

Abstract
1,4-Thienodiazepine-2,5-diones have been synthesized via the Ugi-Deprotection-Cyclization (UDC) approach starting from Gewald 2-aminothiophenes i n a convergent and versatile manner. The resulting scaffold is unprecedented, cyclic and peptidomimetic with four points of diversity introduced from readily available starting materials. In addition to eighteen synthesized and characterized compounds, a virtual compound library was generated and evaluated for chemical space distribution and drug-like properties. A small focused compound library of 1,4-thienodiazepine-2,5-diones has been screened for the activity against p53-Mdm2 interaction. Biological evaluations demonstrated that some compounds exhibited promising antagonistic activity.
Keywords: 1,4-thienodiazepine-2,5-dione; multicomponent reaction; virtual chemical space; p53-Mdm2 interaction
The development of new synthetic scaffolds is of uttermost importance for identifying new lead structures during the drug discovery process. Seven-membered 1,4-diazepine ring based drugs have been found repeatedly as the prototype of a privileged structure with a broad range of biological activities and applications in human medicine (1). Over the last decades, 1,4-benzodiazepines have emerged as a particularly fascinating class of scaffolds in medicinal chemistry because they hit various classes of pharmacologically relevant targets such as GPCRs, ion channels and enzymes (2). For example, anthramycin is a broad-spectrum antineoplastic antibiotic with low toxicity, which binds irreversibly to DNA and inhibits the synthesis of RNA and DNA (3). Recently, 1,4-benzodiazepin-2,5-diones (BDZs) have been widely investigated as potential medicinal agents (4,5). Therefore, preparation and evaluation of libraries based on privileged structures become an important part of the drug discovery process. Thiophene as an effective bioisostere of phenyl ring led to the discovery of a series of 1,4-thienodiazepine drugs, such as the block buster family of olanzapine, clotiazepam, and brotizolam. For example, clotiazepam is structurally similar to diazepam, which is one of the most frequently prescribed medications in the world during the past 40 years (6). Thus, the development of new thiophene scaffolds based on privileged structures, especially 1,4-benzodiazepine family is of great interest.
Multicomponent reaction (MCR) is an alternative strategy in different drug discovery stages including lead discovery and pre-clinical process development (7). MCRs allow the resource and cost effective, fast, and convergent synthesis of diverse compound libraries, and highly improve the efficiency to explore the chemical space with limited synthetic effort (8). The Gewald three-component reaction (G-3CR) is a unique method using elemental sulfur to yield thiophene ring, which builds a platform for the synthesis of new thiophene scaffolds (9). For example, olanzapine is manufactured by Eli Lilly via the formation of its thiophene ring through G-3CR (10). G-3CR provides a convenient way to synthesize 2-aminothiophene 2, which is bioisostere to anthranilic acid 1 (Figure 1) (11). According to the substructure search in currently published protein-ligand cocrystal structures (PDB query and Relibase), most of the retrieved ligands are nonsubstituted anthranilic acid derivatives. Advantageously, the G-3CR accessible thiophene scaffold allows for many more synthetic variations than the substituted anthranilic acids derivatives.
Figure 1.
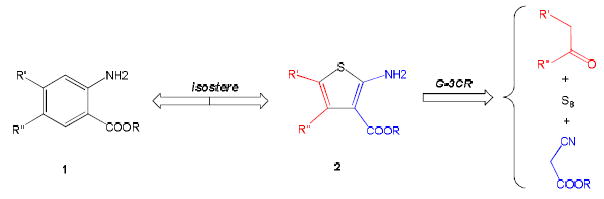
Gewald aminothiophene as a bioisostere of anthranilic acids
Isocyanide-based MCRs (IMCR), such as the Ugi and Passerini reactions, provide a powerful tool for producing arrays of compounds based on multiple scaffolds and with high atom economy (12). The Ugi-Deprotection-Cyclization (UDC) approach generates very useful classes of scaffolds amenable by an initial Ugi reaction of two bifunctional orthogonally protected starting material classes and a secondary reaction to form heterocyclic rings, such as benzimidazole, quinoxalinone, imidazoline, γ-lactame, ketopiperazine and diketopiperazine (13). Notably, UDC strategy is particularly useful for the synthesis of 1,4-benzodiazepine-2,5-diones (14-17). The further enlargement of UDC amenable scaffold classes is highly desirable, and therefore we investigated the development of synthetic routes to 1,4-thienodiazepine-2,5-diones. Herein, we report the preparation of a new 1,4-thienodiazepine-2,5-dione scaffold by a union of Gewald and UDC MCRs (Figure 2).
Figure 2.

Synthesis of the 1,4-thienodiazepine-2,5-dione scaffold via the UDC approach (R1, R2, R3, R4: points of diversity)
Materials and Methods
General
All reagents were purchased from commercial sources and used without further purification. The reactions were conducted under air atmosphere unless otherwise indicated. Analytical thin-layer chromatography (TLC) was performed on SiO2 plates on Alumina available from Whatman. Visualization was accomplished by UV irradiation at 254 nm. Preparative TLC was conducted using Preparative Silica gel TLC plates (1000 μm, 20cm×20cm). Flash column chromatography was performed using SiO2 60 (particle size 0.040-0.055 mm, 230-400 mesh, EM science distributed by Bioman). Proton and carbon NMR spectra were determined on Bruker Avance™ 600 MHz NMR spectrometer. Chemical shifts are reported as δ values in parts per million (ppm) as referenced to residual solvent. 1H NMR spectra are tabulated as follows: chemical shift, number of protons, multiplicity (s = singlet, br.s = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet), and coupling constant. High Resolution Mass spectra were obtained at the University of Pittsburgh Mass Spectrometry facility. LC-MS analysis was performed on an SHIMADZU instrument, using an analytical C18 column (Dionex Acclaim 120 Å, 2.1 × 50 mm, 3.0 μm, 0.2 mL/min). Acetonitrile/water mixtures were used as mobile phase for reverse-phase HPLC coupled to electrospray ionization-mass spectrometry (ESI-MS).
Methyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (2a)
To a mixture of cyclohexanone (0.45 mol, 44.10 g), methyl cyanoacetate (0.3 mol, 28.77 g), sulfur (0.3 mol, 9.60 g), and 50 mL of ethanol, the solution of diethylamine (0.15 mol, 12.75 g) in 25 mL of ethanol was added dropwise with magnetic stirring. After the completion of the addition, the reaction mixture was kept stirring under room temperate overnight. The reaction flask was kept in the freezer for crystallization. Then the precipitate was collected by vacuum filtration, and washed by cold ethanol. After air dry overnight, 42.20 g of yellow solids were obtained (yield: 67%). HPLC/MS: tR = 10.58 min; m/z = 212.1 [M+H]+ 1H NMR (600 MHz, CDCl3): 1.73-1.78 (4H, m), 2.49-2.51 (2H, m), 2.67-2.69 (2H, m), 3.79 (3H, s, OMe), 5.93 (2H, br.s, NH2). 13C NMR (150 MHz, CDCl3): 22.8, 23.3, 24.5, 26.9, 50.6, 105.6, 117.7, 132.4, 161.8, 166.5.
2-(Boc-amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylic acid (3a)
The mixture of 2a (10 mmol, 2.11 g), Boc2O (12.5 mmol, 2.56 g), DMAP (1 mmol, 122 mg) in 25 mL of THF was stirring under reflux overnight. The additional 0.5 g of Boc2O was added, and stirring under reflux overnight. After cooling to RT, the mixture was quenched by water, and extracted by DCM. The organic layer was dried, and concentrated. The residue was dissolved in 20 mL of ethanol, potassium hydroxide (2.24 g) and 20 mL of water was added. The mixture was reflux for 5 hours. The reaction was quenched by water, the aqueous mixture was washed by ether. The aqueous was the adjusted to pH = 6 with 1 M HCl aq. The product was collected by vacum filtration as yellow solids (1.42 g, 48%). HPLC/MS: tR = 7.93 min, m/z = 296.0 [M-H]- HRMS: 297.102519 (found); C14H19NO4S, 297.1035 (calcd.). 1H NMR (600 MHz, CDCl3): 1.57 (9H, s), 1.79-1.82 (4H, m), 2.62-2.63 (2H, m), 2.82-2.83 (2H, m), 10.09 (1H, s). 13C NMR (150 MHz, CDCl3): 22.7, 23.0, 24.2, 26.3, 28.2, 82.3, 109.2, 125.3, 131.7, 151.8, 170.7.
Methyl 2-aminothiophene-3-carboxylate (2b)
Triethylamine (50 mmol, 5.0 mL) was added dropwise to a mixture of 1,4-dithiane-2,5-diol (7.60 g, 50 mmol), methyl cyanoacetate (9.59 g, 100 mmol), and DMF (40 mL). The mixture was stirred at 45 °C for 30 min. After cooled to RT, the reaction mixture was diluted with aqueous acetic acid (0.4 M, 200 mL). The mixture was extracted with ether (4 × 40 mL), and the combined organic layer was washed with water (2 × 40 mL). After dried over sodium sulfate, the organic layer was filtered through silica gel pad. After evaporation, 8.70 g of yellow solids were obtained (yield: 55%). HPLC/MS: tR = 9.01 min; m/z = 158.2 [M+H]+ 1H NMR (600 MHz, CDCl3): 3.83 (3H, s), 5.94 (2H, br.s), 6.20 (1H, d, J = 5.4 Hz), 6.98 (1H, d, J = 6.0 Hz). 13C NMR (150 MHz, CDCl3): 51.0, 106.9, 107.0, 125.8, 162.7, 165.8.
2-(Boc-amino)-3-thiophenecarboxylic acid (3b)
The mixture of 2b (20 mmol, 3.14 g), Boc2O (25 mmol, 5.15 g), DMAP (2 mmol, 244 mg) in 30 mL of THF was stirring under reflux overnight. After evaporation of the solvent, the mixture was quenched by water, and extracted by DCM. The organic layer was dried, and concentrated. The residue was dissolved in 20 mL of ethanol, potassium hydroxide (4.5 g) and 20 mL of water was added. The mixture was reflux for 5 hours. The reaction was quenched by water, the aqueous mixture was washed by ether. The aqueous was the adjusted to pH = 6 with 1 M HCl aq. The product was collected by vacum filtration and further purified by flash chromatography with ethyl acetate, 1.52 g of brown solids were obtained (yield: 31%). HPLC/MS: tR = 10.37 min; m/z = 242.0 [M-H]- HRMS: 243.055945 (found); C10H13NO4S, 243.05653 (calcd.). 1H NMR (600 MHz, CDCl3): 1.58 (9H, s), 6.71 (1H, d, J = 5.4 Hz), 7.23 (1H, d, J = 6.0 Hz), 9.88 (1H, br.s). 13C NMR (150 MHz, d6-DMSO): 28.2, 82.5, 112.5, 115.9, 125.0, 150.1, 151.8, 166.9.
Methyl 2-amino-5-phenylthiophene-3-carboxylate (2c)
Phenyl-acetaldehyde (0.1 mol, 12.0 g), sulfur (0.1 mol, 3.20 g), methyl cyanoacetate (0.1 mol, 8.57 mL) were stirred at RT in absolute ethanol (50 mL). Triethylamine (0.1 mol, 10.0 mL) was added slowly. After the addition was completed, the mixture was heated at reflux for 1 hour. After cooled to RT, the precipitate was collected by vacuum filtration, and washed by cold ethanol. After air dry overnight, 14.6 g of yellow solids were obtained (yield: 63%). HPLC/MS: tR = 11.01 min; m/z = 234.3 [M+H]+ 1H NMR (600 MHz, CDCl3): 3.86 (3H, s), 6.02 (2H, br.s), 7.21-7.26 (2H, m), 7.33-7.46 (2H, m),7.45-7.46 (2H, m). 13C NMR (150 MHz, CDCl3): 51.1, 107.7, 124.7, 125.0, 126.6, 128.8, 133.9, 162.2, 165.8.
2-(Boc-amino)-5-phenylthiophene-3-carboxylic acid (3c)
The mixture of 2c (10 mmol, 2.33 g), Boc2O (15 mmol, 3.10 g), DMAP (1 mmol, 122 mg) in 30 mL of THF was stirring under reflux overnight. After cooling to RT, the mixture was quenched by water, and extracted by DCM. The organic layer was dried, and concentrated. The residue was dissolved in 20 mL of ethanol, potassium hydroxide (2.24 g) and 20 mL of water was added. The mixture was reflux for 1 hour. The reaction was quenched by water, the aqueous mixture was washed by ether. The aqueous was the adjusted to pH = 6 with 1 M HCl aq. The product was collected by vacum filtration as yellow solids (1.50 g, 47%). HPLC/MS: tR = 11.79 min; m/z = 320.1 [M+H]+ HRMS: 319.086854 (found); C16H17NO4S, 319.08783 (calcd.). 1H NMR (600 MHz, CDCl3): 1.60 (9H, s), 7.29-7.31 (1H, m), 7.38- 7.40 (2H, m), 7.45 (1H, s), 7.59-7.60 (2H, m), 9.90 (1H, s). 13C NMR (150 MHz, CDCl3): 28.2, 82.9, 110.9, 119.7, 125.3, 127.4, 129.0, 132.7, 133.5, 152.0, 152.3, 169.1.
Ethyl 2-[[(2-tert-butoxycarbonyl-amino-2,3,4,5,6,7-hexahydro-1-benzothien-3-yl)carbonyl](benzyl)amino]-3-(tert-butyl-amino)-3-oxopropanoate (4a-1)
The mixture of 3a (74.3 mg, 0.25 mmol), benzylamine (0.25 mmol, 27.4 μL), ethyl glyoxylate (0.25 mmol, 49.6 μL), tert-butyl isocyanide (0.25 mmol, 28.3 μL) in 0.5 mL of methanol was stirring under RT for 2 days. The reaction was quenched by water, and extracted by DCM. The organic layer was washed by saturated potassium carbonate (aq), and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (93 mg, yield: 65%). HPLC/MS: tR = 13.21 min; m/z = 572.2 [M+H]+ HRMS: 571.272879 (found); C30H41N3O6S, 571.27161 (calcd.). 1H NMR (600 MHz, CDCl3): 1.24 (3H, t, J = 7.2 Hz), 1.39 (9H, s), 1.55 (9H, s), 1.76-1.82 (4H, m), 2.31 (1H, m), 2.63-2.67 (2H, m), 3.48 (1H, s), 4.09 (2H, m), 4.21 (1H, m), 4.42 (1H, d, J = 15.0 Hz), 4.99 (1H, d, J = 15.0 Hz), 7.27-7.32 (5H, m), 8.19 (1H, s), 9.63 (1H, s). 13C NMR (150 MHz, CDCl3): 13.9, 22.6, 23.4, 23.8, 24.0, 28.3, 28.5, 52.1, 55.1, 61.4, 62.1, 80.7, 115.1, 126.6, 128.0, 128.4, 128.9, 129.4, 135.8, 137.2, 153.0, 164.6, 168.5, 169.0.
Method A for the synthesis of 5a
3,4,6,7,8,9-Hexahydro-3-(N-tert-butyl)-carboxamide-4-benzyl-1H-benzothieno-[2,3-e] -1,4-diazepine-2,5-dione (5a-1)
The mixture of 3a (74.3 mg, 0.25 mmol), benzylamine (0.25 mmol, 27.4 μL), ethyl glyoxylate (0.25 mmol, 49.6 μL), tert-butyl isocyanide (0.25 mmol, 28.3 μL) in 0.5 mL of methanol was stirring under RT for 2 days. The reaction was quenched by water, and extracted by DCM. The organic layer was washed by saturated potassium carbonate (aq), and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by 0.5 mL of TFA, stirring under RT overnight. The reaction was added 10 mL of DCM, then neutralized by saturated potassium carbonate (aq). The mixture was extracted by DCM, the organic layer was combined and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by triethylamine (50 μL) and TBD (10 mg) in 0.5 mL of THF, stirring overnight under 40 °C. 5a-1 was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (35 mg, yield: 33% over three steps). HPLC/MS: tR = 10.95 min, m/z = 426.2 [M+H]+ HRMS: 425.176977 (found); C23H27N3O3S, 425.17731 (calcd.). 1H NMR (600 MHz, CDCl3): 0.86 (9H, s), 1.61 (1H, m), 1.79 (1H, m), 1.86 (1H, m), 1.94 (1H, m), 2.42 (1H, m), 2.60 (2H, m), 3.12 (1H, m), 3.99 (1H, d, J = 14.4 Hz), 4.62 (1H, s), 4.81 (1H, s), 5.63 (1H, d, J = 13.8 Hz), 7.39-7.43 (3H, m), 7.56-7.57 (2H, m). 13C NMR (150 MHz, CDCl3): 22.3, 23.0, 24.5, 26.1, 27.8, 51.3, 52.3, 67.1, 122.2, 128.7, 129.2, 129.6, 129.7, 134.1, 137.1, 104.8, 163.1, 163.7, 168.4.
3,4,6,7,8,9-Hexahydro-3-(N-tert-butyl)-carboxamide-4-phenethyl-1H-benzothieno-[2,3-e]-1,4-diazepine-2,5-dione (5a-2)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (58 mg, yield: 53% over three steps). HPLC/MS: tR = 10.93 min, m/z = 440.2 [M+H]+ HRMS: 439.193180 (found); C24H29N3O3S, 439.19296 (calcd.). 1H NMR (600 MHz, CDCl3): 1.05 (9H, s), 1.61 (1H, m), 1.77 (1H, m), 1.83-1.90 (2H, m), 2.39 (1H, m), 2.58-2.59 (2H, m), 2.96-3.00 (1H, m), 3.01-3.07 (2H, m), 3.95-3.99 (2H, m), 4.60 (1H, s), 5.39 (1H, s), 7.22 (1H, m), 7.28-7.31 (4H, m). 13C NMR (150 MHz, CDCl3): 22.3, 23.0, 24.5, 25.8, 28.0, 34.1, 50.3, 51.8, 68.2, 123.2, 126.8, 128.7, 128.8, 129.9, 134.0, 137.6, 139.7, 163.5, 163.6, 168.9.
3,4,6,7,8,9-Hexahydro-3-(N-tert-butyl)-carboxamide-4-(4-chlorobenzyl)-1H-benzothieno-[2,3-e]-1,4-diazepine-2,5-dione (5a-3)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (32 mg, yield: 28% over three steps). HPLC/MS: tR = 11.32 min, m/z = 460.0 [M+H]+ HRMS: 459.136130 (found); C23H26ClN3O3S, 459.13834 (calcd.). 1H NMR (600 MHz, CDCl3): 0.92 (9H, s), 1.61 (1H, m), 1.78 (1H, m), 1.86 (1H, m), 1.93 (1H, m), 2.40 (1H, m), 2.58-2.60 (2H, m), 3.09 (1H, s), 4.19 (1H, d, J = 14.4 Hz), 4.58 (1H, s), 4.86 (1H, s), 5.37 (1H, d, J = 13.2 Hz), 7.38 (1H, d, J = 7.8 Hz), 7.49 (1H, d, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): 22.3, 22.9, 24.5, 26.0, 27.9, 51.6, 51.9, 67.3, 122.3, 129.6, 129.9, 130.5, 134.1, 134.6, 135.4, 140.3, 163.2, 163.3, 168.4.
3,4,6,7,8,9-Hexahydro-3-(N-cyclopropylmethyl)-carboxamide-4-(4-chlorophenyl)-1H-benzothieno-[2,3-e]-1,4-diazepine-2,5-dione (5a-4)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (18 mg, yield: 16% over three steps). HPLC/MS: tR = 11.10 min, m/z = 446.0 [M+H]+ HRMS: 445.121614 (found); C22H24ClN3O3S, 445.12269 (calcd.). 1H NMR (600 MHz, CDCl3): 1.16 (9H, s), 1.61 (1H, m), 1.78 (1H, m), 1.86-1.88 (2H, m), 2.47 (1H, m), 2.61 (2H, m), 3.00 (1H, m), 4.88 (1H, s), 5.01 (1H, s), 7.33 (2H, d, J = 7.2 Hz), 7.39 (2H, d, J = 6.6 Hz).13C NMR (150 MHz, CDCl3): 22.3, 23.0, 24.6, 25.9, 28.2, 29.7, 52.3, 70.3, 123.4, 127.7, 129.7, 130.2, 133.3, 135.0, 141.3, 162.7, 168.4, 170.7.
3,4,6,7,8,9-Hexahydro-3-(N-cyclopropylmethyl)-carboxamide-4-benzyl-1H-benzothieno-[2,3-e]-1,4-diazepine-2,5-dione (5a-5)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (27 mg, yield: 26% over three steps). HPLC/MS: tR = 10.34 min, m/z = 424.3 [M+H]+ HRMS: 423.161189 (found); C23H25N3O3S, 423.16166 (calcd.). 1H NMR (600 MHz, CDCl3): -0.10 (2H, m), 0.32 (2H, m), 0.43 (1H, m), 1.76 (1H, m), 1.83 (1H, m), 1.89 (1H, m), 2.45 (1H, m), 2.51 (1H, m), 2.58-2.59 (2H, m), 2.77 (1H, m), 3.07 (1H, m), 4.22 (1H, d, J = 14.4 Hz), 4.63 (1H, s), 5.28 (1H, s), 5.45 (1H, d, J = 14.4 Hz), 7.35 (1H, m), 7.39-7.41 (2H, m), 7.53-7.54 (2H, m). 13C NMR (150 MHz, CDCl3): 3.29, 3.33, 10.1, 22.3, 23.0, 24.5, 25.9, 44.5, 52.4, 66.4, 122.1, 128.68, 128.74, 129.0, 129.49, 129.52, 134.3, 136.8, 140.5, 163.3, 164.2, 168.2.
3,4,6,7,8,9-Hexahydro-3-(N-mesityl)-carboxamide-4-(4-chlorobenzyl)-1H-benzothieno-[2,3-e]-1,4-diazepine-2,5-dione (5a-6)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (15 mg, yield: 12% over three steps). HPLC/MS: tR = 11.63 min, m/z = 522.2 [M+H]+ HRMS: 521.153301 (found); C28H28ClN3O3S, 521.15399 (calcd.). 1H NMR (600 MHz, CDCl3): 1.26 (1H, m), 1.58 (6H, s), 1.77 (1H, m), 1.87-1.89 (2H, m), 2.17 (3H, m), 2.50-2.61 (3H, m), 3.08 (1H, m), 4.27 (1H, d, J = 14.4 Hz), 4.83 (1H, s), 5.35 (1H, d, J = 14.4 Hz), 6.58 (1H, s), 6.72 (2H, s), 7.32 (1H, d, J = 7.8 Hz), 7.45 (1H, d, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): 17.1, 20.8, 22.2, 22.9, 24.6, 26.1, 52.3, 67.0, 121.8, 128.9, 129.4, 129.6, 129.7, 130.5, 134.6, 134.7, 135.3, 137.4, 162.7, 163.5, 167.7.
3,4,6,7,8,9-Hexahydro-3-(N-benzyl)-carboxamide-4-(4-chlorobenzyl)-1H-benzothieno-[2,3-e]-1,4-diazepine-2,5-dione (5a-7)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (20 mg, yield: 16% over three steps). HPLC/MS: tR = 11.04 min, m/z = 494.0 [M+H]+ HRMS: 493.122043 (found); C26H24ClN3O3S, 493.12269 (calcd.). 1H NMR (600 MHz, CDCl3): 1.60 (1H, m), 1.77 (1H, m), 1.87 (2H, m), 2.29 (1H, m), 2.59 (1H, m), 2.65 (1H, m), 3.01 (1H, m), 3.94 (1H, m), 4.07 (1H, m), 4.29 (1H, d, J = 14.4 Hz), 4.63 (1H, s), 5.22 (1H, d, J = 14.4 Hz), 5.47 (1H, s), 6.86 (2H, m), 7.20-7.21 (2H, m), 7.26-7.29 (3H, m), 7.34-7.36 (2H, m). 13C NMR (150 MHz, CDCl3): 22.2, 22.9, 24.6, 25.7, 44.1, 51.9, 66.6, 122.1, 127.9, 128.7, 129.5, 130.2, 134.6, 135.0, 136.6, 139.8, 163.3, 163.9, 167.6.
3,4,6,7,8,9-Hexahydro-3-(N-3,4-dichlorobenzyl)-carboxamide-4-(4-chlorobenzyl)-1H-benzothieno-[2,3-e]-1,4-diazepine-2,5-dione (5a-8)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (18 mg, yield: 16% over three steps). HPLC/MS: tR = 11.72 min; m/z = 561.9 [M+H]+ HRMS: 561.044289 (found); C26H22Cl3N3O3S, 561.04475 (calcd.). 1H NMR (600 MHz, CD3OD): 1.35-1.36 (1H, m), 1.66-1.77 (3H, m), 2.12-2.15 (1H, m), 2.51-2.54 (2H, m), 2.87 (1H, m), 4.00 (1H, d, J = 14.4 Hz), 4.27 (1H, d, J = 14.4 Hz), 4.34 (1H, d, J = 15.0 Hz), 7.43 (1H, d, J = 15.0 Hz), 6.98 (1H, m), 7.31-7.33 (5H, m), 7.37 (1H, m). 13C NMR (150 MHz, CD3OD): 21.8, 22.6, 23.8, 25.0, 40.1, 42.0, 51.8, 121.5, 127.8, 128.4, 128.6, 129.6, 130.1, 130.2, 130.7, 131.6, 133.3, 133.9, 135.4, 139.4, 164.1, 165.8, 167.8.
Method B for the synthesis of 5b
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]thien-3-yl}carbonyl)(phenethyl)amino]-3-(tert-butyl-amino)-3-oxopropanoate (4b-1)
The mixture of 3b (74.3 mg, 0.25 mmol), 2-phenylethylamine (0.25 mmol, 31.4 μL), ethyl glyoxylate (0.25 mmol, 49.6 μL), tert-butyl isocyanide (0.25 mmol, 28.3 μL) in 0.5 mL of methanol was stirring under RT for 2 days. 4b-1 was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (74 mg, yield: 56%). HPLC/MS: tR = 12.57 min; m/z = 532.2 [M+H]+ HRMS: 531.238491 (found); C27H37N3O6S, 531.24031 (calcd.). 1H NMR (600 MHz, CDCl3): 1.30 (3H, t, J = 7.2 Hz), 1.41 (9H, s), 1.50 (9H, s), 3.00 (2H, m), 3.81 (2H, m), 4.25-4.31 (2H, m), 4.49 (1H, s), 6.75 (1H, d, J = 5.4 Hz), 6.87 (1H, d, J = 6.0 Hz), 7.14-7.15 (2H, m), 7.20-7.23 (1H, m), 7.26-7.29 (2H, m), 7.84 (1H, br.s), 9.40 (1H, br.s). 13C NMR (150 MHz, CDCl3): 14.0, 28.2, 28.5, 35.4, 51.7, 62.3, 64.7, 73.0, 81.6, 114.1, 115.4, 122.7, 126.7, 128.6, 128.7, 137.8, 146.6, 152.3, 164.3, 168.4, 168.7.
3,4-Dihydro-3-(N-tert-butyl)-carboxamide-4-phenethyl-1H-thieno[2,3-e]-1,4-diazepine-2,5-dione (5b-1)
The mixture of 4b-1 and 0.5 mL of DCM (10% TFA) was stirring under RT overnight. The reaction was added 10 mL of DCM, then neutralized by saturated potassium carbonate (aq). The mixture was extracted by DCM, the organic layer was combined and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by triethylamine (50 μL) and TBD (10 mg) in 0.5 mL of THF, stirring overnight under 40 °C. 5b-1 was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (12 mg, yield: 22% over two steps). HPLC/MS: tR = 9.79 min; m/z = 386.2 [M+H]+ HRMS: 385.145394 (found); C20H23N3O3S, 385.14601 (calcd.). 1H NMR (600 MHz, CDCl3): 1.05 (9H, s), 2.96-3.11 (2H, m), 3.92-4.05 (2H, m), 4.67 (1H, s), 5.35 (1H, br.s), 6.85 (1H, d, J = 5.4 Hz), 7.21- 7.24 (2H, m), 7.28-7.33 (4H, m). 13C NMR (150 MHz, CDCl3): 28.1, 34.3, 51.0, 52.0, 68.4, 118.2, 124.6, 126.8, 127.6, 128.77, 128.79, 137.5, 163.5, 168.5.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]thien-3-yl}carbonyl)(2-(thiophen-3-yl)ethyl) amino]-3-(tert-butyl-amino)-3-oxopropanoate (4b-2)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (60 mg, yield: 45%). HPLC/MS: tR = 12.44 min; m/z = 538.2 [M+H]+ HRMS: 537.194766 (found); C25H35N3O6S2, 537.19673 (calcd.). 1H NMR (600 MHz, CDCl3): 1.30 (3H, t, J = 7.2 Hz), 1.41 (9H, s), 1.51 (9H, s), 3.24 (2H, m), 3.82-3.87 (2H, m), 4.25-4.32 (2H, m), 4.46 (1H, s), 6.75 (1H, m), 6.82 (1H, m), 6.86 (1H, m), 6.92 (1H, m), 7.15 (1H, m), 7.85 (1H, br.s), 9.44 (1H, br.s). 13C NMR (150 MHz, CDCl3): 14.0, 28.2, 28.5, 29.5, 51.7, 62.3, 64.6, 72.9, 81.6, 113.9, 115.5, 122.6, 124.1, 125.5, 127.1, 139.9, 146.9, 152.3, 164.1, 168.3, 168.8.
3,4-Dihydro-3-(N-tert-butyl)-carboxamide-4-(2-(thiophen-3-yl)ethyl)-1H-thieno[2,3-e] -1,4-diazepine-2,5-dione (5b-2)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (19 mg, yield: 43% over two steps). HPLC/MS: tR = 9.64 min; m/z = 392.1 [M+H]+ HRMS: 391.101427 (found); C18H21N3O3S2, 391.10243 (calcd.). 1H NMR (600 MHz, CDCl3): 1.04 (9H, s), 3.21-3.31 (2H, m), 3.99-4.02 (2H, m), 4.68 (1H, s), 5.40 (1H, br.s), 6.83 (1H, m), 6.93 (1H, m), 6.96 (1H, m), 7.18 (1H, m), 7.20 (1H, m). 13C NMR (150 MHz, CDCl3): 28.0, 28.5, 51.0, 52.0, 68.5, 118.1, 124.4, 125.8, 127.2, 127.5, 140.0, 163.5, 163.7, 168.6.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]thien-3-yl}carbonyl)(phenyl)amino]-3-(cyclohexyl-amino)-3-oxopropanoate (4b-3)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (70 mg, yield: 52%). HPLC/MS: tR = 12.75 min; m/z = 544.0 [M+H]+ HRMS: 543.238512 (found); C28H37N3O6S, 543.24031 (calcd.). 1H NMR (600 MHz, CDCl3): 1.27 (2H, m), 1.30 (3H, t, J = 7.2 Hz), 1.37-1.38 (1H, m), 1.39-1.41 (2H, m), 1.52 (9H, s), 1.58 (1H, m), 1.68-1.70 (2H, m), 1.93 (2H, m), 3.83 (1H, s), 4.21-4.24 (2H, m), 4.28-4.29 (1H, m), 4.81 (1H, d, J = 17.4 Hz), 5.08 (1H, d, J = 16.8 Hz), 6.60 (1H, d, J = 6.0 Hz), 6.87 (1H, d, J = 5.4 Hz), 7.32-7.35 (1H, m), 7.39-7.42 (2H, m), 7.51-7.52 (2H, m), 7.92 (1H, br.s), 9.79 (1H, br.s). 13C NMR (150 MHz, CDCl3): 14.0, 24.5, 25.6, 28.2, 32.4, 32.5, 48.4, 62.2, 63.4, 81.8, 112.9, 115.0, 122.8, 127.2, 127.9, 128.9, 135.9, 149.4, 152.3, 164.3, 168.3, 169.0.
3,4-Dihydro-3-(N-cyclohexyl)-carboxamide-4-benzyl-1H-thieno[2,3-e]-1,4-diazepine-2,5-dione (5b-3)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (10 mg, yield: 20% over two steps). HPLC/MS: tR = 10.01 min; m/z = 398.1 [M+H]+ HRMS: 397.145096 (found); C21H23N3O3S, 397.14601 (calcd.). 1H NMR (600 MHz, CDCl3): 0.49 (1H, m), 0.59 (1H, m), 0.98-1.02 (1H, m), 1.09-1.11 (2H, m), 1.24-1.27 (2H, m), 1.46 (3H, m), 3.25 (1H, m), 4.07 (1H, d, J = 13.8 Hz), 4.72 (1H, s), 4.84 (1H, br.s), 5.59 (1H, d, J = 14.4 Hz), 6.84 (1H, d, J = 5.4 Hz), 7.20 (1H, d, J = 5.4 Hz), 7.40-7.44 (3H, m), 7.59-7.60 (2H, m). 13C NMR (150 MHz, CDCl3): 24.4, 24.5, 25.2, 31.9, 32.2, 48.1, 52.8, 66.5, 118.0, 123.6, 127.8, 128.9, 129.4, 129.7, 136.7, 163.1, 163.3, 167.7.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]thien-3-yl}carbonyl)(3,4-dimethoxy phenethyl)amino]-3-(cyclopropylmethyl-amino)-3-oxopropanoate (4b-4)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (53 mg, yield: 36%). HPLC/MS: tR = 11.70 min; m/z = 590.2 [M+H]+ HRMS: 589.248129 (found); C29H39N3O8S, 589.24579 (calcd.). 1H NMR (600 MHz, CDCl3): 0.24-0.26 (2H, m), 0.53-0.54 (2H, m), 1.01 (1H, m), 1.30 (3H, t, J = 7.2 Hz), 1.51 (9H, s), 2.95-2.99 (2H, m), 3.17-3.26 (2H, m), 3.84 (3H, s), 3.85 (3H, s), 4.25-4.34 (2H, m), 4.56 (1H, s), 6.65 (1H, m), 6.68-6.70 (1H, m), 6.75-6.78 (2H, m), 6.92 (1H, d, J = 5.4 Hz), 7.98 (1H, br.s), 9.40 (1H, br.s). 13C NMR (150 MHz, CDCl3): 3.3, 3.4, 10.5, 14.0, 28.2, 34.9, 44.6, 55.86, 55.90, 62.3, 64.2, 81.9, 111.3, 111.8, 113.8, 115.3, 120.6, 122.8, 130.2, 147.5, 147.8, 149.0, 152.3, 165.4, 168.4.
3,4-Dihydro-3-(N-cyclopropylmethyl)-carboxamide-4-(3,4-dimethoxyphenethyl)-1H-thieno[2,3-e]-1,4-diazepine-2,5-dione (5b-4)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (10 mg, yield: 38% over two steps). HPLC/MS: tR = 8.98 min; m/z = 444.0 [M+H]+ HRMS: 443.151569 (found); C22H25N3O5S, 443.15149 (calcd.). 1H NMR (600 MHz, CDCl3): -0.03 (2H, m), 0.35 (2H, m), 0.54 (1H, m), 2.65 (1H, m), 2.94-3.06 (3H, m), 3.85 (3H, s), 3.88 (3H, s), 3.96-4.11 (2H, m), 4.69 (1H, s), 5.60 (1H, br.s), 6.80-6.85 (4H, m), 7.17 (1H, d, J = 5.4 Hz). 13C NMR (150 MHz, CDCl3): 3.35, 3.41, 10.4, 33.6, 44.7, 55.88, 55.92, 111.4, 111.9, 113.9, 118.2, 120.8, 127.7, 129.7, 141.7, 147.8, 149.0, 163.4, 163.9, 167.6.
Method C for the synthesis of 5c
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]-5-phenylthien-3-yl}carbonyl) (cyclopropyl methyl)amino]-3-(tert-butyl-amino)-3-oxopropanoate (4c-1)
The mixture of 3c (0.2 mmol, 63.8 mg), cyclopropyl methamine (0.2 mmol, 17.3 μL), ethyl glyoxylate (0.2 mmol, 39.7 μL), tert-butyl isocyanide (0.2 mmol, 22.6 μL) in 0.5 mL of methanol was stirring under RT for 2 days. 4c-1 was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (46 mg, yield: 41%). HPLC/MS: tR = 13.10 min; m/z = 558.2 [M+H]+ HRMS: 557.255949 (found); C29H39N3O6S, 557.25596 (calcd.). 1H NMR (600 MHz, CDCl3): 0.19-0.28 (2H, m), 0.60-0.65 (2H, m), 1.10-1.12 (1H, m), 1.31 (3H, t, J = 7.2 Hz), 1.41 (9H, s), 1.53 (9H, s), 3.38-3.42 (1H, m), 3.69-3.72 (1H, m), 4.29 (2H, q, J = 7.2 Hz), 4.65 (1H, s), 7.19 (1H, s), 7.25-7.27 (1H, m), 7.36-7.38 (2H, m), 7.56-7.57 (2H, m), 8.06 (1H, s), 9.53 (1H, s). 13C NMR (150 MHz, CDCl3): 3.4, 4.5, 10.0, 14.0, 28.2, 28.6, 51.6, 62.2, 63.4, 81.8, 115.2, 118.7, 125.2, 127.2, 129.0, 133.3, 133.9, 145.6, 152.4, 164.7, 167.8, 169.1.
7-Phenyl-3,4-dihydro-3-(N-cyclopropylmethyl)-carboxamide-4-cyclopropylmethyl-1H-thieno[2,3-e]-1,4-diazepine-2,5-dione (5c-1)
The mixture of 4c-1 and 0.5 mL of DCM (10% TFA) was stirring under RT overnight. After the evaporation of the solvent, the residue was treated by triethylamine (100 μL) and TBD (10 mg) in 0.5 mL of THF, stirring overnight under 40 °C. 5c-1 was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (12 mg, yield: 35% over two steps). HPLC/MS: tR = 10.45 min; m/z = 412.2 [M+H]+ HRMS: 411.161429 (found); C22H25N3O3S, 411.16166 (calcd.). 1H NMR (600 MHz, CDCl3): 0.47 (1H, m), 0.54 (1H, m), 0.62 (1H, m), 0.67 (1H, m), 1.15 (9H, s), 1.25 (1H, m), 3.33 (1H, m), 3.90 (1H, m), 4.81 (1H, s), 5.91 (1H, s), 7.29 (1H, m), 7.35-7.38 (2H, m), 7.43 (1H, s), 7.48-7.49 (2H, m). 13C NMR (150 MHz, CDCl3): 3.7, 4.4, 10.2, 28.2, 52.1, 53.7, 67.9, 122.6, 124.9, 125.5, 128.1, 129.1, 132.8, 136.7, 141.7, 163.2, 164.1, 168.2.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]-5-phenylthien-3-yl}carbonyl)(2-methoxyethyl)amino]-3-(tert-butyl-amino)-3-oxopropanoate (4c-2)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellow oil (55 mg, yield: 49%). HPLC/MS: tR = 12.81 min; m/z = 562.1 [M+H]+ HRMS: 561.250182 (found); C28H39N3O7S, 561.25087 (calcd.). 1H NMR (600 MHz, CDCl3): 1.31 (3H, t, J = 7.2 Hz), 1.41 (9H, s), 1.52 (9H, s), 3.36 (3H, s), 3.65-3.71 (2H, m), 3.81 (2H, m), 4.27 (2H, m), 4.72 (1H, s), 7.21 (1H, s), 7.23-7.26 (1H, m), 7.34-7.36 (2H, m), 7.53-7.55 (2H, m), 7.93 (1H, br.s), 9.54 (1H, br.s). 13C NMR (150 MHz, CDCl3): 14.1, 28.2, 28.5, 51.6, 59.0, 62.1, 64.5, 71.2, 81.8, 115.1, 119.0, 125.1, 127.2, 128.9, 133.2, 133.9, 145.6, 152.4, 164.3, 168.2, 168.9.
7-Phenyl-3,4-dihydro-3-(N-tert-butyl)-carboxamide-4-(2-methoxyethyl)-1H-thieno [2,3-e]-1,4-diazepine-2,5-dione (5c-2)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (11 mg, yield: 27% over two steps). HPLC/MS: tR = 10.12 min; m/z = 416.0 [M+H]+ HRMS: 415.155023 (found); C21H25N3O4S, 415.15658 (calcd.). 1H NMR (600 MHz, CDCl3): 1.11 (9H, s), 3.20-3.24 (1H, m), 3.43 (3H, s), 3.63-3.66 (1H, m), 3.89-3.91 (1H, m), 4.53-4.57 (1H, m), 4.71 (1H, s), 6.91 (1H, s), 7.28-7.29 (2H, m), 7.33-7.35 (2H, m), 7.38 (1H, s), 7.45-7.47 (2H, m). 13C NMR (150 MHz, CDCl3): 28.2, 48.9, 51.4, 58.7, 66.4, 68.4, 122.5, 124.6, 125.5, 127.9, 129.0, 133.1, 136.2, 163.8, 164.7, 168.7.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]-5-phenylthien-3-yl}carbonyl)(phenethyl) amino]-3-(tert-butyl-amino)-3-oxopropanoate (4c-3)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (63 mg, yield: 52%). HPLC/MS: tR = 13.43 min; m/z = 608.1 [M+H]+ HRMS: 607.270091 (found); C33H41N3O6S, 607.27161 (calcd.). 1H NMR (600 MHz, CDCl3): 1.34 (3H, t, J = 7.2 Hz), 1.44 (9H, s), 1.53 (9H, s), 3.06 (2H, m), 3.89 (2H, m), 4.28-4.34 (2H, m), 4.53 (1H, s), 7.10 (1H, s), 7.19-7.20 (2H, m), 7.22-7.29 (4H, m), 7.35-7.37 (2H, m), 7.50-7.51 (2H, m), 7.88 (1H, br.s), 9.48 (1H, br.s).13C NMR (150 MHz, CDCl3): 14.1, 28.2, 28.6, 35.4, 51.7, 62.4, 64.7, 81.9, 114.9, 118.4, 125.2, 126.8, 127.2, 128.7, 128.8, 128.9, 133.3, 133.8, 137.9, 145.8, 152.3, 168.2, 168.8.
7-Phenyl-3,4-dihydro-3-(N-tert-butyl)-carboxamide-4-phenethyl-1H-thieno[2,3-e]-1,4-diazepine-2,5-dione (5c-3)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (18 mg, yield: 38% over two steps). HPLC/MS: tR = 10.89 min; m/z = 462.0 [M+H]+ HRMS: 461.175973 (found); C26H27N3O3S, 461.17731 (calcd.). 1H NMR (600 MHz, CDCl3): 1.04 (9H, s), 2.95-3.10 (2H, m), 3.92-4.03 (2H, m), 4,71 (1H, s), 5.45 (1H, br.s), 7.19-7.21 (1H, m), 7.26-7.28 (5H, m), 7.30-7.32 (2H, m), 7.40-7.44 (3H, m). 13C NMR (150 MHz, CDCl3): 28.1, 34.3, 50.9, 52.1, 68.5, 122.5, 125.1, 125.5, 126.8, 128.0, 128.8, 129.0, 132.9, 137.5, 163.6, 164.0, 168.8.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]-5-phenylthien-3-yl}carbonyl)(2-methoxyethyl)amino]-3-(cyclopropylmethyl-amino)-3-oxopropanoate (4c-4)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (42 mg, yield: 38%). HPLC/MS: tR = 12.47 min; m/z = 560.1 [M+H]+ HRMS: 559.236369 (found); C28H37N3O7S, 559.23522 (calcd.). 1H NMR (600 MHz, CDCl3): 0.27-0.29 (2H, m), 0.54-0.55 (2H, m), 1.04 (1H, m), 1.32 (3H, t, J = 7.2 Hz), 1.54 (9H, s), 3.22-3.28 (2H, m), 3.39 (3H, s), 3.70-3.75 (2H, m), 3.83-3.92 (2H, m), 4.27-4.32 (2H, m), 4.84 (1H, s), 7.24-7.28 (2H, m), 7.35-7.38 (2H, m), 7.54-7.55 (2H, m), 8.18 (1H, s), 9.51 (1H, s). 13C NMR (150 MHz, CDCl3): 3.37, 3.40, 10.5, 14.1, 28.2, 39.1, 44.5, 59.1, 62.2, 71.2, 82.0, 114.7, 119.1, 125.2, 127.2, 128.4, 128.9, 133.1, 133.9, 152.3, 165.3, 168.3, 168.7.
7-Phenyl-3,4-dihydro-3-(N-cyclopropylmethyl)-carboxamide-4-(2-methoxyethyl)-1H-thieno[2,3-e]-1,4-diazepine-2,5-dione (5c-4)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (9 mg, yield: 29% over two steps). HPLC/MS: tR = 9.69 min; m/z = 414.3 [M+H]+ HRMS: 413.140096 (found); C21H23N3O4S, 413.14093 (calcd.). 1H NMR (600 MHz, CDCl3): 0.01-0.05 (2H, m), 0.30-0.38 (2H, m), 0.69-0.71 (1H, m), 2.84-2.86 (1H, m), 2.94-2.98 (1H, m), 3.29-3.32 (1H, m), 3.45 (3H, s), 3.65-3.67 (1H, m), 3.94 (1H, m), 4.51-4.54 (1H, m), 4.80 (1H, s), 7.24-7.27 (1H, m), 7.30-7.32 (2H, m), 7.38 (2H, m), 7.43-7.45 (2H, m). 13C NMR (150 MHz, CDCl3): 3.37, 3.39, 10.5, 44.6, 49.1, 58.7, 67.9, 69.5, 122.4, 124.4, 125.4, 127.9, 129.0, 132.9, 136.2, 142.8, 163.8, 165.4, 168.3.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]-5-phenylthien-3-yl}carbonyl)(2-methoxyethyl)amino]-3-(cyclohexyl-amino)-3-oxopropanoate (4c-5)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (82 mg, yield: 70%). HPLC/MS: tR = 13.04 min; m/z = 588.3 [M+H]+ HRMS: 587.265023 (found); C30H41N3O7S, 587.26652 (calcd.). 1H NMR (600 MHz, CDCl3): 1.21- 1.25 (3H, m), 1.29 (3H, t, J = 7.2 Hz), 1.35-1.42 (2H, m), 1.52 (9H, s), 1.58-1.60 (2H, m), 1.71 (2H, m), 1.94-1.96 (2H, m), 3.36 (3H, s), 3.67 (1H, m), 3.73 (1H, m), 3.80-3.82 (3H, m), 4.27 (2H, m), 4.80 (1H, s), 7.21-7.25 (2H, m), 7.33-7.35 (2H, m), 7.52-7.53 (2H, m), 7.97 (1H, d, J = 7.2 Hz), 9.47 (1H, s). 13C NMR (150 MHz, CDCl3): 14.1, 24.6, 25.6, 28.2, 32.5, 32.6, 48.6, 59.0, 62.1, 64.3, 71.0, 81.9, 114.9, 119.0, 125.1, 127.2, 128.9, 133.2, 133.9, 146.2, 152.3, 164.2, 168.4, 168.5.
7-Phenyl-3,4-dihydro-3-(N-cyclohexyl)-carboxamide-4-(2-methoxyethyl)-1H-thieno [2,3-e]-1,4-diazepine-2,5-dione (5c-5)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (12 mg, yield: 19% over two steps). HPLC/MS: tR = 10.44 min; m/z = 442.1 [M+H]+ HRMS: 441.171348 (found); C23H27N3O4S, 441.17223 (calcd.). 1H NMR (600 MHz, CDCl3): 0.85-0.87 (1H, m), 1.06-1.16 (3H, m), 1.27-1.28 (1H, m), 1.44-1.51 (2H, m), 1.64-1.66 (1H, m), 1.76-1.77 (1H, m), 3.27-3.28 (1H, m), 3.45 (3H, s), 3.54-3.56 (1H, m), 3.64-3.66 (1H, m), 3.96 (1H, m), 4.54-4.57 (1H, m), 4.76 (1H, s), 7.07 (1H, d, J = 7.2 Hz), 7.28-7.30 (1H, m), 7.33- 7.36 (2H, m), 7.45-7.46 (2H, m). 13C NMR (150 MHz, CDCl3): 24.5, 24.7, 25.4, 32.4, 32.7, 48.3, 49.2, 58.7, 67.9, 69.5, 122.6, 124.5, 125.5, 127.9, 129.0, 133.0, 136.3, 151.6, 163.7, 164.5, 168.3.
Ethyl 2-[({2-[(tert-butoxycarbonyl)amino]-5-phenylthien-3-yl}carbonyl)(2-cyclopropylmethyl)amino]-3-(cyclohexyl-amino)-3-oxopropanoate (4c-6)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1) as yellowish solids (70 mg, yield: 60%). HPLC/MS: tR = 13.36 min; m/z = 584.2 [M+H]+ HRMS: 583.271707 (found); C31H41N3O6S, 583.27161 (calcd.). 1H NMR (600 MHz, CDCl3): 0.19-0.24 (2H, m), 0.62-0.63 (2H, m), 1.11 (1H, m), 1.25 (1H, m), 1.29 (3H, t, J = 7.2 Hz), 1.31-1.34 (2H, m), 1.38-1.42 (2H, m), 1.52 (9H, s), 1.55-1.59 (1H, m), 1.70 (2H, m), 1.93 (2H, m), 3.42-3.44 (1H, m), 3.67-3.70 (1H, m), 3.81-3.87 (1H, m), 4.28 (2H, q, J = 7.2 Hz), 4.74 (1H, s), 7.19 (1H, s), 7.24-7.27 (1H, m), 7.35-7.37 (2H, m), 7.54-7.56 (2H, m), 8.05 (1H, d, J = 7.2 Hz), 9.46 (1H, br.s). 13C NMR (150 MHz, CDCl3): 3.6, 4.5, 10.0, 14.1, 24.5, 25.6, 28.2, 32.4, 32.6, 48.5, 55.5, 62.2, 63.2, 82.0, 115.0, 118.8, 125.2, 127.3, 129.0, 133.3, 133.9, 146.3, 152.3, 165.0, 168.0, 168.7.
7-Phenyl-3,4-dihydro-3-(N-cyclohexyl)-carboxamide-4-cyclopropylmethyl-1H-thieno[2,3-e]-1,4-diazepine-2,5-dione (5c-6)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (13 mg, yield: 25% over two steps). HPLC/MS: tR = 10.67 min; m/z = 438.2 [M+H]+ HRMS: 437.176794 (found); C24H27N3O3S, 437.17731 (calcd.). 1H NMR (600 MHz, CDCl3): 0.46-0.53 (2H, m), 0.60-0.63 (2H, m), 0.87-0.91 (1H, m), 1.05-1.10 (2H, m), 1.15-1.32 (4H, m), 1.46-1.50 (2H, m), 1.61-1.63 (1H, m), 1.72-1.74 (1H, m), 3.31-3.34 (1H, m), 3.60-3.62 (1H, m), 3.88-3.92 (1H, m), 4.86 (1H, s), 6.02 (1H, d, J = 7.2 Hz), 7.29 (1H, m), 7.33-7.35 (2H, m), 7.39 (1H, s), 7.45-7.46 (2H, m). 13C NMR (150 MHz, CDCl3): 3.7, 4.4, 10.3, 24.5, 24.7, 25.3, 32.5, 32.6, 48.5, 53.8, 67.6, 122.8, 124.4, 125.4, 127.9, 129.0, 133.0, 136.2, 163.4, 164.3, 168.2.
NMR and FP binding assays
The Human recombinant Mdm2 (residues 1-118) and p53 (residues 1-321) were expressed and purified as described in Bista et al., 2009 (18). Uniform 15N labeling was achieved by growing the Escherichia coli BL21(DE3) RIL in M9 minimal medium containing 15NH4Cl as nitrogen source (19). The SEI-AIDA experiment was performed according to Bista et al., 2009 (18). 14 μM Mdm2/p53 complex was mixed with compounds in 1:1 molar ratio; amount of p53 released from the complex by the compound was estimated from 1D spectrum and Kd of Mdm2-inhibitor interaction was calculated according to Krajewski et al., 2007 (20). Fluorescence polarization (FP) binding assays were performed according to Czarna et al., 2009 (21). Binding constant and inhibition curves were fitted using the SigmaPlot (SPSS Science Software).
1H-15N HSQC spectra were acquired using the fast-HSQC pulse sequence (Mori et al., 1995) (22). For the 1H-15N HSQC spectrum, a total of 2048 complex points in t2 and 192 t1 increments were acquired. NMR data were processed using the Bruker program Xwin-NMR version 3.5. Titration experiments were performed using a series of 1H-15N HSQC of labeled Mdm2 or unlabeled Mdm2 along with the unlabelled partner. 10% D2O was added to NMR samples to provide lock signal. 100mM stock solutions of the compounds were prepared by dissolving them in perdeuterated DMSO. For HSQC titration, 0.2 mM 15N-labeled Mdm2 was mixed with compounds in 1:1 and 1:5 protein: compound molar ratio. Normalized chemical shift perturbations were calculated according to equation (Stoll et al., 2001) (23):
Virtual Library Generation
The virtual library of Gewald-UDC thienodiazepinediones was created using the Reactor software as described in the manual (24). As starting materials, we used commercially available 2-aminothiophenes (N = 120), 349 primary amines and 322 isocyanides. The isocyanides are either commercially available or can be synthesized in 1-2 steps using the classical Hoffmann or Ugi syntheses (25). Consequently the theoretical chemical space of this virtual library is 120 × 349 × 322 = 13,485,360 (not including stereoisomers), which is almost unmanageable when calculating the physicochemical properties. As the father of modern MCR chemistry, Ivar Ugi pointed out in 1971 “if, for example, 40 each of the different components are reacted with one another, the result is 404 = 2,560,000 reaction products, which is quite a high figure considering that it is the same order of magnitude as the total number of chemical compounds described to date” (25). Nowadays more than 1,000 isocyanides are commercially available, thus the U-4CR space increased to 1,0004 = 1011 compounds which is out of reach of every current super computers. Thus, in order to investigate such large chemical spaces the program RandReactor was written to provide smaller random sublibraries of such very large chemical spaces. The program code is freely available and described in the Supplemental Information.
Results & Discussion
Synthesis of 1,4-thienodiazepine-2,5-dione scaffold
The synthesis of the thienodiazepinedione scaffold 5 was designed by employing the Ugi four-component reaction (U-4CR) and a subsequent deprotection and ring closure. In order to access a new scaffold with high variability the thiophene carboxylic acid was synthesized by another versatile MCR the Gewald-3CR. We synthesized three different exemplary substituted thiophene carboxylic acids to investigate scope and limitations and the influence of this particular starting material class in the subsequent UDC procedure. Cyclohexane-fused aminothiophene 2a was prepared via the G-3CR of cyclohexanone, sulfur and methyl cyanoacetate (26). 4,5-Unsubstituted aminothiophene 2b was synthesized from the Gewald reaction of 1,4-dithiane-2,5-diol and methyl cyanoacetate (27). Phenyl-substitued aminothiophene 2c was obtained from the G-3CR of phenylacetaldehyde, sulfur and methyl cyanoacetate (27). Then we initiated the preparation of thiophene carboxylic acid 3a-c by Boc-protection of 2a-c, followed by hydrolysis (Scheme 1). After pH adjustment to 6, compounds 3a-c were precipitated out and collected by filtration, which are ready to be applied according to UDC strategy.
Scheme 1.

Synthesis of 3a-c.
Therefore, N-Boc thiophene carboxylic acid 3a was employed as the bifunctional mono protected starting material for the synthesis of 1,4-thienodiazepine-2,5-diones 5a. Interestingly, there is no previous report on the compatibility of Gewald aminothiophene derivatives employed in the Ugi four-component reaction (U-4CR). A preliminary investigation by the reaction of thiophene carboxylic acid 3a, benzylamine, tert-butyl isocyanide, and ethyl glyoxalate, gave the desired condensation product 4a-1 in 65% yield (Scheme 2). The previous work in our lab demonstrated that TBD is an efficient organocatalyst for amidations of primary and secondary amines under mild conditions (28). As expected, compound 5a-1 was obtained by the deprotection of 4a-1 under a 10% solution of TFA in dichloromethane and following cyclization using catalytic amount of TBD in 62% yield over two steps.
Scheme 2.

Ugi four-component reaction of 3a. Regents and conditions: MeOH, RT, 2 days, 65%.
Under further investigation, we found that it is not necessary to isolate the Ugi product as the intermediate for next steps. The following deprotection of the crude Ugi product and subsequent cyclization gave the corresponding thienodiazepinedione 5a-1 in 33% yield over three steps. Encouraged by these results, a small focused library of thienodiazepinediones 5a was synthesized in order to enlarge the diversity of reactants (Table 1). The thienodiazepinedione products were isolated by chromatography without isolation of the intermediate. This protocol works well by employing different substituted amines and isocyanides as well. Overall, the yield is around 50-80% in average for each step of the transformation. Thus we predict that the procedure isolating and purifying (e.g. by mass directed HPLC) only the final product will be useful for automated library generation for the production of large libraries.
Table 1.
Synthesis of 1,4-thienodiazepine-2,5-diones 5a

| entry | R1 | R2 | compound | yield (%)a |
|---|---|---|---|---|
| 1 | t-Bu | benzyl | 5a-1 | 33 |
| 2 | t-Bu | 2-phenyl ethyl | 5a-2 | 53 |
| 3 | t-Bu | 4-chlorobenzyl | 5a-3 | 28 |
| 4 | t-Bu | 4-chlorophenyl | 5a-4 | 16 |
| 5 | cyclopropyl methyl | benzyl | 5a-5 | 26 |
| 6 | 2,4,6-(CH3)3C6H2 | 4-chlorobenzyl | 5a-6 | 12 |
| 7 | benzyl | 4-chlorobenzyl | 5a-7 | 16 |
| 8 | 3,4-dichlorobenzyl | 4-chlorobenzyl | 5a-8 | 16 |
isolated yields, over three steps (Method A).
The Ugi reaction of N-Boc thiophene carboxylic acid 3b, 2-phenyl ethylamine, tert-butyl isocyanide, and ethyl glyoxalate, gave the corresponding condensation product 4b-1 in 56% yield, isolated by chromatography. A mild condition was adopted for the deprotection of Ugi product 5b-1 using a 10% solution of TFA in dichloromethane. However, the ‘three-step, one-separation’ procedure for the synthesis of 5b-1 is unsatisfactory by using the crude Ugi product 6a without separation. The transformation of 4b-1 after purification afforded the corresponding 1,4-thienodiazepine-2,5-dione 5b-1. N-Boc thiophene carboxylic acid 3b was employed as the bifunctional starting material for the synthesis of 1,4-thienodiazepine-2,5-diones 5b under Ugi-deprotection-cyclization sequence (Table 2). Meanwhile, 1,4-thienodiazepine-2,5-diones 5c were synthesized from N-Boc thiophene carboxylic acid 3c under similar conditions. The Ugi reaction of 3c and ethyl glyoxalate with different amines and isocyanides gave the condensation products 4c in 38-70% yield. Then, a series of 1,4-thienodiazepine-2,5-diones 5c were obtained by deprotection and subsequent cyclization of the corresponding Ugi products (Table 2).
Table 2.
UDC approach for the synthesis of 5b and 5c

| entry | R1 | R2 | compound | yield (%)a |
|---|---|---|---|---|
| 1 | t-Bu | 2-phenyl ethyl | 4b-1 | 56 |
| 5b-1 | 22b | |||
| 2 | t-Bu | 3-thienyl ethyl | 4b-2 | 45 |
| 5b-2 | 43b | |||
| 3 | cyclohexyl | benzyl | 4b-3 | 52 |
| 5b-3 | 20b | |||
| 4 | cyclopropyl methyl | 3,4-dimethoxy phenyl ethyl | 4b-4 | 36 |
| 5b-4 | 38b | |||
| 5 | t-Bu | cyclopropyl methyl | 4c-1 | 41 |
| 5c-1 | 35c | |||
| 6 | t-Bu | 2-methoxy ethyl | 4c-2 | 49 |
| 5c-2 | 27c | |||
| 7 | t-Bu | phenyl ethyl | 4c-3 | 52 |
| 5c-3 | 38c | |||
| 8 | cyclopropyl methyl | 2-methoxy ethyl | 4c-4 | 38 |
| 5c-4 | 29c | |||
| 9 | cyclohexyl | 2-methoxy ethyl | 4c-5 | 70 |
| 5c-5 | 19c | |||
| 10 | cyclohexyl | cyclopropyl methyl | 4c-6 | 60 |
| 5c-6 | 25c |
Isolated yields;
over two steps (Method B);
over two steps (Method C).
In summary, we have demonstrated that TDZ scaffold can be achieved by the union of two sequential multicomponent reactions (29). This approach takes advantages of the maximal points of diversity offered by the G-3CR and U-4CR. A wide range of amines and isocyanides, as well as thiophene carboxylic acids are tolerated under the reaction procedure. All compounds are racemic, because the Ugi reaction yields a new stereocenter at C-3 position. Our synthetic strategy allows convenient preparation of these compounds to avoid the limitations imposed by traditional methods involving the condensation of amino acid derivatives.
Cheminformatics of virtual libraries
In order to evaluate the chemical space of TDZ scaffold, a virtual library was created based on the synthetic route (refer to Figure 2). We developed a random module software for the program Reactor (JChem 5.2.2, 2009, www.chemaxon.com) allowing us to generate a random virtual library (N = 50,000), which is freely available and described in the Supplemental Information. The compounds from the virtual library were introduced into Instant JChem (Instant JChem 2.5.1, 2009, www.chemaxon.com) to calculate some main physical properties. In addition, the compound library of commercial available benzodiazepines (substructure search from eMolecules, N = 2,498) was also analyzed by Instant JChem for comparison. The distribution of benzodiazepine library and a random virtual library of 1,4-thienodiazepine-2,5-diones (N = 50,000) was presented in term of the major descriptors molecular weight, logP and TPSA (Figure 3). The virtual library based on this new scaffold enlarges the chemical space of the benzodiazepine family. We foresee this scaffold to be useful in virtual screening and lead discovery, due to its largely unexplored chemical space.
Figure 3.

The distribution of molecular weight, logP and TPSA: (A) benzodiazepine library of a substructure search from eMolecules (N = 2,498); (B) a random virtual library of 1,4-thienodiazepine-2,5-diones (N = 50,000).
The physical properties of the virtual library were analyzed by frequency distributions in PASW Statistics 18 (Figure S1 in Supplemental Information). The range of molecular weight (between 253.3 Da. and 569.7 Da. with a mean value of 469.7 Da.) is broad due to the diversity of reactant components. The molecular weight of the compounds we physically synthesized is less than 500 Da. The mean of logP is 2.1 with standard deviation 2.3 indicating the drug-like property. Total polar surface area (TPSA) is a major descriptor relevant for example for cell membrane permeability (30). TPSA of the majority of compounds is between 126.8 Å2 and 190.4 Å2. The number of rotatable bonds (NRB), a very good descriptor of oral bioavailability, has a mean value of 7 with the standard deviation 2. The number of HBA (hydrogen bond acceptors) ranges from 4 to 13, with an average number of 7. The number of HBD (hydrogen bond donors) ranges from 2 to 8, with an average number of 3. In terms of drug likeness, 67.0% of 50,000 compounds obey Lipinski’s rule. And 85.8% of them are predicted to be bioavailable (mass ≤ 500, LogP ≤ 5, HBD ≤ 5, HBA ≤ 10, PSA ≤ 200, NRB ≤ 10, and fused aromatic rings ≤ 5). Noteworthy, the overall shape of the compounds based on the thienodiazepinedione scaffold is non-planar. The presence of stereochemistry and sp3 centers has been recognized to correlate with success as compounds transition from discovery, through clinical testing, to drugs (31). Based on the commonly accepted descriptors, it is likely that lead-like compounds with novel and potent biological activity will be obtained based on this scaffold.
We also create a virtual library of 3-D conformers, which can be used as an input for docking and other virtual analysis software (Supplemental Information). Omega (Omega 2.3.2, 2009, www.eyesopen.com) was used to generate 3-D structures of a random virtual library (N = 5,000). For example, the tricyclic backbone (6-, 5-, 7-membered rings) in 3-D structures of compound 5a-7 has the same conformation, while two side chains are oriented oppositely with certain rotation flexibility (Figure S2 in Supplemental Information).
Antagonist activities of p53-Mdm2 interaction
Since quite sometimes we are interested in the design of small molecular weight p53-Mdm2 antagonists and new scaffolds desirable for potent and selective candidates (32-34). The tumor suppressor p53 is well recognized as a therapeutic target for new anticancer interventions (35). The activation of wild-type p53 in human tumors by antagonizing murine double minute 2 (Mdm2) is a promising and potentially non toxic therapeutic strategy (36). The disruption of the p53-Mdm2 interaction can be accomplished by mimicking the p53 fragment with particular emphasis on the Mdm2 binding site using peptides, foldamers and peptoids (α-helical transactivation domain) (37). However, small-molecule libraries are favorable for the design of Mdm2 antagonists in terms of the desirable bioavailability and stability (33). The first class of potent and selective small-molecule Mdm2 antagonists, nutlins were identified from cis-imidazoline compounds (38). Next, benzodiazepinedione compound 6 has been identified as a potent Mdm2 antagonist (Kd = 80 nM) (39). The cocrystal structure of Hdm2 complex shows that compound 6 binds to the p53-binding site (side chains Phe 19, Trp 23, and Leu 26) of Hdm2 (Figure 4). Such benzodiazepinedione inhibitors were found to suppress human tumor cell proliferation in vitro and sensitize tumors to doxorubicin in vivo (40). Further optimization and SAR were investigated in order to improve the potency and cellular activity of BDZ antagonists (41-44). Although bioisosterism is a lead modification approach to attenuate the biological properties of the lead compounds, 1,4-thienodiazepine-2,5-dione scaffold (TDZs) as a potential pharmacophore has been rarely investigated so far. Thus we hypothesized that the peptidomic 1,4-thienodiazepine-2,5-dione scaffold could also act as α-helix mimetics to disturb the p53-Mdm2 interaction.
Figure 4.

Cocrystal structure of benzodiazepinedione Hdm2 antagonist (PDB code: 1T4E). Compound 6 is shown in yellow sticks, three water molecules as turquoise balls, the receptor amino acid side chains are labeled around the binding site. The hydrogen bonding network is shown in red dash line.
Since thienodiazepinediones are bioisosteric to benzodiazepindiones, we screened the compound library by our recently developed fluorescence polarization (FP) assay.(21) This robust FP assay uses fluorescent p53-like peptide and recombinant Mdm2 to measure the potency of p53-Mdm2 antagonist. To our delight, 5a-7 and 5a-8 have shown antagonist activities of p53-Mdm2 interaction through FP assay screening. Both antagonists have dose dependent effect to compete with p53-like peptide, as shown in Figure 5. The measured inhibition constant (Ki) values of Mdm2 with 5a-7 and 5a-8 were determined as 40 μM and 45 μM, respectively.
Figure 5.

Some thienodiazepinediones are competitive inhibitors of the p53-Mdm2 interaction: (A) FP binding curve of compound 5a-7, (B) FP binding curve of compound 5a-8.
Compound 5a-7 was docked into the p53 binding site of Mdm2 in order to understand the possible binding mode of small molecular antagonists (Figure 6). We were using the modeling/docking software MOLOC (Gerber, P.; www.moloc.ch) (45,46). Compound 5a-6 also has weak interaction with Mdm2, which indicates that p-chlorobenzyl fragment might be crucial for the binding. This is consistent with other reported scaffolds, such as benzodiazepinedione 6, as well as nutlins, chromenones and spirooxindoles (23,38,47). Our recently described 3-finger pharmacophore model can be applied here and suggests that the compounds bind such that the p-chlorobenzyl group mimics Trp23, the cyclohexyl and the benzyl moiety mimics Leu26 and Phe19, respectively (33). This is consistent with the docking model suggesting that the 4-chlorobenzyl group points deeply into tryptophan binding site.
Figure 6.

Compound 5a-7 (yellow sticks) docked into p53 binding site of Mdm2 (PDB code: 1YCR). Two low energy poses are shown (yellow sticks) as well as the side chains of the p53 hot spot FWL (pink sticks). In the first model the thiophene annulated cyclohexyl ring and the benzyl group mimics Phe19 and Leu26, respectively; whereas in the second model the cyclohexylthiophene fragment lays on top of the Leu26 binding site and the benzyl group points into the Phe19 binding site. Both poses suggest that there is not perfect shape and electrostatic complementarity which accounts for the moderate affinity for Mdm2.
Next, we utilized NMR to investigate the antagonistic behavior of compounds 5a-7 and 5a-8 towards the p53-Mdm2 interaction. For this purpose, we used the NMR-based antagonist induced dissociation assay (AIDA) that we developed recently (18,20). This NMR competition assay is performed on the Mdm2/p53 complex and the release of p53 from the complex is monitored as a function of the increased addition of an inhibitor. Therefore, AIDA-NMR indicates not only whether the compound is able to inhibit the protein-protein interaction in vitro, but also the dissociation constant (Kd) of the compound-Mdm2 interaction. The assay shows that the compounds are able to dissociate the Mdm2/p53 complex, and Kd values are 30 ± 20 μM and 10 ± 6 μM for compound 5a-7 and 5a-8, respectively.
Furthermore, we used the heteronuclear single quantum coherence (HSQC) NMR spectroscopy, which has been intensively used to monitor the biophysical properties of p53-Mdm2 antagonists (23,48,49). 2D HSQC spectra, showing binary titrations of 15N-Mdm2 with compounds 5a-7 and 5a-8, are shown in Figure 7 and Figure 8, respectively. Although compound 5a-8 shows a stronger binding affinity, both antagonists have similar modes of binding to Mdm2. The biggest chemical shift perturbations occur in proximity of the p53 binding site, as shown in Figure 7.
Figure 7.

2D 1H-15N HSQC spectrum of Mdm2 titrated with compound 5a-7. (Blue: human Mdm2(1-118) mixed molar with excess of compound 5a-7 (Mdm2:compound 5a-7 ratio 1:5); Red: the reference spectrum of free Mdm2)
Figure 8.

2D 1H-15N HSQC spectrum of Mdm2 titrated with compound 5a-8. (Blue: human Mdm2(1-118) mixed molar with excess of compound 5a-8 (Mdm2:compound 5a-8 ratio 1:5); Red: the reference spectrum of free Mdm2)
Conclusions and Future Directions
We have described the synthesis of the new scaffold 1,4-thienodiazepine-2,5-dione using the unprecedented union of Gewald and Ugi multicomponent reactions and incorporating the Ugi-Deprotection-Cyclization (UDC) approach. The compounds accessible can be varied at four points in the molecular skeleton. The synthesis can be easily adapted for high throughput chemistry or medicinal chemistry for high-dimensional combinatorial libraries for screening purpose. The resulting compounds are in general drug-like by means of the Pfizer rules. Some compounds were shown to antagonize the p53-Mdm2 protein-protein interaction by two complementary screening techniques, and can provide a good starting point for further design and optimization. Moreover, we provide a useful program to generate the random virtual library for computational chemistry applications. Current efforts in our laboratories focus on the optimization of the potency of p53-Mdm2 antagonists based on this interesting new class of compounds.
Supplementary Material
Figure 9.

NMR mapping of binding site of compound 5a-8. Atoms experiencing very large (Δδ > 0.08 ppm) and large (Δδ > 0.04 ppm) differences in chemical shifts upon addition of molar excess of the compound are marked red and orange, respectively. Assignment of the N-terminal domain of Mdm2 was published previously (23).
Acknowledgments
This work was supported the NIH grant 1R21GM087617-01 (to AD), Deutsche Krebshilfe, Grant 108354 (to TAH) and is part of a NCI-RAND program.
Abbreviations
- AIDA
antagonist induced dissociation assay
- BDZ
1,4-benzodiazepin-2,5-dione
- FP
fluorescence polarization
- G-3CR
Gewald three-component reaction
- HSQC
heteronuclear single quantum coherence
- MCR
multicomponent reaction
- Mdm2
murine double minute 2
- TBD
1,5,7-triazabicyclo[4,4,0]dec-5-ene
- TDZ
1,4-thienodiazepine-2,5-dione
- UDC
Ugi-Deprotection-Cyclization
- U-4CR
Ugi four-component reaction
Footnotes
Supporting Information Additional supporting information may be found in the online version of this article.
Figure S1: Statistical distributions of physical properties of a random virtual library (N = 50,000).
Figure S2: 3-D conformers of compound 4g generated by Omega2.
References
- 1.Ramajayam R, Giridhar R, Yadav MR. Current scenario of 1,4-diazepines as potent biomolecules - A mini review. Mini-Rev Med Chem. 2007;7:793–812. doi: 10.2174/138955707781387876. [DOI] [PubMed] [Google Scholar]
- 2.Sternbach LH. The benzodiazepine story. J Med Chem. 1979;22:1–7. doi: 10.1021/jm00187a001. [DOI] [PubMed] [Google Scholar]
- 3.Kopka ML, Goodsell DS, Baikalov I, Grzeskowiak K, Cascio D, Dickerson RE. Crystal structure of a covalent DNA-drug adduct: anthramycin bound to C-C-A-A-C-G-T-T-G-G and a molecular explanation of specificity. Biochemistry. 2002;33:13593–13610. doi: 10.1021/bi00250a011. [DOI] [PubMed] [Google Scholar]
- 4.Karp GM, Manfredi MC, Guaciaro MA, Ortlip CL, Marc P, Szamosi IT. Synthesis and herbicidal activity of 1H-1,4-benzodiazepine-2,5-diones. J Agric Food Chem. 1997;45:493–500. [Google Scholar]
- 5.Joseph CG, Wilson KR, Wood MS, Sorenson NB, Phan DV, Xiang ZM, et al. The 1,4-benzodiazepine-2,5-dione small molecule template results in melanocortin receptor agonists with nanomolar potencies. J Med Chem. 2008;51:1423–1431. doi: 10.1021/jm701303z. [DOI] [PubMed] [Google Scholar]
- 6.Nakanishi M, Tahara T, Araki K, Shiroki M, Tsumagari T, Takigawa Y. Psychotropic drugs. 18. Synthesis and structure-activity relations of 5-phenyl-1,3-dihydro-2H-thieno[2,3-e][1,4]diazepin-2-ones. J Med Chem. 2002;16:214–219. doi: 10.1021/jm00261a010. [DOI] [PubMed] [Google Scholar]
- 7.Hulme C. Applications of multicomponent reactions in drug discovery - lead generation to process development. In: Zhu J, Bienaymé H, editors. Multicomponent Reactions. Wiley-VCH; Weinheim: 2005. pp. 311–341. [Google Scholar]
- 8.Dömling A. Recent advances in isocyanide-based multicomponent chemistry. Curr Opin Chem Biol. 2002;6:306–313. doi: 10.1016/s1367-5931(02)00328-9. [DOI] [PubMed] [Google Scholar]
- 9.Sabnis RW, Rangnekar DW, Sonawane ND. 2-Aminothiophenes by the Gewald reaction. J Heterocycl Chem. 1999;36:333–345. [Google Scholar]
- 10.Chakrabarti JK, Hicks TA, Hotten TM, Tupper DE. Heteroarene fused benzodiazepines. Part 1. Synthesis of thieno[2,3-b][1,5]-, -[3,2-b][1,5]-, and -[3,4-b][1,5]benzodiazepines. J Chem Soc, Perkin Trans. 1978;1:937–941. [Google Scholar]
- 11.Huang Y, Dömling A. The Gewald multicomponent reaction. Mol Divers. 2010;14 doi: 10.1007/s11030-010-9229-6. in press. [DOI] [PubMed] [Google Scholar]
- 12.Dömling A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- 13.Hulme C, Gore V. “Multi-component reactions: emerging chemistry in drug discovery” ‘from xylocain to crixivan’. Curr Med Chem. 2003;10:51–80. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]
- 14.Keating TA, Armstrong RW. A remarkable two-Step synthesis of diverse 1,4-benzodiazepine-2,5-diones using the Ugi four-component condensation. J Org Chem. 1996;61:8935–8939. doi: 10.1021/jo961517p. [DOI] [PubMed] [Google Scholar]
- 15.Hulme C, Cherrier MP. Novel applications of ethyl glyoxalate with the Ugi MCR. Tetrahedron Lett. 1999;40:5295–5299. [Google Scholar]
- 16.Hulme C, Ma L, Kumar NV, Krolikowski PH, Allen AC, Labaudiniere R. Novel applications of resin bound alpha-amino acids for the synthesis of benzodiazepines (via Wang resin) and ketopiperazines (via hydroxymethyl resin) Tetrahedron Lett. 2000;41:1509–1514. [Google Scholar]
- 17.Hulme C, Peng J, Tang SY, Burns CJ, Morize I, Labaudiniere R. Improved procedure for the solution phase preparation of 1,4-benzodiazepine-2,5-dione libraries via Armstrong’s convertible isonitrile and the Ugi reaction. J Org Chem. 1998;63:8021–8023. [Google Scholar]
- 18.Bista M, Kowalska K, Janczyk W, Dömling A, Holak TA. Robust NMR Screening for Lead Compounds Using Tryptophan-Containing Proteins. J Am Chem Soc. 2009;131:7500–7501. doi: 10.1021/ja901863h. [DOI] [PubMed] [Google Scholar]
- 19.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1972. [Google Scholar]
- 20.Krajewski M, Rothweiler U, D’Silva L, Majumdar S, Klein C, Holak TA. An NMR-based antagonist induced dissociation assay for targeting the ligand-protein and protein-protein interactions in competition binding experiments. J Med Chem. 2007;50:4382–437. doi: 10.1021/jm070365v. [DOI] [PubMed] [Google Scholar]
- 21.Czarna A, Popowicz GM, Pecak A, Wolf S, Dubin G, Holak TA. High affinity interaction of the p53 peptide-analogue with human Mdm2 and Mdmx. Cell Cycle. 2009;8:1176–1184. doi: 10.4161/cc.8.8.8185. [DOI] [PubMed] [Google Scholar]
- 22.Mori S, Abeygunawardana C, Johnson MO, Vanzijl PCM. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J Magn Reson B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 23.Stoll R, Renner C, Hansen S, Palme S, Klein C, Belling A, et al. Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry. 2001;40:336–344. doi: 10.1021/bi000930v. [DOI] [PubMed] [Google Scholar]
- 24.Pirok G, Mate N, Varga J, Szegezdi J, Vargyas M, Dorant S, et al. Making “Real” Molecules in Virtual Space. J Chem Inf Model. 2006;46:563–568. doi: 10.1021/ci050373p. [DOI] [PubMed] [Google Scholar]
- 25.Ugi I. Isonitrile Chemistry. Academic Press; New York: 1971. [Google Scholar]
- 26.Fondjo ES, Dopp D, Henkel G. Reactions of some anellated 2-aminothiophenes with electron poor acetylenes. Tetrahedron. 2006;62:7121–7131. [Google Scholar]
- 27.Hwang KJ, Lee TS, Kim KW, Kim BT, Lee CM, Park EY, et al. 4-Hydroxy-6-oxo-6,7-dihydro-thieno[2,3-b] pyrimidine derivatives: Synthesis and their biological evaluation for the glycine site acting on the N-methyl-D-aspartate (NMDA) receptor. Arch Pharm Res. 2001;24:270–275. doi: 10.1007/BF02975090. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Dömling A. Efficient synthesis of arrays of amino acid derived Ugi products with subsequent amidation. J Comb Chem. 2009;11:403–409. doi: 10.1021/cc9000136. [DOI] [PubMed] [Google Scholar]
- 29.Dömling A, Ugi I. The 7-componet reaction. Angew Chem Int Ed. 1993;32:563–564. [Google Scholar]
- 30.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 31.Lovering F, Bikker J, Humblet C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava S, Beck B, Wang W, Czarna A, Holak TA, Dömling A. Rapid and efficient hydrophilicity tuning of p53/mdm2 antagonists. J Comb Chem. 2009;11:631–639. doi: 10.1021/cc9000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dömling A. Small molecular weight protein-protein interaction antagonists--an insurmountable challenge? Curr Opin Chem Biol. 2008;12:281–291. doi: 10.1016/j.cbpa.2008.04.603. [DOI] [PubMed] [Google Scholar]
- 34.Dömling A. Selective and dual-action p53/mdm2/mdm4 antagonists. 2008 WO2008130614. [Google Scholar]
- 35.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 36.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Robinson JA. Design of protein-protein interaction inhibitors based on protein epitope mimetics. ChemBioChem. 2009;10:971–973. doi: 10.1002/cbic.200900055. [DOI] [PubMed] [Google Scholar]
- 38.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 39.Grasberger BL, Lu TB, Schubert C, Parks DJ, Carver TE, Koblish HK, et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem. 2005;48:909–912. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 40.Koblish HK, Zhao SY, Franks CF, Donatelli RR, Tominovich RM, LaFrance LV, et al. Benzodiazepinedione inhibitors of the Hdm2: p53 complex suppress human tumor cell proliferation in vitro and sensitize tumors to doxorubicin in vivo. Mol Cancer Ther. 2006;5:160–169. doi: 10.1158/1535-7163.MCT-05-0199. [DOI] [PubMed] [Google Scholar]
- 41.Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Gupta V, Lattanze J, et al. 1,4-benzodiazepine-2,5-diones as small molecule antagonists of the HDM2-p53 interaction: discovery and SAR. Bioorg Med Chem Lett. 2005;15:765–770. doi: 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Leonard K, Marugan JJ, Raboisson P, Calvo R, Gushue JM, Koblish HK, et al. Novel 1,4-benzodiazepine-2,5-diones as Hdm2 antagonists with improved cellular activity. Bioorg Med Chem Lett. 2006;16:3463–3468. doi: 10.1016/j.bmcl.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 43.Marugan JJ, Leonard K, Raboisson P, Gushue JM, Calvo R, Koblish HK, et al. Enantiomerically pure 1,4-benzodiazepine-2,5-diones as Hdm2 antagonists. Bioorg Med Chem Lett. 2006;16:3115–20. doi: 10.1016/j.bmcl.2006.03.067. [DOI] [PubMed] [Google Scholar]
- 44.Cummings MD, Schubert C, Parks DJ, Calvo RR, LaFrance LV, Lattanze J, et al. Substituted 1,4-Benzodiazepine-2,5-diones as alpha-Helix mimetic antagonists of the HDM2-p53 protein-protein interaction. Chem Biol Drug Des. 2006;67:201–205. doi: 10.1111/j.1747-0285.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 45.Gerber PR, Muller K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J Comput Aided Mol Des. 1995;9:251–268. doi: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- 46.Gerber PR. Charge distribution from a simple molecular orbital type calculation and non-bonding interaction terms in the force field MAB. J Comput Aided Mol Des. 1998;12:37–51. doi: 10.1023/a:1007902804814. [DOI] [PubMed] [Google Scholar]
- 47.Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 48.D’Silva L, Ozdowy P, Krajewski M, Rothweiler U, Singh M, Holak TA. Monitoring the effects of antagonists on protein-protein interactions with NMR spectroscopy. J Am Chem Soc. 2005;127:13220–13226. doi: 10.1021/ja052143x. [DOI] [PubMed] [Google Scholar]
- 49.Krajewski M, Ozdowy P, D’Silva L, Rothweiler U, Holak TA. NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro. Nat Med. 2005;11:1135–1136. doi: 10.1038/nm1105-1135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.