety of porphyrins and nonporphyrin compounds have been conjugated with carbohydrates to serve as substrates for cellular lectins (e.g., galectins),7 known for their higher expression in many tumor cells. Such conjugates have shown some potential in tumor imaging and PDT.8 Encouraged by these reports and results from our group,9,10 we conjugated PS with carbohydrates known for their high affinity to galectin-3. We selected the rigidified multivalent lactose derivatives reported by Vrasidas et al.11 that display significantly different binding to galectin-1 and galectin-3. For example, derivatives with rigidified multivalent lactose bound with 4300-fold higher affinity to galectin-3 than to galectin-1. Therefore, to determine the effect of multivalent lactose in adding tumor-specificity, we have linked such carbohydrate moieties to our PS12 with the expectation of enhancing specificity for galectin-3-expressing tumors.
A second aspect that could potentially influence and even limit the antitumor efficacy of porphyrin PS is the presence or absence in the tumor tissue of ATP-dependent transporters, such as ABCG2, also known as BCRP (breast cancer resistance protein).12 Such transporters have been recognized to export a diverse array of compounds, including certain PS.13–15 However, by conjugation with carbohydrates, porphyrins appear to lose their substrate property for ABCG2 and, hence, may be retained more effectively by cells thereby contributing to improved PDT.16
The present manuscript describes a detailed investigation showing the synthesis and in vitro and in vivo photodynamic effects of a series of carbohydrate conjugates of 2-(1′-hexyloxyethyl) pyropheophorbide-a (HPPH), by itself an effective photosensitizer currently under phase I/II clinical trials at Roswell Park Cancer Institute, Buffalo NY.17–20 We discovered that carbohydrate modifications altered the cell biology of PS uptake and PDT action, and that these changes appeared to be independent of galectin-3 expression by the target cells.
Results and Discussion
Synthesis of the HPPH–Carbohydrate Conjugates
To investigate the impact of β-galactose carbohydrates on tumor-selectivity and photosensitizing efficacy, galactose and lactose (Galβ1→4Glc) moieties were individually conjugated with 3-devinyl-3-(1′-hexyloxy)ethyl-pyropheophorbide-a (HPPH). The related non-β-galactose carbohydrates, glucose and cellobiose (Glcβ1→4Glc), were similarly conjugated to HPPH in order to obtain reagents to assess the relative contribution of galectin binding to uptake and action. The pairs of HPPH conjugates with either mono- or disaccharides possess similar overall lipophilicity (Figure 1). To determine the impact of linker(s) with multivalent carbohydrate units in the conjugates, certain rigidified multivalent lactose molecules were also synthesized. For the preparation of HPPH–carbohydrate conjugates without a rigid linker, the acetoxy- group at position-1 (anomeric carbon atom) of the commercially available acetyl protected carbohydrate was converted into the corresponding 1-bromo analogue, which was then converted into the azido derivative by following known methodology.21 Hydrogenation of the azido analogues in the presence of Pd/C at room temperature produced the required 1-aminotetraacetogalactose (1), 1-aminotetraacetoglucose (2), 1-aminoheptaacetolactose (3), and 1-aminoheptaacetocellobiose (4) in good yields. A typical example is illustrated in Scheme 1.
Figure 1.
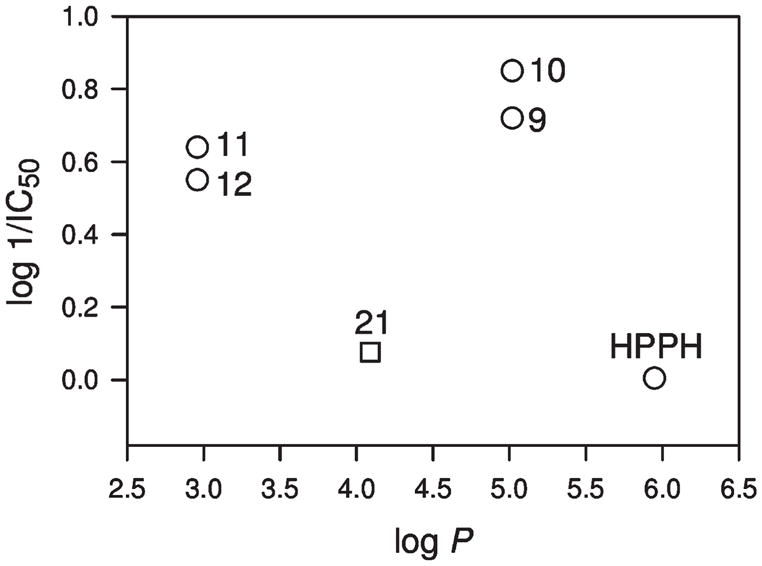
IC50 (μM)of PS in RIF cells (derived from Figure 3A)as a function of log P (determined using computer software PALLAS): 9 (HPPH-Gal), 10 (HPPH-Glu), 11 (HPPH-Lac), 12 (HPPH-Cell), 21 (HPPH-Monolac). The log P values for compounds 22 (HPPH-Dilac) and 23 (HPPH-Tetralac) could not be directly calculated due to software limitations (see text).
Scheme 1.

Synthesis of Carbohybrate Scheme 4
Reagents: (a) HBr/AcOH; (b) NaN3, Bu4NHSO4; (c) H2, Pd/C.
Reaction of carbohydrates 1–4 with HPPH using 1-ethyl-3-(3′-dimethylaminopropyl) carbodiimide (EDCI) as a coupling reagent produced the corresponding intermediates 5–8 which under standard deprotection conditions (NaOMe in dichloromethane/methanol) afforded the final conjugates 9–12 (Scheme 2).
Scheme 2.

Synthesis of HPPH–Carbohydrate Conjugatesa
aReagents: (a) EDCI, DMAP; (b) NaOMe/MeOH.
For the preparation of the mono- and multivalent rigidified lactose conjugates, the acetyl protected lactose with a rigidified linker was synthesized first by following known methodology.11 In brief, intermediate 13, which upon stirring with TFA yielded 14, which was then coupled with the rigidified lactose moieties (15–17) using (benzotriazol-1-yloxy)tris(dimethylamino) phosphonium hexafluorophosphate (BOP) as a coupling reagent to produce the intermediates 18–20, which on subsequent deacetylation afforded the conjugates 21–23, respectively (see Scheme 3). The electronic absorption spectra of all the conjugates were similar to that of HPPH.
Scheme 3.

HPPH Linked with Various Carbohydrates with Rigid Linkersa
a Reagents and conditions: (a) N-Boc-ethylenediamine, BOP, Et3N; (b) TFA; (c) BOP, Et3N; (d) NaOMe/MeOH.
Rationale for the Synthesis of Carbohydrate Conjugates with Similar Lipophilicity
The HPPH–carbohydrate conjugates were designed to evaluate the importance of the carbohydrate moieties to enhance target-specificity. For example, in conjugates 9 and 10, HPPH was linked to galactose and glucose moieties, respectively. Both compounds exhibit similar lipophilicity (Figure 1) but differ in the orientation of the hydroxy-group at position-4 of the hexose that determines the binding to lectins. If lipophilicity is the predominant criterion that defines cellular uptake and action, then both conjugates might be expected to produce similar PDT efficacy. Similarly, the conjugates 11 and 12, containing the lactose (galactose–glucose) and cellobiose (glucose–glucose), respectively, were prepared. These disaccharide conjugates were used to determine the effects of the orientation of the 4-OH group when presented in the context of a glycosidically linked galactose in a disaccharide and also as a part of a compound with reduced overall lipophilicity when compared to the mono- (9 and 10) and disaccharide–HPPH conjugates (11 and 12).
In general, lipophilicity has proven to be an important factor in directing the pharmacokinetic and pharmacodynamic properties of many PS because it influences the biodistribution and clearance and thus the bioactivity of drugs.4 Lipophilicity is indicated by the logarithm of the partition coefficient, log P, which reflects the equilibrium partitioning of a molecule between a nonpolar and a polar phase, such as n-octanol and water.22 Recently, several theoretical methods have been developed to predict lipophilicity. We applied a program module of PALLAS to determine the lipophilicity of the compounds synthesized, and the results are shown in Figure 1. The log P values of conjugates 22 and 23 could not be calculated because their molecular weight exceeded the program’s limitation but must be below that of conjugates 21 because (i) of the additive–constitutive nature of substituted functional groups and (ii) the very strongly negative estimated (Pallas) log P of moieties 15 (log P ~ −3) and 16 (log P ~ −5).
In Vitro Photosensitizing Activity
HPPH and the corresponding carbohydrate conjugates were evaluated for in vitro photosensitizing efficacy in the murine radiation-induced fibrosarcoma (RIF) and colon carcinoma (Colon26) cell lines. These cell lines express closely similar levels of the β-galactose-recognizing proteins galectin-1 and galectin-3 (Figure 2). They differ, however, in immune-detectable levels of the ATP-dependent transporter ABCG2 that is able to mediate cellular export of HPPH,16 with RIF cells expressing high levels while Colon26 cells are essentially devoid of ABCG2 expression.
Figure 2.
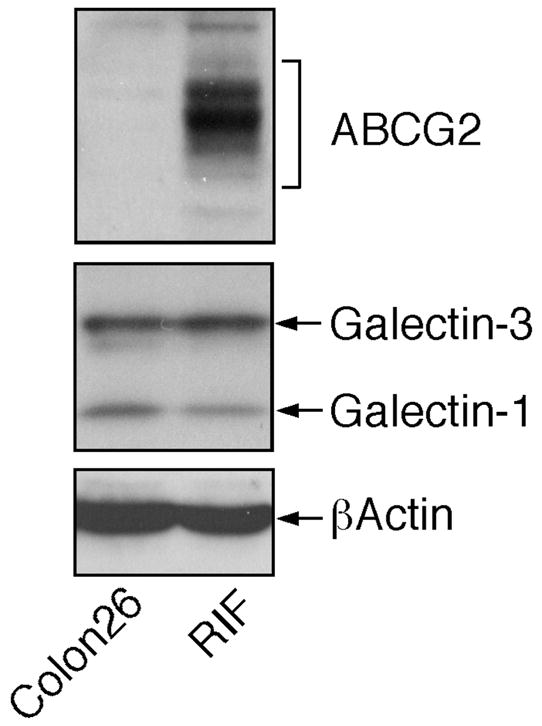
Expression of galectin-1, galectin-3, and ABCG2 in RIF and Colon26 cells in vitro. Equal amounts of cell extracts were analyzed by Western blotting for the indicated proteins.
Cells were incubated with increasing concentrations of the PS for 24 h, followed by exposure to 665 nm light and MTT assays performed 48 h later. Neither RIF nor Colon26 cells showed any significant dark cytotoxicity up to 1 μM PS (Figure 3A,B, upper panels). RIF cells showed drug- and concentration-dependent phototoxicity (Figure 3A, lower panel). All mono- and disaccharide conjugates (9, 10, 11, 12) produced higher PDT efficacy than HPPH. Among the conjugates, those with galactose (9) and glucose (10) were approximately twice as effective as those with lactose (11) and cellobiose (12). Compared to the lactose analogue (11), which was linked to HPPH with an amide bond, the lactose derivative with a rigid linker (21) was several-fold less effective and was comparable to HPPH. The conjugates with the same rigid linker but with an increasing number of saccharide moieties, e.g., the tetramer (22) and the octamer (23), were ineffective. No appreciable differences were observed between the conjugates, which could be attributed to the differing orientation of the 4-OH group in the hexose (galactose and glucose), thus ruling out a measurable contribution of galectin-3 to the PS efficacy of the galactose conjugates.
Figure 3.

In vitro photoreaction with HPPH, 9 (HPPH-Gal), 10 (HPPH-Glu), 11 (HPPH-Lac), 12 (HPPH-Cel), 21 (HPPH-Monolac), 22 (HPPH-Dilac), and 23 (HPPH-Tetralac) in RIF (A) and Colon26 cells (B). Cells were incubated with the indicated PS in 10% serum-containing medium for 24 h and then exposed to light (665 nm at 3.2 mW/cm2) for a total fluence of 0.5 J/cm2 (lower panels). Cells exposed to the PS without light are shown in the upper panels. After treatment, the cells were incubated in growth medium for 48 h. Surviving cell population was measured by MTT assay. The data are expressed as mean of three experiments. Standard deviations are <6.1% and for most values are not indicated for clarity.
In contrast, Colon26 cells, analyzed for their response to HPPH, the corresponding galactose analogue 9 and glucose derivative 10 only showed equal photosensitivity to all three agents when exposed to them for 24 h. When exposure conditions were reduced to 4 h, HPPH, 9, and 10 showed similar responses to the 24 h incubation in RIF cells.
In our earlier QSAR study in a congeneric series of HPPH analogues without carbohydrate modifications, we showed the importance of lipophilicity for in vitro and in vivo PS efficacy, with HPPH possessing optimal values with a log P of ~6.4 Although HPPH and its carbohydrate conjugates are not strictly a congeneric series, a number of observations can be made with regard to lipophilicity (Figure 1). HPPH, as well as compounds 9 and 10, which differ by less than one log value in lipophilicity, displayed equivalent activity in Colon26 cells after 24 h of drug uptake. In RIF cells, compounds 9 and 10 showed similar activity to that seen in Colon26 cells; however, relatively lower activity was detected for HPPH in RIF cells, suggesting that either cellular uptake and accumulation, or the photoreaction in these cells, are altered. Altered cellular accumulation, in part, could be due to the action of the ABCG2 pump (Figure 2) for which HPPH is a substrate.16
The lower activity of the compounds 11 and 12, and in particular of the compounds 21, 22, and 23, is not clearly attributable to lipophilicity alone and may be influenced by the presence of a larger carbohydrate moiety on the molecules and the rigidity of the linkage, either of which could potentially affect the mode by which the compounds are taken up and intracellularly distributed
In Vitro Uptake of PS
To assess whether carbohydrate-dependent changes in phototoxicity were due to altered PS levels in cells at the time of light treatment, PS uptake was determined. Initial measurements by fluorescence spectroscopy of the PS uptake in RIF cells indicated increased accumulation over 3 or 24 h of the mono- and disaccharide analogues (9–12) over HPPH. However, the conjugates with an increasing number of lactose moieties with an extended linker showed uptake similar to HPPH for 21 and minimal uptake for 22 and 23. The failure to be bound and/or be internalized explains the inactivity for compounds 22 and 23 in the cytotoxity assay (Figure 3A). The same distributions, albeit with slightly lower levels, were observed after 3 h PS exposure. To compare the PS uptake in RIF and Colon26 cells and to avoid potential pitfalls of fluorescence measurements such as PS aggregation etc., 14C-labeled HPPH and HPPH-Gal (9), as the representative carbohydrate conjugate, were prepared by following the methodology shown in Scheme 2 in which 14C was introduced into HPPH by reacting pyropheophorbide with 14C-labeled hexanol23 (for synthetic details see Experimental Section). It is apparent from Figures 4B,C that accumulation of 9 in RIF cells exceeded that of HPPH at both exposure periods, while there was no significant difference in accumulation between the two PS in Colon26 cells. These observations suggest a role for the ABCG2 transporter in determining intracellular level of HPPH in RIF cells.
Figure 4.

In vitro uptake of HPPH and HPPH carbohydrate conjugates. (A) Replicate cultures were incubated with 0.25 μM PS in medium containing 10% fetal calf serum for varying time periods at 37 °C; PS = HPPH, 9 (HPPH-Gal), 10 (HPPH-Glu), 11 (HPPH-Lac), 12 (HPPH-Cel), 21 (HPPH-Monolac), 22 (HPPH-Dilac), and 23 (HPPH-Tetralac). PS content was determined by fluorescence spectroscopy and expressed as fluorescent units per mg protein; each determination was done in triplicate. (B,C) Uptake of [14C]-HPPH and [14C]-HPPH-Gal by RIF and Colon26 cells; Replicate cultures were incubated in medium containing 10% fetal calf serum and 0.8 μM 14C-labeled compounds at 37 °C. At the times indicated, triplicate cultures were washed, counted, and the cell-associated radioactivity determined. The results are expressed as mean and standard deviation.
We have shown earlier that HPPH is a substrate for the ABCG2 transporter.16 Retention of HPPH by RIF cells is improved by treatment of the cells with the tyrosine kinase inhibitor imatinib mesylate that inhibits, among others, the ATP-dependent transport function of ABCG2 and thus HPPH efflux. We have further reported that 9 does not appear to be a substrate for ABCG2. Therefore, 9 accumulation is not measurably affected by imatinib mesylate in RIF cells.16 Neither HPPH nor 9 accumulation are affected by imatinib mesylate in ABCG2 nonexpressing Colon26 cells (data not shown).
Subcellular Localization
Previous studies with various types of PS, including the alkyl ether analogues of pyropheophorbide-a, showed that the most effective forms localized to mitochondria.24,25 Fluorescence microscopy confirmed the predominant mitochondrial location of HPPH (Figure 5). In contrast, the subcellular localization of the representative galactose-conjugated HPPH in RIF cells differed from HPPH by being predominantly lysosomal (Figure 5). Similarly, all other mono- and disaccharide conjugates were located in lysosomes. The lactose conjugate (11) deviated somewhat from the above pattern in that also a fraction overlapped with the Golgi marker. The low level accumulation of the tetra- and octasaccharide conjugates appeared to have broad localization with fluorescence detectable in the three subcellular compartments tested: mitochondria, lysosomes, and golgi (summarized in Table 3, Supporting Information). A similar localization was found for each PS when monitored after 3 and 24 h of incubation. In the case of carbohydrate conjugates, an initial binding to the cell surface followed by internalization and progressive lysosomal accumulation was evident. Lysosomal localization became most pronounced after 24 h of incubation, probably in part due to sequestration into the organelle. The change from mitochondria to lysosomes suggests that with the carbohydrate conjugation, HPPH has been subjected to a different mode of uptake and intracellular distribution. The predominance of the compounds in lysosomes suggests the involvement of endocytosis, i.e., through phagocytosis/pinocytosis. However, neither the β-galactose conjugates (9, 11) nor the related non-β-galactose carbohydrates (10, 12) showed specificity for galectin-3 that could be inhibited by inclusion of even >100-fold excess of corresponding free carbohydrates during in vitro or in vivo evaluation of the conjugates (data not shown).
Figure 5.

Subcellular localization of HPPH and 9 (HPPH-Gal) in RIF cells. The cells were incubated for 3 h in medium containing 10% FCS and 2 μM PS (red) and the indicated trackers (green) for mitochondria, lysosomes, or golgi. Images were merged to indicate the overlap in fluorescence. (See Table 1 and Figure 3 of Supporting Information) for a summary of localization of other HPPH–carbohydrate conjugates.)
From this we conclude that the photosensitizing action of HPPH and its carbohydrate conjugates is appreciably determined by the subcellular location. This suggests that the altered subcellular localization, i.e., to the lysosome, triggers mechanisms that differ from those of the predominantly mitochondrially localizing HPPH and that the effectiveness of these mechanisms shows considerable cell type specificity.
STAT-3 Dimerization in RIF and Colon26 Cells
An immediate consequence of the PDT reaction is the generation of singlet oxygen, which, through propagation of redox reactions, results in the oxidation of intracellular proteins.26 One of the specific targets of the photoreaction is the covalent cross-linking of the latent signal-transducing protein STAT-3 in the cytoplasm.27
The degree of STAT-3 cross-linking is a direct measure of the PS-dependent reaction and in part correlates with the level of stress reactions leading to cell killing. Of note is that STAT-3 cross-linking itself is not causative in cell death. The STAT-3 cross-linking reactions in RIF and Colon26 cells (Figure 6) reported a PS type and dose dependence that mirrored the relative activities of the PS to cause cell killing (Figure 3), namely a several-fold difference between HPPH and both galactose and glucose-conjugated HPPH, a difference that was characteristic for RIF cells but not for Colon26 cells. Furthermore, the results correspond well with the PS uptake data shown in Figure 4, with RIF cells accumulating significantly greater amounts of 9 and 10 than HPPH, while Colon26 cells contained nearly equal levels of HPPH and 9. Also, RIF cells appear less sensitive to HPPH than Colon26 cells despite nearly equal HPPH content (Figure 4). These results indicate that not only the relative amount of total PS taken up by cells but also the cell type and local production of singlet oxygen determines the level of oxidative reactions within the PS-harboring organelles as well as the cytoplasmic compartment in which subsequent propagation of redox reactions occur.
Figure 6.
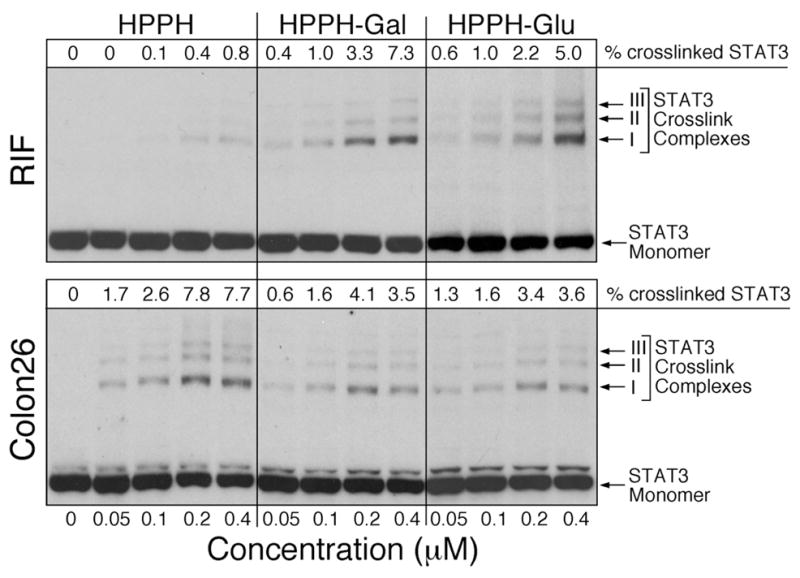
Covalent cross-linking of STAT-3 as a function of PS action. RIF and Colon26 cells were incubated with the indicated concentrations of HPPH or HPPH conjugates in medium containing 10% FCS. After 4 h of drug incubation, the cells were exposed to 665 nm light at 3 J/cm2 and immediately extracted. STAT-3 cross-linking was identified by Western blotting. The relative level of cross-linking was determined by densitometry and the values indicated at the top of the immunoblot images.
In Vivo Biodistribution of HPPH and HPPH–Galactose
The PS efficacy of HPPH conjugates was evaluated in vivo using tumor bearing mice. The biodistribution of the PS was determined with the same preparations of [14C]-HPPH and [14C]-HPPH-galactose derivative 9 as applied in vitro (Figure 4). The relative uptake of the PS by the different organs in C3H mice bearing RIF tumor and BALB/c mice bearing Colon26 tumor were comparable (Figure 7). The uptake of PS by the tumors was in the range of major organs with a tendency for tumor values to be ~2-fold higher than skin. HPPH values in tumors tended slightly higher than 9 with appreciable retention of both PS lasting at least 48 h. The notable exception in the biodistribution was the several-fold higher level for 9 in the liver. The prominent liver localization of 9 was expected and might be attributed to clearance through the galactose receptor system on hepatic parenchymal cells.
Figure 7.

The in vivo biodistribution of 14C-labeled HPPH and 9 (HPPH-Gal) in C3H and Balb/c mice: the 14C-labeled photosensitizers (0.2 mL/5 μCi) were administered to 6 mice/group (Balb/c mice bearing Colon26 tumors and C3H mice bearing RIF tumors). At 24 and 48 h after injection, three mice/time point were sacrificed. The organs of interest (skin, muscle, tumor, heart, lung, liver, kidney, and spleen) were removed and the radioactivity was measured (see the text). The raw data were then converted to counts/g tissue weight and plotted using SigmaPlot 10.0 software.
In Vivo Photosensitizing Activity of HPPH and HPPH Conjugates
The in vivo photosensitizing efficacy of HP-PH and selected HPPH–carbohydrate conjugates was determined in RIF and Colon26 tumors. To maximize the detection of potential PDT improvement with carbohydrate conjugates, a low fluence regimen of 48 J/cm2 was first chosen that produces no tumor cures with an HPPH dose of 0.47 μmol/kg (Figure 8A,B). In contrast to HPPH, 9, under the same conditions, achieved long-term tumor control of ~40% of RIF and 30% of Colon26 tumors. To validate the in vitro results for inactive carbohydrate conjugates, the dilactose conjugate (22) was tested in RIF tumors and also was found to be inactive in vivo. With increased fluence (135 J/cm2), the efficacy of the active PS tested in RIF tumors improved such that HPPH yielded approximately 50% cures and cure rates with the galactose derivative 9 were raised to ~90% (data not shown).
Figure 8.
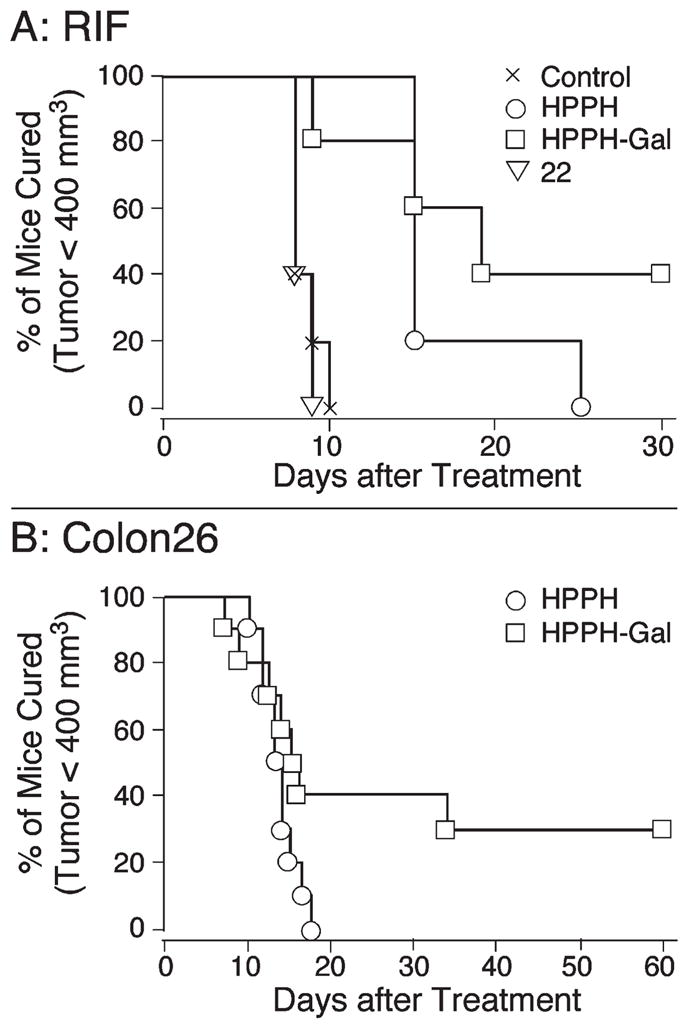
In vivo photosensitizing efficacy of HPPH and 9 (HPPH-Gal) (drug dose: 0.47 μmol/kg; fluence: 48 J/cm2) in C3H (RIF tumors) and Balb/c (Colon26 tumors) mice (10 mice/group, light delivered 24 h post PS injection); 22 (HPPH-Dilac).
The similarity of cure data in Figure 8 and tumor drug distribution data in Figure 7 between the ABCG2 positive RIF and ABCG2 negative Colon26 tumor lines suggests that in vivo drug delivery to the tumor cells and tumor response may be governed primarily by conditions in the tumor microenvironment such as coresident stromal cells and the prevalent vascular leakiness rather than tumor cell-specific uptake mechanisms. Nevertheless, the functional properties identified for HPPH and HPPH conjugates in RIF and Colon26 cells also are likely to apply to stromal and endothelial cell types that contribute to the tumor tissue.
Skin Phototoxicity of HPPH and the Corresponding Galactose Derivative (9)
One of the problems with most porphyrin-based compounds is long-lasting skin phototoxicity. Therefore, the skin phototoxicity of HPPH–Gal was compared with HPPH in C3H mice bearing RIF tumors at the therapeutic doses of PS. At various times after PS administration, mice were restrained without anesthesia in Plexiglas holders designed to allow one hind leg be exposed to therapeutic light. The contralateral hind leg served as a control.28 The results are summarized in Figure 9. While normal skin phototoxicity subsided within 4 days with both PS, as indicated by a lack of response when feet were exposed to light at 96 h after drug injection, the initial erythema responses were stronger with HPPH than 9. In part, the skin reaction was in agreement with the detectable presence of HPPH and conjugates in skin tissue (Figure 7). In no case did the response exceed pronounced erythema and/or progress to exudation and/or necrosis.
Figure 9.
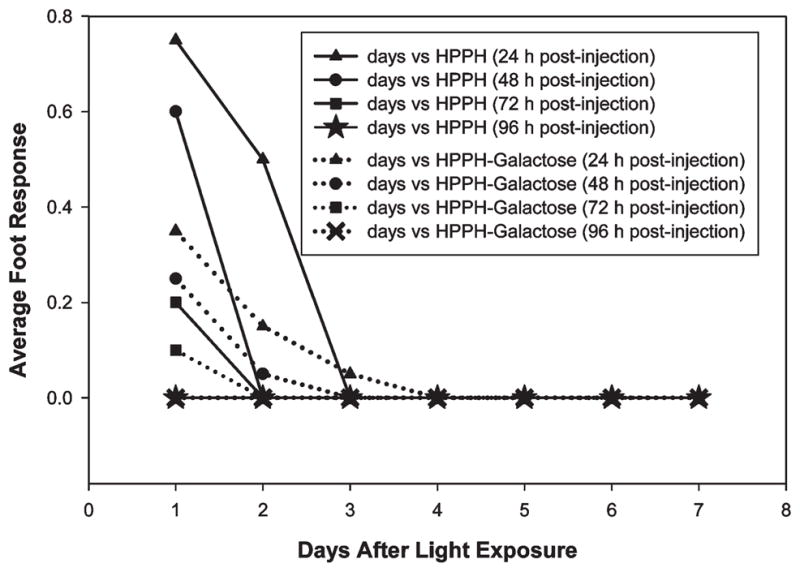
Foot response (skin phototoxicity) of HPPH and 9 (HPPH-Gal) was measured in C3H mice bearing RIF tumors at the therapeutic dose. Foot response was judged using a 0–3 scale.28 0–0.1 = no apparent difference from normal, 0.3 = slight edema, 0.5 = (moderate edema), 0.75 = large edema, 1.0-large erythema with exudate, 1.2 = moderate edema with slight scaly or crusty appearance, 1.5 = definite erythema and definite scaly or crusty appearance.
Conclusions
In an effort to enhance the efficacy and selectivity of photosensitizers currently under development by our group, we conjugated the clinically effective antitumor photosensitizer 3-devinyl-3-(1′-hexyloxyethyl) pyropheophorbide-a (HPPH) with mono-, di-, or tetrasaccharides. We hoped that these conjugates would target carbohydrate-binding molecules known to be differentially expressed on the surface of many tumor cells such as the carbohydrate-binding lectins galectin-1 and -3. Carbohydrate conjugates and unconjugated HPPH had similar singlet oxygen yields. Several of the resulting carbohydrate conjugates exhibited superior antitumor activity. This could not be explained by enhanced photosensitizer binding to cellular galectins. Drug lipophilicity and lack of conjugate transport by the ABCG2 transporter may be contributing factors. PDT efficacy of carbohydrate-conjugated HPPH may be determined by subcellular localization and the signaling mechanisms initiated by the photoreaction at the site of photosensitizer accumulation. Further definition of these signaling mechanisms as well as optimization of the carbohydrate conjugates at variable treatment parameters in vivo are currently underway.
Experimental Section
All the chemicals and reagents were purchased from chemical vendors and used as received. The solvents were dried before using in various reactions by following the standard procedures. UV–visible spectra were recorded in dichloromethane. NMR spectra were recorded in deuterated chloroform (CDCl3) or pyridine-d5 containing tetramethylsilane (TMS) as an internal standard at 303 K. Chemical shifts are reported as parts per million (ppm) with respect to TMS (δ 0.00) or most deshielded peak of pyridine- d5 (δ 8.74). High-resolution mass spectroscopy (HRMS) analyses were performed at the Michigan State University (MSU)–NIH Mass Spectrometry Facility, East Lansing, MI. The purity of the compounds was ascertained by HPLC analyses and was >95%. The reactions were monitored using small strips of precoated silica thin layer chromatography (TLC) (250 μm thickness) plates. Preparative TLC was performed on 20 cm × 20 cm silica gel TLC plates (1 mm thickness).
Synthesis of 1-α-Bromo-cellobiose Pentaacetate (4b)
Cellobiose octaacetate (1 g, 1.47 mmol) was treated with HBr/AcOH (5 mL) under ice-cold conditions for 4 h. The reaction mixture was poured into aqueous saturated sodium bicarbonate solution, and the precipitate so obtained was filtered and washed with cold water (50 mL) to yield 997 mg (97%) of the title product 4b and was used as such in the next step without any further purification; mp 185–187 °C, reported 184 °C.29
Synthesis of 1-β-Azido-cellobiose Heptaacetate (4c)
To a suspension of the 1-α-bromo cellobiose heptaacetate 4b (997 mg), sodium azide (465 mg, 7.15 mmol) and tetrabutyl ammonium hydrogen sulfate (486 mg, 1.43 mmol) in ethyl acetate (10 mL) and aqueous saturated sodium bicarbonate solution (10 mL) were added. The reaction mixture was stirred vigorously at room temperature overnight. It was then diluted with ethyl acetate (40 mL) and washed with water (3 × 50 mL). The organic layer was dried over anhydrous sodium sulfate and filtered. The solvents were evaporated, and the residue was purified over a silica column using 50% ethyl acetate/hexane as eluant to yield 789 mg (84%) of pure product 4c. 1H NMR (600 MHz, CDCl3, δ ppm): 5.15 (m, 1H, 3-H), 5.11 (m, 1H, 3′-H), 5.02 (m, 1H, 4′-H), 4.88 (m, 1H, 2′-H), 4.82 (m, 1H, 2-H), 4.59 (d, J = 8.56Hz, 1H, 1′-H), 4.49 (m, 2H, 1-H and 6-Ha), 4.32 (m, 1H, 6′-Ha), 4.09 (m, 1H, 6-Hb), 4.01 (m, 1H, 6′-Hb), 3.76 (t, J = 9.30 Hz, 1H, 4-H), 3.65 (m, 2H, 4′-H and 5-H), 2.10, 2.05, 2.02, 1.99, 1.98, 1.97, and 1.94 (each s, 3H, 6 × CH3CO2); mp 180–182 °C, reported 182–182.5 °C.30
Synthesis of 1-β-Amino-cellobiose Heptaacetate (4)
The 1-β-azido-cellobiose heptaacetate (789 mg) in methanol (10 mL) was hydrogenated in the presence of palladium/carbon (10% w/w) at room temperature for 2 h. The reaction mixture was filtered over celite to remove palladium/carbon particles, which upon evaporating the solvent afforded 4 in quantitative yield. H NMR (600 MHz, CDCl3, δ ppm): 5.21 (m, 1H, 3-H), 5.14 (m, 1H, 3′-H), 5.06 (m, 1H, 4′-H), 4.92 (m, 1H, 2′-H), 4.73 (m, 1H, 2-H), 4.50 (m, 2H, 1′-H and 6-Ha), 4.36 (m, 1H, 6′-Ha), 4.32 (m, 1H, 1-H), 4.06 (m, 2H, 6-Hb and 6′-Hb), 3.71 (m, 1H, 4-H), 3.65 (m, 1H, 5-H), 3.58 (m, 1H, 4′-H), 2.16, 2.08, 2.05, 2.02, 2.01, 2.00, and 1.98 (each s, 3H, 7 × CH3CO2). Mass calcd for C26H38NO17: 636.2139 (MH+); HRMS found: 636.2142.
By following a similar synthetic approach other carbohydrate analogues 1-β-amino-galactose tetraacetate, 1-β-amino-glucose tetraacetate and 1-β-amino-lactose pentaacetate were synthesized in good yields from galactose pentaacetate, glucose pentaacetate, and lactose octaacetate, respectively.
General Procedure for the Synthesis of Peracetylated Carbohydrate–HPPH Conjugates Linked with Amide Bond (5–8)
Methyl3-(1′-hexyloxyethyl)-3-devinylpyropheophorbide-a (HPPH) was synthesized by following the methodology developed in our laboratory.17 To a solution of HPPH (1 equiv) and 1-β-amino-carbohydrates 1–4 (2 equiv) in 10 mL of dry dichloromethane, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDCI, 2 equiv), and 4-(dimethylamino)pyridine (DMAP, 2 equiv) were added and the reaction mixture was stirred at room temperature under N2 atmosphere overnight. It was then diluted with dichloromethane (40 mL), washed with water (3 × 50 mL), dried over anhydrous sodium sulfate, and concentrated to yield a crude product, which was first partially purified by alumina column using 2% methanol in dichloromethane as eluant and finally by silica column using 3% methanol in dichloromethane to obtain pure final precursors 5–8 in fair to good yield.
Peracetylated Galactose–HPPH Conjugate (5)
The title compound was obtained in 66% yield. H NMR (600 MHz, CDCl3, δ ppm): 9.80 (d, J = 2.1 Hz, 1H, meso-H), 9.48 and 8.53 (each s, 1H, meso-H), 6.11 (m, 1H, CONH), 5.92 (m, 1H, 31-CH-(O-hexyl)CH3), 5.40 (m, galactose-H), 5.30–4.96 (m, total 5H, 132-CH2, 3 × galactose-H), 4.49 and 4.37 (each m, 1H, 17- and 18-H), 4.10–3.96 (m, 3H, galactose-H), 3.70 (m, 4H, 81-CH2 and 32 -OCH2CH2CH2CH2CH2CH3), 3.62, 3.40, and 3.28 (each s, 3H, 2-,7- and 12-CH3), 2.69, 2.38, and 2.18 (each m, total 4H, 171- and 172-CH2), 2.13 (m, 3H, 31-CH(O-hexyl)CH3), 2.06, 2.00, 1.96, and 1.93 (each s, 3H, 4 × galactose CH3CO2), 1.82 (d, J = 6.40 Hz, 3H, 18-CH3), 1.73 (m, 3H, 82-CH3), 1.50–1.20(m, 8H, 32-OCH2CH2CH2CH2CH2CH3), 0.79 (m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.46 and −1.69 (each br, 1H, NH). Mass calcd for C53H68N5O12: 966.4864 (MH+); HRMS found: 966.4869.
Peracetylated Glucose–HPPH Conjugate (6)
The title compound was obtained in 73% yield. 1H NMR (400 MHz, CDCl3, δ ppm): 9.79 (s, 1H, meso-H), 9.46 (d, J = 2.2 Hz, 1H, meso-H) and 8.51 (s, 1H, meso-H), 6.07 (m, 1H, CONH), 5.90 (q, J = 6.7, 1H, 31-CH(O-hexyl)CH3), 5.30–4.95 (m, total 4H, 132-CH2 and 2 × glucose-H), 4.82 and 4.75 (each m, 1H, 2 × glucose-H), 4.46 and 4.35 (each m, 1H, 17- and 18-H), 4.30–4.00 (m, 3H, 3 × glucose-H), 3.76 (m, 1H, glucose-H), 3.68 (m, 4H, 81-CH2 and 32-OCH2(CH2)4CH3), 3.58, 3.39, and 3.27 (each s, 3H, 2-, 7- and 12-CH3), 2.64, 2.36, and 2.16 (each m, total 4H, 171- and 172-CH2), 2.11 (m, 3H, 31-CH(O-hexyl)CH3), 2.09, 2.07, 2.03, and 2.01 (each s, 3H, 4 × glucose CH3CO2), 1.80 (d, J = 7.30 Hz, 3H, 18-CH3), 1.67 (m, 3H, 82-CH3), 1.42–1.22 (m, 8H, 32-OCH2CH2CH2CH2CH2CH3), 0.75 (m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.45 and −1.71 (each br, 1H, NH). Mass calcd for C53H68N5O12: 966.4864 (MH+); HRMS found: 966.4867.
Peracetylated Lactose–HPPH Conjugate (7)
The title compound was obtained in 70% yield. UV–vis λmax (CH2Cl2, nm): 660 (4.75 × 104 ), 605 (0.78 × 104), 537 (0.95 × 104), 505 (0.94 × 104), 410 (10.39 × 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.79 (s, 1H, meso-H), 9.41 (d, J = 3.2 Hz, 1H, meso-H) and 8.51 (s, 1H, meso-H), 6.05 (m, 1H, CONH), 5.91 (q, J = 6.6 Hz, 1H, 31-CH(O-hexyl)CH3), 5.33 (m, 1H, lactose-H), 5.29–5.05 (m, total 4H, 132-CH2 and 2 × lactose -H), 4.92 (m, 1H, lactose-H), 4.68(m, 1H, lactose-H), 4.50–4.34 (m, total 5H, 17-H, 18-H and 3 × lactose-H), 4.15–4.00 (m, 3H, 3 × lactose-H), 3.83 (t, J = 6.6, 1H, lactose-H), 3.65–3.60 (m, total 4H, 81-CH2 and 32-OCH2CH2CH2CH2CH2CH3), 3.68, 3.39, and 3.27 (each s, 3H, 2-, 7- and 12-CH3), 2.67 and 2.36 (each m, total 3H, 171- and 172-CH2), 2.18–1.95 (m, total 22H, 171-CH2, 31-CH (O-hexyl)CH3, 6 × lactose CH3CO2), 1.90 (s, 3H, 18-CH3), 1.79 (m, 3H, lactose CH3CO2), 1.73–1.66 (m, total 6H, 82-CH3 and 32-OCH2CH2CH2CH2CH2CH3), 1.20 (m, total 8H, 32-OCH2CH2CH2CH2CH2CH3), 0.78(m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.48 and −1.70 (each br, 1H, NH). Mass calcd for C65H84N5O20: 1254.5709 (MH+); HRMS found: 1254.5712.
Peracetylated Cellobiose–HPPH Conjugate (8)
The title compound was obtained in 38% yield. 1H NMR (600 MHz, CDCl3, δ ppm): 9.80 (s, 1H, meso-H), 9.26 (d, J = 10.3 Hz, meso-H) and 8.53 (s, 1H, meso-H), 6.21 (m, 1H, CONH), 5.93 (m, 1H, 31-CH(O-hexyl)CH3), 5.30–5.04 (m, 5H, 132-CH2, 3 × cellobiose-H), 4.90 (m, 1H, cellobiose-H), 4.70 (m, 1H, cellobiose-H), 4.52–4.30 (m, 6H, 17-H, 18-H and 4 × cellobiose-H), 4.07 (m, 1H, cellobiose-H), 4.01 (m, 1H, cellobiose-H), 3.71–3.57 (m, total 7H, 32-OCH2CH2CH2CH2CH2CH3, 81-CH2 and 3 × cellobiose-H), 3.69, 3.39, and 3.28 (each s, 3H, 2-, 7- and 12-CH3), 2.68 and 2.38 (each m, total 3H, 171- and 172-CH2), 2.13 (m, 3H, 31-CH(O-hexyl)CH3), 2.11 (m, 1H, 171-CH2), 2.07–1.93 (each s, 3H, 7 × cellobiose CH3CO2), 1.81 (d, J = 7.90 Hz, 3H, 18-CH3), 1.61 (m, 3H, 82-CH3), 1.50–1.20 (m, 8H, 32-OCH2CH2CH2CH2CH2CH3), 0.78(m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.48 and −1.70 (each br, 1H, NH). Mass calcd. for C65H84N5O20: 1254.5709 (MH+); HRMS found: 1254.5711.
General Procedure for the Deacetylation of Peracetylated Carbohydrate–HPPH Conjugates Linked with Amide Bond (9–12)
To a solution of 5–8 in dry dichloromethane (10 mL) and methanol (1 mL), sodium methoxide in methanol (1 M, 200 μL) was added and the reaction mixture was stirred for 30 min under N2 atmosphere at room temperature. It was then neutralized with acetic acid, evaporated under vacuum, and the crude product thus obtained was purified by silica column using gradient 2–8% methanol in dichloromethane as eluant to obtain compounds 9–12 in good yield.
Galactose–HPPH Conjugate (9)
The title compound was obtained in 94% yield. UV–vis λmax (THF, nm): 661 (4.75 × 104 ), 604 (0.71 × 104 ), 536 (0.88 × 104 ), 505 (0.87 × 104 ), 410 (8.37 × 104), 317 (1.89 × 104). 1H NMR (400 MHz, pyridine-d5, δ ppm): 10.25 (d, J = 5.6 Hz, 1H, meso-H), 9.95 (s, 1H, meso-H), 9.62 (d, J = 9.2,1H, CONH), 8.80 (s, 1H, meso-H), 6.14(m, 1H, 31-CH(O-hexyl)CH3), 5.94 (t, J = 9.0, 1H, NHCO), 5.27 (ABX, 2H, 132-CH2), 4.89 (brs, 4H, 4 × galactose–OH), 4.60–4.15 (m, total 9H, 7 × galactose-H, 17- and 18-H), 3.85–3.80 (m, 4H, 81-CH2 and 31-OCH2CH2CH2CH2CH2CH3), 3.75, 3.46, and 3.36 (each s, 3H, 2-, 7- and 12-CH3), 2.96–2.32 (each m, total 4H, 171- and 172-CH2), 2.28 (d, J = 6.6 Hz, 3H, 31-CH(O-hexyl)CH3), 1.85 (m, 2H, 32-OCH2CH2CH2CH2CH2CH3), 1.82 (d, J = 7.2 Hz, 3H, 18-CH3), 1.74 (t, J = 7.6 Hz, 3H, 82-CH3), 1.60–1.20 (m, 6H, 32-OCH2CH2CH2CH2CH2CH3), 0.75 (m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.43 and −1.65 (each br, 1H, NH). Mass calcd for C45H59N5O8: 798.4442 (MH+); HRMS found: 798.4440.
Glucose–HPPH Conjugate (10)
The title compound was obtained in 96% yield. UV–vis (THF, nm): 661 (4.75 × 104), 604(0.68 × 104), 536(0.85 × 104), 506(0.84 × 104), 411 (9.19 × 104 ), 318 (1.84 × 104). 1H NMR (400 MHz, pyridine-d5, δ ppm): 10.25 (d, J = 5.2 Hz, 1H, meso-H), 9.96 and 8.80 (each s, 1H, meso-H), 9.59 (d, J = 9.0,1H, CONH), 6.14 (m, 1H, 31-CH)O-hexyl)CH3), 6.02 (t, J = 9.0 Hz, 1H, NHCO), 5.36 (br, 6H, 132-CH2 and 4 × glucose–OH), 3.84–3.79 (m, 5H, 81-CH2, 32-OCH2CH2CH2CH2CH2CH3), 3.76, 3.46, and 3.37 (each s, 3H, 2-, 7-, and 12-CH3), 2.97–2.45 (each m, total 4H, 171- and 172-CH2), 2.28 (d, J = 6.6 Hz, 3H, 31-CH(O-hexyl)CH3), 1.87 (m, 2H, 32-OCH2CH2CH2CH2CH2CH3), 1.85 (d, J = 7.1 Hz, 3H, 18-CH3), 1.75 (t, J = 7.4, 3H, 82-CH3), 1.55–1.18 (m, 6H, 32-OCH2CH2CH2CH2CH2CH3), 0.75 (m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.43 and −1.64 (each br, 1H, NH). Mass calcd for C45H59N5O8: 798.4442 (MH+); HRMS found: 798.4441.
Lactose–HPPH Conjugate (11)
The title compound was obtained in 85% yield. UV–vis (THF, nm): 661 (4.75 × 104 ), 536 (0.95 × 104 ), 505 (0.94 × 104), 409 (9.84 × 104). 1H NMR (400 MHz, pyridine-d5, δ ppm): 10.24 (d, J = 4.7 Hz, meso-H), 9.94 and 8.78 (each s, 1H, meso-H), 9.66 (d, J = 9.0 Hz, 1H, NHCO), 6.13 (m, 1H, 31-CH(O-hexyl)CH3), 5.94 (t, J = 9.0 Hz, 1H, NHCO), 5.24 (ABX, 2H, 132-CH2), 4.94 (br, 12H, 17-H, 18-H, 5 × lactose-H and 7 × lactose–OH), 4.55–3.95 (m, 11H, 17-H, 18-H and 9 × lactose-H), 3.83–3.76 (m, 4H, 81-CH3 and 31-OCH2CH2CH2CH2CH2CH3), 3.74, 3.45, and 3.36 (each s, 3H, 2-, 7-, and 12-CH3), 3.00–2.43 (each m, total 4H, 171- and 172-CH2), 2.27 (d, J = 6.8 Hz, 3H, 31-CH(O-hexyl)CH3), 1.84 (m, 2H, 32-OCH2CH2CH2CH2CH2CH3), 1.81 (d, J = 7.4 Hz, 3H, 18-CH3), 1.73 (t, J = 7.5 Hz, 3H, 18-CH3), 1.55–1.20 (m, 6H, 32-OCH2CH2CH2CH2CH2CH3), 0.74 (m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.48 and −1.70 (each br, 1H, NH). Mass calcd for C51H69N5O13: 960.4970 (MH+); HRMS found: 960.4966.
Cellobiose–HPPH Conjugate (12)
The title compound was obtained in 90% yield. UV–vis (THF, nm): 661 (4.75 × 104), 604 (0.72 × 104), 536 (0.89 × 104), 505 (0.89 × 104), 409 (9.34 × 104), 318 (1.97 × 104 ). 1H NMR (400 MHz, pyridine-d5, δ ppm): 10.25 (d, J = 4.9 Hz, 1H, meso-H), 9.96 and 8.79 (each s, 1H, meso-H), 9.67(dd, J = 2.0 and 9.2, 1H, NHCO), 6.14 (m, 1H, 31-CH(O-hexyl)CH3), 5.96 (t, J = 9.1, 1H, NHCO), 5.35 (m, 1H, 132-CH2), 5.15 (m, 2H, 132-CH2 and cellobiose-H), 4.88 (brs, 7H, 7 × cellobiose–OH), 4.58–3.96 (m, 14H, 17-H, 18-H and 12 × cellobiose-H), 3.86–3.76 (m, 4H, 81-CH2 and 32-OCH2CH2CH2CH2CH2CH3), 3.77, 3.46, and 3.37 (each s, 3H, 2-, 7-, and 12-CH3), 3.00–2.43 (each m, total 4H, 171-and 172-CH2),2.28 (d, J= 6.5Hz, 3H, 31-CH(O-hexyl) CH3), 1.85 (m, 2H, 32-OCH2 CH2CH2CH2CH2CH3), 1.82 (d, J = 7.2 Hz, 3H, 18-CH3), 1.75 (t, J = 7.6 Hz, 3H, 82-CH3), 1.50–1.20 (m, 6H, 32-OCH2CH2CH2CH2CH2CH3), 0.74 (m, 3H, 32-OCH2CH2CH2CH2CH2CH3), 0.46 and −1.69 (each br, 1H, NH). Mass calcd for C51H69N5O13: 960.4970 (MH+); HRMS found: 960.4972.
HPPH with Boc Protected Amino Side Chain (13)
To a stirred solution of HPPH (600 mg) and N-Boc-ethylenediamine (200 μL) in dry dichloromethane (10 mL), BOP (440 mg), and dry triethylamine (1 mL) were added and the reaction mixture was stirred under N2 atmosphere overnight. Solvents were removed under reduced pressure, and the crude residue was purified by silica column using 4% methanol in dichloromethane as eluant to obtain title compound 13 in 95% yield. 1H NMR (400 MHz, CDCl3, δ ppm): 9.79 (d, J = 3.2 Hz, 1H, meso-H), 9.15 (d, J = 9.8 Hz, 1H, meso-H), 8.52 (s, 3H, meso-H), 6.19 (br, 1H, CONH), 5.92 (m, 1H, 31-CH(O-hexyl)CH3), 5.16 (ABX, 2H, 132-CH2), 4.89 (brs, 1H, NHBoc),4.52 and 4.30(each m, 1H, 17-and 18-H), 3.72–3.52 (m, 4H, 81-CH2 and 32-OCH2CH2CH2CH2CH2CH3), 3.39 and 3.28 (each s, 3H, 2- and 7-CH3), 3.24–3.09 (m, 7H, 12-CH3 and CONHCH2CH2NHBoc), 2.70–2.32 (each m, total 4H, 171- and 172-CH2), 2.13 (d, J = 6.6 Hz, 3H, 31-CH(O-hexyl)CH3), 1.79 (d, J = 7.7 Hz, 3H, 18-CH3), 1.75 (m, 2H, 32-OCH2CH2CH2CH2CH2CH3), 1.62 (m, 3H, 82-CH3), 1.26–1.20 (m, 15H, (CH3)3COO and 32-OCH2CH2CH2CH2CH2CH3), 0.78 (m, 3H, OCH2CH2CH2CH2CH2CH3), 0.50 and −1.64 (each br, 1H, NH). Mass calcd for C46H63N6O5: 779.4860 (MH+); HRMS found: 779.4863.
HPPH with Free Amino Side Chain (14)
Compound 13 (36.8 mg) in dichloromethane (5 mL) was treated with trifluoroacetic acid (1 mL). After 30 min, solvents were removed under reduced pressure and the residue was used as such in the next step without further purification.
General Approach for the Synthesis of Peracetylated Carbohydrate–HPPH Conjugates Linked with Rigidified Linker (18–20)
Peracetylated multivalent lactose analogues 15–17 were prepared by following the reported procedure. For the synthesis of the title compounds, compound 14 (2 equiv), BOP (2 equiv), and triethylamine (3 equiv) were dissolved in 10 mL of dry dichloromethane under N2 atmosphere and reacted individually to multivalent lactose analogues 15–17 at room temperature overnight. After the standard workup, the residue was purified by silica column chromatography. The major band was collected (eluted with 2–4% methanol in dichloromethane) to give 18–20 in modest yields. The NMR data were similar to those reported in the literature.
Monovalent Lactose–HPPH Conjugate (21)
To a solution of 18 (60 mg) in dry dichloromethane (10 mL) and methanol (1 mL), sodium methoxide (1 M, 200 μL) in methanol was added and the reaction mixture was stirred for 30 min under the N2 atmosphere. It was then neutralized by strong cationic resin (Dowex), and after filtering out the resin, the reaction mixture was concentrated to dryness and the crude solid thus obtained was chromatographed on silica column using 10% methanol in dichloromethane as eluant to yield 40 mg (83%) of title compound. UV–vis λmax (THF, nm): 661 (4.75 × 104), 604 (0.79 × 104), 536 (0.95 × 104), 506 (0.99 × 104), 410 (10.00 × 104), 317 (3.17 × 104). Mass calcd for C64H82N8O14S: 1219.5749 (MH+); HRMS found: 1219.5740.
Divalent Lactose–HPPH Conjugate (22)
To a solution of 19 (40 mg) in dry dichloromethane (10 mL) and methanol (1 mL), 1 M NaOMe in methanol (200 μL) was added and the reaction mixture was stirred for 1 h under a N2 atmosphere. It was then neutralized by strong cationic resin (Dowex), and after filtering out the resin, the reaction mixture was evaporated to dryness. The residue was washed with dichloromethane (2 × 50 mL) and water (2 × 50 mL). Evaporation of the solvent gave the title compound in 8%% (25 mg) yield. UV–vis λmax(THF, nm): 664 (4.615 × 104), 606 (0.99 × 104), 541 (1.13 × 104), 509(1.13 × 104), 414 (9.70 × 104). Mass calcd for C80H106N10O24S2: 1655.6900 (MH+); HRMS found: 1655.6910.
Tetravalent Lactose–HPPH Conjugate (23)
To a solution of 20 (18 mg) in dry dichloromethane (10 mL) and methanol (1 mL), NaOMe (1 M) in methanol (250 μL) was added and the reaction mixture was stirred for 4 h under a N2 atmosphere. A homogeneous solution was obtained after neutralization with Dowex resin. After removal of the resin by filtration, the solvent was evaporated under vacuum to dryness and washed with dichloromethane. Yield 10 mg (79%) of the title compound. UV–vis λmax (THF, nm): 663 (1.475 × 104), 540 (0.42 × 1040.164), 508 (0.41 × 104), 398 (3.31 × 104). Mass calcd for C132H168N16O46S4: 2842.0259 (MH+); HRMS found: 2842.0202.
Synthesis of 14C HPPH Methyl Ester
Methyl pyropheophorbide-a 50 mg (0.091 mmol) was reacted with 33%HBr/acetic acid (3.0 mL) for 2 h at room temperature. Evaporation of the acids under high vacuum gave a residue, which was not isolated and was immediately reacted with n-hexanol [1-14C] (250 μL, 2 mmol, specific activity-5 mCi/mmol, American Radiolabeled Chemicals, cat. no. ARC0225) in dry dichloromethane (3 mL). Anhydrous potassium carbonate (20 mg) was added, and the reaction mixture was stirred for 45 min under a nitrogen atmosphere. It was then diluted with dichloromethane (20 mL) and washed with water (2 × 50 mL). The organic layer was separated and dried over anhydrous sodium sulfate. The crude residue obtained after evaporating the solvent was purified by preparative plates (Silica), eluting with 2% methanol/dichloromethane. Evaporation of the solvents gave the title compound in 33% yield (20 mg).
Synthesis of 14C-HPPH
The foregoing 14C HPPH methyl ester was dissolved in a solution of methanol (4 mL), distilled peroxide-free tetrahydrofuran (3 mL), and lithium hydroxide (32 mg in 3 mL water) and stirred for 2 h until the hydrolysis was complete (monitored by TLC). After the standard workup, the desired product was purified by silica prep plate (1000 um), eluted with 7% methanol/dichloromethane). Yield (determined spectrophotometrically): 17 mg (87%), specific activity: 5.0 mCi/mmol. Purity was ascertained by HPLC (98% pure).
Synthesis of 14C-Labeled Galactose–HPPH Conjugate
To a solution of aminogalactose pentaacetate (5.6 mg), 14C labeled HPPH (5 mg, 0.00517 mmol), and BOP (6.6 mg) in dry DMF (2 mL), 40 μL of triethylamine were added. The reaction mixture was stirred at room temperature under the N2 atmosphere for 7 h. Evaporation of the solvent under vacuum gave a crude product, which was purified by silica plates (5% MeOH in dichloromethane). The product was dissolved in a mixture of dichloromethane (5 mL) and 20 μL of NaOMe (in MeOH) was added, and the reaction mixture was stirred for 30 min, then 0.8 g of Dowex resin was added to quench the reaction. The solution was collected after removing the resin, concentrated and purified by a silica column (eluent: 1 – 10% MeOH in dichloromethane). The appropriate fractions were collected. Evaporation of the solvent gave pure 14C labeled galactose–HPPH conjugate in quantitative yield (determined spectrophotometrically: 6.3 mg (quantitative yield) with specific activity 4.6 mCi/mmol.
HPLC Analysis
The purity of the final products was ascertained by HPLC using a Waters Delta 600 system consisting of the 600 controller, 600 fluid handling unit, and 996 photodiode array detector equipped with a Waters Symmetry C18 column, 5 μm particle size, with dimensions of 4.6 mm × 150 mm. The mobile phase composition was 95% methanol/5% water, run isocratically at a flow of 1.0 mL/min. The retention time and percent purity of each compound is tabulated in Table 2 (Supporting Information); the component percentages are based on the area counts of the peaks from the 408 nm channel. Under these HPLC conditions, the conjugates 22 and 24 could not be eluted. However, their corresponding acetoxy-derivatives were >95% pure.
In Vitro Photocytotoxicity Assay
RIF and Colon26 cells were grown in α-MEM with 10% fetal calf serum, L-glutamine, penicillin, and streptomycin at 37 °C in 5% CO2, 95% air, and 100% humidity. Cells were seeded in 96-well plates at a 5 × 103 cells per well (five replicates) in complete medium. After overnight incubation at 37 °C, photosensitizers were added at variable concentrations for 24 h in the dark. Drug-containing medium was replaced with fresh medium and cells were illuminated with 0.5 J/cm2 delivered by an argon-pumped dye laser (665 nm at a dose rate of 3.2 mW/cm2). After PDT, the cells were incubated for 48 h at 37 °C in the dark and phototoxicity (relative growth compared to untreated cells of each compound tested) was determined by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide) assay. Dose–response cell survival curves were generated (using Microcal Origin 6.0), and the LD50 values were determined based on Gaussian and Sigmoidal fit. Each data point represents the mean from three separate experiments, and the error bars are the standard deviation. Similar experiments were later performed with Colon-26 with HPPH and 9 for comparison and for both cell types with drugs (HPPH and the galactose derivative 9) incubated for 4 h.
In Vitro PS Uptake Determined by Fluorescence
RIF cells were seeded at 1.5 × 105 cells per well in 6-well plates. After overnight incubation at 37 °C, PS at 0.25 μM were added to triplicate wells and incubated in the dark at 37 °C for 3 or 24 h. Drug-containing medium was removed and cells were washed with PBS and were solubilized by incubating with gentle rocking in the dark at 37 °C for 2 h with 1 mL Solvable (PerkinElmer Life and Analytical Sciences). The cell solutions were diluted with 2 mL of ddH2O, and the fluorescence of each sample was measured (excitation 415 nm; emission 667 nm) on a FluoroMax-2 (ISA, JOBIN YVON-SPEX, Horiba Group). The protein content of the sample was determined using the Bio-Rad DC Assay. The data was plotted as fluorescence units/mL of protein.
Intracellular Localization
Cells were grown in 6-well plates on poly-L-lysine coated coverslips, coincubated with PS and organelle-specific fluorescent dyes (3–4 h and 24 h), washed with PBS, and imaged at 40× on an inverted fluorescence microscope (Zeiss Axiovert 200W, Germany) with a charged-coupled device camera (Dage Zeiss AxioCam MRm) using an AxioCam MR-MRGrab Framegrabber and AxioVision LE 4.1 imaging software. For organelle staining, cells were incubated with MitoTracker Green (1 μM for 1 h, mitochondria), Lyso-Tracker Green, or carboxylate-modified 0.1 μm diameter yellow–green Fluospheres (0.5 μM for 0.5 h or 1/100000 dilution overnight, respectively, lysosomes), or Bodipy C5 ceramide–albumin complexes (5 μM, Golgi apparatus) according to manufacturer’s directions (Invitrogen). Filter combinations were as follows: for HPPH-containing compounds Ex BP D410/40 nm, BeamSplitter FT 505dcxvu, and Em BP 675/50 nm; for LysoTracker Green and Fluospheres Ex 390/22 nm, FT 420 nm and Em BP4 60/50 nm; and for Bodipy C5 ceramide–albumin Ex BP 565/30 nm, BeamSplitter FT 585 nm and Em BP 520/60.
Uptake of 14C-Labeled HPPH or the Corresponding Galactose Derivative (9)
For uptake by 14C-labeled HPPH or HPPH-Gal (specific activity 5 Ci/mol and 4.6 Ci/mol, respectively), RIF and Colon26 cells were seeded at 3 × 104 cells per cm2 culture area in 24-well or 6-well cluster plates. After one day, the medium was replaced by 0.2 mL per cm2 culture area with medium containing either 0.5% or 10% fetal calf serum and 0.8 μM 14C-labeled PS and incubated in the dark at 37 °C. At different time points up to 24 h, the drug-containing medium was removed, cells were washed with phosphate buffered saline and either released by trypsin digestion and counted in a hematocytometer or solubilized within the culture well. The solubilized cell samples were transferred to scintillation vials, UniverSol scintillation fluid added, and the radioactivity counted. The uptake of radioactivity was corrected for background counts and expressed in pmol PS per 1 × 106 cells. Experiments were carried out in triplicate.
Test for ABCG2 Pump Specificity
In the same experimental setting as used for PS uptake measurements, RIF or Colon26 were preincubated at 37 °C with or without 10 μM imatinib mesylate for 0.5 h, followed by addition of 14C-labeled PS or unlabeled PS. For flowcytometric analyses of PS accumulation, the cells incubated with nonradioactive PS were released from culture wells and resuspended in cytometric tubes in cold medium with 2% FCS. Tubes were thereafter kept in the dark on ice to inhibit ABCG2 efflux activity. Fluorescence of the photosensitizer was measured by flow cytometry on a FACS-Caliber flow cytometer, BD Invitrogen), with excitation at 488 nm and emission at 680/20 nm. After subtracting autofluorescence (untreated or inhibitor only cells), the (percentage) change in the fluorescence mediated by the inhibitor was calculated. An increase of more than 15% fluorescence was considered significant (p < 0.05, students 2-tailed t test) and indicated that the photosensitizer was a substrate of the ABCG2 pump.
In Vivo Photosensitizing Efficacy
The in vivo antitumor photosensitizing efficacies of HPPH–carbohydrate conjugates were evaluated in mice with tumors implanted on the shoulder. C3H mice were implanted with RIF tumors and BALB/c mice with Colon26 tumors. Groups of five C3H or 10 BALB/c mice were injected with the compounds (0.47μmol/kg) intravenously. The tumors were exposed to laser light (665 nm) at various fluence and fluence rates at 24 h post injection. Two light dose conditions were applied 135 J/cm2, 75 mW/cm2, and 48 J/cm2, 7 mW/cm2 ). After in vivo PDT, the tumor volume V = (L × W2) × 0.5 (L = length (the longest dimension) and W = width (perpendicular to the long axis) was measured with calipers. The hours-to-end point (HTE) the time for the tumor to grow to 400 mm3 was calculated by linear interpolation using Microsoft Excel-based software. Mice were sacrificed just after their tumors reach the end point. For each experimental group, a Kaplan–Meier hours-to-end point (HTE) curve was generated (using Prism 4, GraphPad Software, Inc.), and the median Kaplan–Meier tumor growth HTE time was estimated. To test for significant differences between pairs of HTE curves, the Cox–Mantel test was used.31
In Vivo Biodistribution Studies
The 14C-labeled photosensitizers (0.2 mL/5 μCi) were individually administered to mice through a lateral tail vein (three mice/group (BALB/c mice bearing Colon26 tumors and C3H mice bearing RIF tumors)). At 24, 48 h after injection, three mice/time point were sacrificed using CO2 followed by cervical dislocation as per Institute’s approved protocol. The blood was collected into heparinized syringes, kept ice cold, and immediately centrifuged to obtain plasma. The plasma was transferred to a scintillation vial for C-14 counting. The organs of interest (skin, muscle, tumor, heart, lung, liver, kidney, and spleen) were removed. Sample weights were recorded, and 1 mL of Solvable was added to each sample and incubated overnight at 53 °C. The samples were allowed to dissolve before adding 80 μL of peroxide (20 μL at a time). After samples were sufficiently bleached, 15 mL of Ultima Gold scintillation fluid was added to each and the samples were run on a Beckman LS 6000LL scintillation counter. The raw data were then converted to count s/g tissue weight and plotted using SigmaPlot 10.0 software.
Supplementary Material
Acknowledgments
Financial assistance from the NIH (CA 55791) and shared resources of the RPCI support grant (P30CA16056) is highly appreciated. We thank Adam Sumlin for his assistance in evaluating HPPH and HPPH-Gal for skin phototoxicity.
Footnotes
Dedicated to (late) Dr. Allan R. Oseroff, Chief, Department of Dermatology, RPCI, Buffalo, New York.
Abbreviations: PDT, photodynamic therapy; QSAR, quantitative structure–activity relationship; PS, photosensitizer; ABCG2, ATP-binding cassette, subfamily G, member 2; STAT3, signal transducers and activator of transcription proteins.
Supporting Information Available: NMR and the HPLC analysis of the final products and the intracellular localization characteristics of compounds 9–12 and 21–23. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Brown SB, Brown EA, Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5:497–508. doi: 10.1016/S1470-2045(04)01529-3. [DOI] [PubMed] [Google Scholar]; (b) Pandey RK, Goswami LN, Chen Y, Gryshuk A, Missert JR, Oseroff A, Dougherty TJ. Nature: A rich source for developing multifunctional agents. Tumor imaging and photodynamic therapy. Laser Surg Med. 2006;38:445–467. doi: 10.1002/lsm.20352. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dougherty TJ, Levy JG. Clinical Applications of Photodynamic Therapy. In: Horspool W, Lenci F, editors. CRC Handbook of Organic Photochemistry and Photobiology. 2. CRC Press LLC; Boca Raton, FL: 2004. pp. 147-1–147-17. [Google Scholar]; (b) Dolmans DE, JGJ, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lang K, Mosinger J, Wagnerova DM. Photophysical properties of porphyrinoid sensitizers noncovalently bound to host molecule; models for photodynamic therapy. Coord Chem Rev. 2004;248:321–350. [Google Scholar]; (b) Schastak S, Jean B, Handzel R, Kostenich G, Hermann R, Sack U, Orenstein A, Wang YS, Wiedemann P. Improved pharmacokinetics, biodistribution and necrosis in vivo using a new near infrared photosensitizer: tetrahydroporphyrin tetratosylat. J Photochem Photobiol, B. 2005;78:203–213. doi: 10.1016/j.jphotobiol.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 4.(a) Potter WR, Henderson BW, Bellnier DA, Pandey RK, Vaughan LA, Weishaupt KR, Dougherty TJ. Parabolic quantitative structure–activity relationships and photodynamic therapy: application of a three-compartment model with clearance to the in vivo quantitative structure–activity relationships of a congeneric series of pyropheophorbide derivatives used as photosensitizers for photodynamic therapy. Photochem Photobiol. 1999;70:781–788. [PubMed] [Google Scholar]; (b) Henderson BW, Bellnier DA, Greco WR, Sharma A, Pandey RK, Vaughan L, Weishaupt KR, Dougherty TJ. A quantitative structure activity relationship for a congeneric series of pyropheophorbide derivatives as photosensitizers for photodynamic therapy. Cancer Res. 1997;57:4000–4007. [PubMed] [Google Scholar]
- 5.Sharman WM, van Lier JE, Allen CM. Targeted photodynamic therapy via receptor mediated delivery systems. Adv Drug Delivery Rev. 2004;56:53–76. doi: 10.1016/j.addr.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Savellano MD, Hasan T. Photochemical targeting of epidermal growth factor receptor: a mechanistic study. Clin Cancer Res. 2005;11:1658–1668. doi: 10.1158/1078-0432.CCR-04-1902. [DOI] [PubMed] [Google Scholar]
- 7.(a) Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]; (b) Nangia-Makker P, Conklin J, Hogan V, Raz A. Carbohydrate-binding proteins in cancer, and their ligands as therapeutic agents. Trends Mol Med. 2002;8:187–192. doi: 10.1016/s1471-4914(02)02295-5. [DOI] [PubMed] [Google Scholar]
- 8.(a) Alper J. Searching for Medicine’s Sweet Spot. Science. 2001;291:2338–2343. doi: 10.1126/science.291.5512.2338. [DOI] [PubMed] [Google Scholar]; (b) Kaldapa C, Blais JC, Carre V, Granet R, Sol V, Guilloton M, Spiro M, Krausz P. Synthesis of new glycosylated neutral and cationic porphyrins dimers. Tetrahedron Lett. 2000;41:331–335. [Google Scholar]; (c) Sol V, Blais JC, Carre V, Granet R, Guilloton M, Spiro M, Krausz P. Synthesis, spectroscopy, and photocytotoxicity of glycosylated amino acid porphyrin derivatives as promising molecules for cancer phototherapy. J Org Chem. 1999;64:4431–4444. [Google Scholar]; (d) Sylvain I, Zerrouki R, Granet R, Huang YM, Lagorce JF, Guilloton M, Blais JC, Krausz P. Synthesis and biological evaluation of thioglycosylated porphyrins for an application in photodynamic therapy. Bioorg Med Chem. 2002;10:57–69. doi: 10.1016/s0968-0896(01)00255-3. [DOI] [PubMed] [Google Scholar]; (e) Laville I, Figueiredo T, Loock B, Pigaglio S, Maillard P, Grierson DS, Carrez D, Croisy A, Blais J. Synthesis, cellular internalization and photodynamic activity of glucoconjugated derivatives of tri and tetra(meta-hydroxyphenyl)chlorins. Bioorg Med Chem. 2003;11:1643–1652. doi: 10.1016/s0968-0896(03)00050-6. [DOI] [PubMed] [Google Scholar]; (f) Chen X, Hui L, Foster DA, Drain CM. Efficient Synthesis and Photodynamic Activity of Porphyrin-Saccharide Conjugates: Targeting and Incapacitating Cancer Cells. Biochemistry. 2004;43:10918–10929. doi: 10.1021/bi049272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Zheng G, Graham A, Shibata M, Missert JR, Oseroff AR, Dougherty TJ, Pandey RK. Synthesis of β-galactose-conjugated chlorins derived by enyne metathesis as galectin-specific photosensitizers for photodynamic therapy. J Org Chem. 2001;66:8709–8716. doi: 10.1021/jo0105080. [DOI] [PubMed] [Google Scholar]; (b) Zheng X, Pandey RK. Porphyrin–carbohydrate conjugates: Impact of carbohydrate moieties in PDT. Anticancer Agents Med Chem. 2008;8:241–268. doi: 10.2174/187152008783961897. [DOI] [PubMed] [Google Scholar]
- 10.(a) Li G, Pandey SK, Graham A, Dobhal MP, Mehta R, Chen Y, Gryshuk A, Rittenhouse-Olson K, Oseroff A, Pandey RK. Functionalization of OEP-based benzochlorins to develop carbohydrate-conjugated photosensitizers. Attempt to target β-galactoside-recognized proteins. J Org Chem. 2004;69:158–172. doi: 10.1021/jo030280b. [DOI] [PubMed] [Google Scholar]; (b) Pandey SK, Zheng X, Morgan J, Missert JR, Liu TH, Shibata D, Bellnier DA, Oseroff AR, Henderson BW, Dougherty TJ, Pandey RK. Purpurinimide carbohydrate conjugates: Effect of the position of the carbohydrate moiety in photosensitizing efficacy. Mol Pharmaceutics. 2007;4:448–464. doi: 10.1021/mp060135x. [DOI] [PubMed] [Google Scholar]; (c) Pandey SK, Sajjad M, Chen Y, Yao R, Missert JR, Batt C, Nabi HN, Oseroff AR, Pandey RK. Comparative positron-emission tomography (PET) imaging and phototherapeutic potential of I-labeled methyl-3-(1′-iodonenxyloxyethyl)pyropheophorbide-a vs the corresponding glucose and galactose conjugates. J Med Chem. 2009;52:445–455. doi: 10.1021/jm8012213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vrasidas I, Andre S, Valentini P, Bock C, Lensch M, Kaltner H, Liskamp RMJ, Gabius HJ, Pieters RJ. Rigidified multivalent lactose molecules and their interactions with mammalian galectins: a route to selective inhibitors. Org Biomol Chem. 2003;1:803–810. doi: 10.1039/b210923a. [DOI] [PubMed] [Google Scholar]
- 12.Zheng X, Pandey SK, Camacho S, Morgan J, Bellnier DA, Pandey RK. Pyropheophorbide-carbohydrate conjugates as galectin-specific photosensitizers for photodynamic therapy. Third International Conference on Porphyrins and Phthalocyanines (ICCP-3); New Orleans, LA. July 11–16, 2004. [Google Scholar]
- 13.Robey RW, Steadman K, Polgar O, Bates SE. ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther. 2005;4:187–194. [PubMed] [Google Scholar]
- 14.Jonker JW, Buitelaar M, Wagenaar E, van der Valk MA, Scheffer GL, Scheper RJ, Plosch T, Kuipers F, Elferink RPJO, Rosing H, Beijnen JH, Schinkel AH. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA. 2002;99:15649–15654. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, Bates SE. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64:1242–1246. doi: 10.1158/0008-5472.can-03-3298. [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Baer MR, Bowman M, Pera P, Zheng X, Morgan J, Pandey RK, Oseroff AR. The Tyrosine Kinase Inhibitor Imatinib Mesylate Enhances the Efficacy of Photodynamic Therapy by Inhibiting ABCG2. Clin Cancer Res. 2007;13(8):2463–2470. doi: 10.1158/1078-0432.CCR-06-1599. [DOI] [PubMed] [Google Scholar]
- 17.(a) Pandey RK, Sumlin AB, Constantine S, Aoudia M, Potter WR, Bellnier DA, Henderson BW, Rodgers MA, Smith KM, Dougherty TJ. Alkyl ether analogs of chlorophyll-a derivatives, Part 1: synthesis, photophysical properties and photodynamic efficacy. Photochem Photobiol. 1996;64:194–204. doi: 10.1111/j.1751-1097.1996.tb02442.x. [DOI] [PubMed] [Google Scholar]; (b) Shibata R, Tamiaki H. Self-aggregation of synthetic zinc chlorophyll derivative possessing a perfluoroalkyl group in a fluorinated solvent. Biorg Med Chem. 2006;14:2235–2241. doi: 10.1016/j.bmc.2005.11.007. [DOI] [PubMed] [Google Scholar]; (c) Shibata R, Mizoguchi T, Inazu T, Tamiaki H. Self-aggregation of synthetic zinc chlorophyll derivatives possessing multi-perfluoroalkyl chains in perfluorinated solvents. Photochem Photobiol Sci. 2007;6:749–757. doi: 10.1039/b702866k. [DOI] [PubMed] [Google Scholar]
- 18.Bellnier DA, Greco WR, Nava H, Loewen GM, Oseroff AR. Mild skin photosensitivity in cancer patients following injection of Photochlor for photodynamic therapy. Cancer Chemother Pharmacol. 2005;57:40–45. doi: 10.1007/s00280-005-0015-6. [DOI] [PubMed] [Google Scholar]
- 19.Anderson TR, Dougherty TJ, Tan D, Sumlin A, Schlossin JM, Kanter PM. Photodynamic therapy for sarcoma pulmonary metastases. Anticancer Res. 2003;23:3713–3718. [PubMed] [Google Scholar]
- 20.(a) Bellnier DA, Greco WR, Loewen GM, Nava H, Oseroff AR, Pandey RK, Tsuchida T, Dougherty TJ. Population pharmacokinetics of the photodynamic therapy agent 2-(1′-hexyloxyethyl)-2-devinylpyropheophorbide-a in cancer patients. Cancer Res. 2003;63:1806–1813. [PubMed] [Google Scholar]; (b) Loewen GM, Pandey R, Belliner D, Henderson B, Dougherty T. Endobronchial photodynamic therapy for lung cancer. Lasers Surg Med. 2006;38:364–370. doi: 10.1002/lsm.20354. [DOI] [PubMed] [Google Scholar]
- 21.Tropper FD, Anderson FO, Braun S, Roy R. Phase transfer catalysis as a general and steroselective entry into glycosyl azides from glycosyl halides. Synthesis. 1992:618–620. [Google Scholar]
- 22.(a) Lao A, Hansch C, Elkins D. Partition coefficient and their uses. Chem Rev. 1971;71:525–616. [Google Scholar]; (b) Zheng G, Potter WR, Camacho SH, Missert JR, Wang G, Bellnier DA, Henderson BW, Rodgers MAJ, Dougherty TJ, Pandey RK. Synthesis, photophysical properties, tumor uptake, and preliminary in vivo photosensitizing efficacy of a homologous series of 2-(1′-alkoxyethyl)3-devinylpurpurin-18-N-alkylimides with variable lipophilicity. J Med Chem. 2001;44:1540–1559. doi: 10.1021/jm0005510. [DOI] [PubMed] [Google Scholar]
- 23.Bellnier DA, Henderson BW, Pandey RK, Potter WR, Dougherty TJ. Murine pharmacokinetics and antitumor efficacy of the photodynamic sensitizer 2–91-hexyloxyethyl)-2-devinylpyropheophorbide-a. J Photochem Photobiol, B. 1993;20:55–61. doi: 10.1016/1011-1344(93)80131-r. [DOI] [PubMed] [Google Scholar]
- 24.(a) Kessel D, Luo Y. Intracellular sites of photodamage as a factor in apoptotic cell death. J Porphyrins Phthalocyanines. 2001;5:181–184. [Google Scholar]; (c) Morgan J, Potter WR, Oseroff A. Comparison of photodynamic targets in a carcinoma cell line and its mitochondrial DNA-deficient derivative. Photochem Photobiol. 2000;70:747–757. doi: 10.1562/0031-8655(2000)071<0747:coptia>2.0.co;2. [DOI] [PubMed] [Google Scholar]; (b) Kessel D, Luo Y, Deng Y, Chang CK. The role of subcellular localization in inhibition of apoptosis by photodynamic therapy. Photochem Photobiol. 1997;65(3):422–426. doi: 10.1111/j.1751-1097.1997.tb08581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgan J, Oseroff AR, Weissig, Torchilin VP. Mitochondrial-based anti-cancer photodynamic therapy. Adv Drug Delivery Rev. 2001;49:71–86. doi: 10.1016/s0169-409x(01)00126-0. [DOI] [PubMed] [Google Scholar]
- 26.Tsaytler PAM, Sakharov DV, Krijgsveld J, Egmond MR. Immediate protein targets of photodynamic treatment in carcinoma cells. J Proteome Res. 2008;7(9):3868–78. doi: 10.1021/pr800189q. [DOI] [PubMed] [Google Scholar]
- 27.(a) Henderson BW, Daroqui C, Tracy E, Vaughan LA, Loewen GM, Cooper MT, Baumann H. Cross-Linking of Signal Transducer and Activator of Transcription 3-A Molecular Marker for the Photodynamic Reaction in Cells and Tumors. Clin Cancer Res. 2007;13(11):3156–3163. doi: 10.1158/1078-0432.CCR-06-2950. [DOI] [PubMed] [Google Scholar]; (b) Liu W, Oseroff AR, Baumann H. Photodynamic therapy causes cross-linking of signal transducer and activator of transcription proteins and attenuation of interleukin-6 cytokine responsiveness in epithelial cells. Cancer Res. 2004;64:6579–6587. doi: 10.1158/0008-5472.CAN-04-1580. [DOI] [PubMed] [Google Scholar]; (c) Chen Y, Ohkubo K, Zhang M, Wenbo E, Liu W, Pandey SK, Ciesielski M, Baumann H, Erin T, Fukuzumi S, Kadish KM, Fenstermaker R, Oseroff A, Pandey RK. Photophysical, electrochemical characteristics and cross-linking of STAT-3 protein by an efficient bifunctional agent for fluorescence image-guided photodynamic therapy. J Photochem Photobiol Sci. 2007;6(12):1257–1267. doi: 10.1039/b710395f. [DOI] [PubMed] [Google Scholar]
- 28.(a) Pandey RK, Bellnier DA, Smith KM, Dougherty TJ. Porphyrin and chlorin derivatives as potential photosensitizers in photodynamic therapy. Photochem Photobiol. 1991;53:65–72. doi: 10.1111/j.1751-1097.1991.tb08468.x. [DOI] [PubMed] [Google Scholar]; (b) Shibata R, Tamiaki H. Self-aggregation of synthetic zinc chlorophyll derivative possessing a perfluoroalkyl group in a fluorinated solvent. Biorg Med Chem. 2006;14:2235–2241. doi: 10.1016/j.bmc.2005.11.007. [DOI] [PubMed] [Google Scholar]; (c) Shibata R, Mizoguchi T, Inazu T, Tamiaki H. Self-aggregation of synthetic zinc chlorophyll derivatives possessing multi-perfluoroalkyl chains in perfluorinated solvents. J Photochem Photobiol Sci. 2007;6:749–757. doi: 10.1039/b702866k. [DOI] [PubMed] [Google Scholar]
- 29.Brundish DE, Baddiley J. Synthesis of glucosylglycerols and diglucoglycerols and their identification in small amounts. Carbohydr Res. 1968;8:308–3016. [Google Scholar]
- 30.Bertho V, Uber A. 1-Azido und 1-aminoderivative acetylierter monosen und biosen. Liebigs Ann. 1949;562:229–239. [Google Scholar]
- 31.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies. J Natl Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.