

Abstract
Over the past few decades, our expanding knowledge of the mammalian immune system – how it is developed, activated, and regulated – has fostered hope that it may be harnessed in the future to successfully treat human cancer. The immune system activated by cancer vaccines may have the unique ability to selectively eradicate tumor cells at multiple sites in the body without inflicting damage on normal tissue. However, progress in the development of cancer vaccines that effectively capitalize on this ability has been limited and slow. The immune system is restrained by complex, negative feedback mechanisms that evolved to protect the host against autoimmunity and may also prevent antitumor immunity. In addition, tumor cells exploit a plethora of strategies to evade detection and elimination by the immune system. For these reasons, the field of cancer immunotherapy has suffered considerable setbacks in the past and faces great challenges at the present time. Some of these challenges may be overcome through the use of RNA interference, a process by which gene expression can be efficiently and specifically “knocked down” in cells. This chapter focuses on the current status and future prospects in the application of small interfering RNA and microRNA, two main forms of RNA interference, to treat cancer by curtailing mechanisms that attenuate the host immune response.
Keywords: RNA interference (RNAi), Small interfering RNA (siRNA), MicroRNA (miRNA), Cancer, Tumor, Immunotherapy, Vaccine, Dendritic cell, T cell
1. Introduction
1.1. The Promises and Pitfalls of Cancer Immunotherapy
Despite decades of persistent and intense research effort, the treatment of late-stage cancer in the clinic has achieved limited success and remains elusive. Standard chemotherapeutic regimens frequently fail to control the growth of large, disseminated tumors without causing severe side effects in patients. In addition, surgical and radiological methods are unable to eradicate metastatic or minimal residual disease, and the recurrence of malignancy after treatment by these approaches continues to be a virtually insurmountable obstacle. As a result, the current survival periods for late-stage cancer patients are dismal and in urgent need of improvement.
The generation of tumor-specific CD4+ and CD8+ T cell-mediated immunities by cancer vaccines represents a potentially promising route to the successful control of advanced cancer. Type 1-helper CD4+ T (Th1) cells are able to efficiently stimulate and maintain the effector function of cytotoxic CD8+ T cells. Together, these two arms of the adaptive immune system have the specificity and potency to kill cancerous cells at multiple sites in the body without inflicting significant damage on normal tissue. Furthermore, the establishment of immunological memory after the tumor has been cleared may provide complete and long-term protection against disease relapse. Over the past decade, vaccination with defined antigens has emerged as one of the most attractive approaches to generate antigen-specific CD4+ and CD8+ T cell-mediated immunities and therefore provides an attractive alternative strategy to cancer treatment. However, these vaccines have limited effectiveness, and thus, their promise has not yet been realized in the clinic.
1.2. The Process of RNA Interference
As part of the homeostasis of the host, the immune system is constantly held in check by mechanisms that limit the duration and magnitude of an acute inflammatory response or maintain peripheral tolerance to self-antigen. Furthermore, the tumor microenvironment is rich in immunosuppressive molecules that inhibit the survival, activation, and function of infiltrating T cells either directly or through the recruitment of regulatory immune cells. RNA interference (RNAi) – a remarkable phenomenon first observed in the late 1980s that later evolved into a technology with immense biomedical applications – provides the unique and unparalleled ability to specifically silence expression of target genes (for reviews, see (1–3)). RNAi has thus recently emerged as a powerful addition to the arsenal of cancer immunotherapy that could overcome many of the obstacles it currently faces.
RNAi can be mediated either by small interfering RNA (siRNA) or microRNA (miRNA) molecules. Figure 1 illustrates the molecular steps involved in the RNAi process. In siRNA-mediated gene silencing, cytoplasmic double-stranded RNA (dsRNA) is first cleaved into 21–28 nucleotide duplexes by the RNase-III enzyme Dicer. These duplexes are incorporated into the multiprotein RNA-inducing silencing complex (RISC) and serve as a template that binds to complementary target mRNA, which is degraded by the RISC enzyme Slicer. Multiple types of dsRNA have been delivered into a wide variety of cells and organisms, demonstrating broadly that siRNA is an effective tool for specific gene knockdown (for review, see (4)).
Fig. 1.
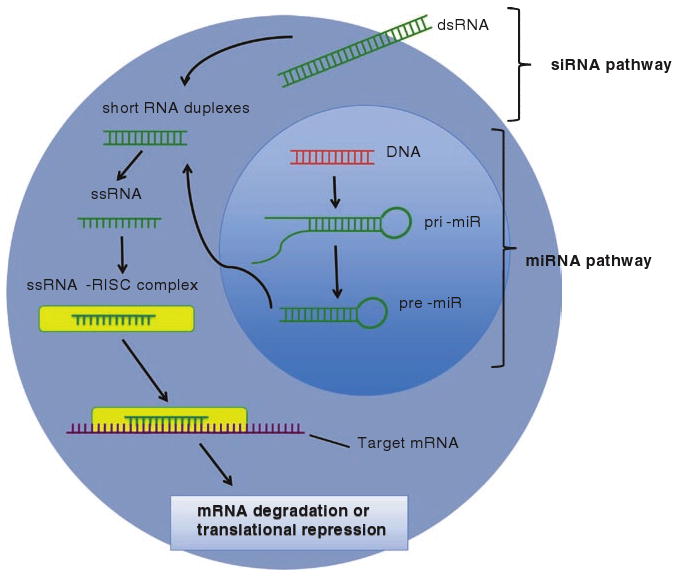
The processes of siRNA and miRNA-mediated gene silencing. In the miRNA pathway, primary RNA transcripts (pri-miR) are processed by the enzyme Drosha into 60 base pair precursors (pre-miR) in the nucleus. Following export into the cytoplasm, the pre-miR is cleaved into single-stranded 22 nucleotide sequences by the enzyme Dicer and then incorporated into the RISC. The mature miRNA guides the RISC to target mRNA sequences, where it induces either degradation or translational inhibition. The siRNA pathway is similar to the miRNA pathway, with the exception that long, exogenous double-stranded RNA is the initial agent that is processed into a silencing complex
In the early 1990s, it was discovered that multiple aspects of cellular function are influenced by endogenous miRNA, a diverse class of small (∼22 nucleotide) and copious RNA species that, similar to siRNA, knocks down a vast array of genes at the post-transcriptional stage. Collectively, these molecules are predicted by computer algorithms to modulate the expression of as much as 90% of human genes. miRNA is initially produced as a ∼60 nucleotide precursor with a defined stem-loop structure. Following nuclear export, it is processed by the enzyme Dicer into a ∼22 nucleotide duplex, and a single strand is then preferentially incorporated into RISC. The miRNA–RISC binds to partially complementary sites in the 5′ or 3′ untranslated regions of target mRNA molecules, where it induces either translational repression or mRNA degradation (for review, see (5)).
1.3. Applications of RNA Interference to Cancer Immunotherapy
In this chapter, we summarize the current progress and future prospects for the application of siRNA and miRNA in cancer immunotherapy. In this context, the use of these molecules to target specific genes in dendritic cells (DCs) and T cells – essential players in adaptive immunity – will be discussed. Specifically, we will examine how RNAi can be employed to empower the immune system, to abolish intrinsic inhibitory pathways that restrict its function, and to render it insensitive to the suppressive nature of the tumor microenvironment. The majority of experiments on this topic have been conducted in cell culture and animal models, with few studies translated into the clinic. Undeniably, several obstacles – including targeted delivery, incomplete gene silencing, non-specific immune responses, and off-target effects – must be overcome before RNAi can be successfully delivered into humans. Some of these obstacles are outlined together with potential solutions. Altogether, we hope to show that siRNA and miRNA, while small in size, are large in medical significance. These molecules are powerful and emerging additions to the clinical toolkit, which may one day propel immunotherapy to the forefront of remedies for cancer and transform the way it is delivered to patients.
2. RNA Interference in Dendritic Cells
Among all the professional antigen-presenting cells (APCs), mature DCs unquestionably have the strongest capacity to present antigen to and prime either naïve CD8+ or CD4+ T cells. Therefore, the delivery of tumor-associated antigen (TAA) to DCs is a cornerstone of cancer immunotherapy. In the major histocompatibility complex (MHC) class I-restricted pathway for antigen presentation, cytoplasmic antigen is processed by proteasomes and loaded onto MHC class I molecules in the endoplasmic reticulum, where it is eventually shuttled to the DC surface and can interact with the T cell receptor (TCR) on cognate CD8+ T cells. In comparison, in the MHC class II-restricted pathway, extracellular antigen is taken up into DCs by endocytosis into endosomes, where the acidic environment and the presence of proteases degrade the antigen for loading onto membrane-bound MHC class II molecules, which are transported to the cell surface where they interact with the TCR of CD4+ T cells. Efficient T cell activation requires binding between antigen-MHC complexes and the cognate TCR, as well as engagement of costimulatory molecules on DCs.
Mature DCs have high numbers of MHC and costimulatory molecules and are capable of migrating into secondary lymphoid organs, making them excellent activators of naïve T cells and, consequently, ideal instigators of tumor-specific immune responses. Many vaccination strategies developed thus far have focused on engineering DCs with molecules that would enhance the immunostimulatory capacity of these cells. However, the functions of DCs are also negatively regulated by feedback mechanisms, which prevent hyperactivation of the adaptive immune system. These mechanisms are currently being unraveled at a swift pace, and their inhibition offers a new and potentially effective approach to unleash the unbridled antigen presentation ability of DCs. RNAi technology, as delivered through siRNA or miRNA, provides an ideal way to achieve this purpose. Specifically, it may be of interest to use this technology to suppress the major immunosuppressive pathways, to bias CD4+ T cell differentiation towards a Th1 response, and to prolong the lifespan of DCs.
2.1. Suppression of Immunosuppressive Pathways in Dendritic Cells
DCs express a variety of molecules that may suppress antigen presentation or T cell activation and function. Two such important molecules are A20 and suppressor of cytokine signaling 1 (SOCS1). A20 is an ubiquitin ligase involved in the attenuation of T cell-mediated responses (6, 7). SOCS1 is a broadly immuno-suppressive protein that inhibits signaling through the interferon (IFN)-γ, interleukin (IL)-2, IL-6, IL-7, IL-12, and IL-15 pathways, all of which contribute to T cell expansion (8). In addition, SOCS1 can directly suppress antigen presentation to T cells (9). Studies have shown that antigen-loaded DCs, silenced for either A20 (10) or SOCS1 (8, 11, 12) by siRNA, are able to activate large numbers of effector T cells, which correlates with inhibition of tumor growth in mice. Therefore, knockdown of both of these molecules by RNAi represents an effective strategy for cancer immunotherapy.
Recent studies have shown that the upregulation of SOCS1 in DCs is at least in part due to signaling through the Tyro3/Axl/Mer (TAM) family of receptor tyrosine kinases (13). These molecules signal through the STAT1 pathway to induce expression of SOCS1 (13). Notably, transgenic mice that lack the TAM receptors develop profound, broad-spectrum autoimmune disease, suggesting that these receptors play a critical role in controlling the magnitude of the immune response (14). It was recently demonstrated that the TAM component Mer blocks the NF-κB pathway and is required for apoptotic cell-induced T cell tolerance (15). Therefore, RNAi-mediated silencing of TAM, and in particular Mer, represents a potentially effective strategy for therapeutic vaccination against cancer. We have recently found that in vivo knockdown of Mer in epidermal DCs by shRNA dramatically enhances the number of T cells specific for the E7 oncoprotein of human papillomavirus type-16, generated by an E7 DNA vaccine (Mao et al., personal communication). In the future, it will be of interest to explore the suppression of each of the TAM receptors in DCs individually as well as in combination with one another.
In addition to the molecules mentioned above, which suppress antigen presentation, DCs also secrete a variety of proteins that directly inhibit the activation, survival, and function of cognate T cells. For instance, the tryptophan-degrading enzyme indoleamine-2,3-dioxygenase (IDO) is secreted abundantly by plasmacytoid DCs and depletes tryptophan availability in tumor-draining lymph nodes (16). This induces profound anergy in T cells, as they rely heavily on tryptophan for proliferation and function (17). Therefore, reduction of IDO expression by RNAi may improve the overall cytotoxic activity of T cells in the tumor microenvironment.
Molecules expressed on the surface of DCs also have direct suppressive effects on T cells and represent excellent targets for cancer immunotherapy. For example, the surface proteins programmed death-1 ligand (PD-L1) and PD-L2 – members of the B7 family – recognize and bind to the PD-1 receptor on T cells. Signaling through PD-1 inhibits T cell activation through suppression of the IL-2 and IFN-γ pathways (18) and through induction of cell cycle arrest (19). Antibody blockade of PD-L1 and PD-L2 on DCs has been shown to improve proliferation of and cytokine production by CD4+ T cells (20), leading to improved control of ovarian carcinoma in a preclinical model (21). This finding suggests that siRNA-mediated knockdown of PD-L1 might achieve a similar immunostimulatory effect. In addition, the surface molecules immunoglobulin receptor 2 (DIgR2) (22) and Notch ligands (Delta1, Jagged1, Jagged2) (23–25) expressed by DCs have all been shown to deliver suppressive signals to T cells. Importantly, vaccination of mice with tumor antigen-loaded DCs transfected with DIgR2 siRNA, compared to control siRNA, generated higher levels of tumor-specific CD4+ and CD8+ T cells as well as protective immunity against tumor challenge (22). Furthermore, CD4+ T cells, incubated with allogeneic DCs transfected with siRNA targeting the Notch ligands, displayed higher levels of IFN-γ production (26). These studies indicate that DIgR2 and Notch ligands play a suppressive role in DC biology, and that the downregulation of these molecules is an attractive approach for generating therapeutic immunity against cancer.
DCs have unparalleled capacity to present antigen to and to prime naïve T cells, but express abundant amounts of the proapoptotic surface molecule Fas ligand (FasL) (27). Interactions between FasL and the Fas death receptor on the membrane of T cells may curb the magnitude of the adaptive immune response. Therefore, knockdown of FasL in DCs by RNAi might prevent activation-induced T cell apoptosis upon vaccination and thus bolster antitumor immunity. Huang et al. have previously developed an in vivo gene delivery system using gene gun that could efficiently deliver DNA-encoding siRNA to silence the expression of specific molecules on DCs (27). With this system, they demonstrated that a short hairpin RNA construct targeting FasL was able to significantly enhance the potency of a DNA vaccine against the E7 oncoprotein of human papillomavirus type-16, a model cervical cancer antigen (27). These immunological effects correlated with the eradication of subcutaneously implanted E7-expressing tumors in mice (27).
DCs also secrete factors that attract regulatory T (Treg) cells to the site of antigen presentation in order to tame the immune response. Iellem et al. showed that DCs recruit these cells principally by secreting the chemokines CCL17 and CCL22, which bind to the CCR4 and CCR8 receptors on Treg cells, respectively (28). In this context, it is conceivable that the silencing of CCL17 and CCL22 by RNAi would be a possible way to reduce Treg cell migration to the lymphoid compartments and thereby prevent the suppression of T cell activation.
Finally, it has been revealed that miRNA molecules have an important role in control of the immune system (29). It has been shown that mice deficient in Bic, the gene that codes for miR-155, have poorly functional immune systems characterized by reduced cytokine and antibody production as well as heightened susceptibility to bacterial challenge (30, 31). We have recently found that in DCs, miR-155 appears to have a suppressive role (Mao et al., personal communication). We showed in a mouse model that miR-155 is induced in bone marrow-derived DCs upon stimulation with Toll-like receptor agonists. In addition, in vivo biolistic transfection of mice with DNA-encoding miR-155 via gene gun suppressed antigen-specific T cell-mediated immunity, whereas transfection with a partially antisense inhibitor of the miRNA reversed this effect. Our current findings suggest that miR-155 plays a suppressive role in DC biology, likely through post-transcriptional silencing of critical mediators of the NF-κB pathway such as IKKε. A miRNA that may have a similar function in DCs is miR-146a, an activation-induced molecule shown to silence TRAF6 and IRAK1, key components of the Toll-like receptor pathway (32). Thus, introduction of an inhibitor of miR-155 or miR-146a into DCs may represent an effective way to enhance the antigen presentation ability of these cells and augment tumor-specific immunity.
2.2. Induction of Th1-Polarizing Pathways in Dendritic Cells
It is now clear that, in addition to cytotoxic CD8+ T cells, helper CD4+ T cells are also critical in the development of robust antitumor immunity. During antigen presentation, cytokines secreted by DCs profoundly influence the developmental fate of naïve CD4+ T cells. Based on our current understanding, T cells are programmed through this interaction to adopt the Th1, Th2, Th17, or Treg lineages, each of which is distinguished by a characteristic cytokine secretion profile and has a distinct role in the immune response against cancer. It has been shown that cytokines produced by Th1 cells, including IFN-γ and TNF-α, promote the proliferation and function of activated CD8+ T cells. In this respect, Th1 cells are important for the generation of optimal tumor-specific immunity. Thus, strategies that improve the Th1-polarizing ability of DCs may represent a promising direction in cancer vaccine design.
A delicate balance between exposure to the DC-secreted cytokines, IL-10 and IL-12, determines naïve CD4+ T cell fate: IL-12 induces Th1 differentiation while IL-10 inhibits it. Therefore, decreasing the amount of IL-10 relative to IL-12 in DCs provides a potential method for promoting Th1 responses. Liu et al. have shown that transfection with siRNA targeting IL-10 reduces levels of this cytokine and elevates production of IL-12 in DCs (33). Furthermore, these DCs preferentially induced the development of naïve CD4+ T cells into the Th1 phenotype (33). In addition, it has been found that IL-6 stimulates Th2 differentiation at the expense of the Th1 response (for review, see (34)). Therefore, silencing of IL-6 by siRNA, either alone or in combination with IL-10 knockdown, may further augment Th1-mediated immunity. It will be important to explore the therapeutic efficacy of these approaches in animal as well as clinical models of cancer in the future.
2.3. Attenuation of Apoptotic Pathways in Dendritic Cells
When CD8+ T cells are activated following antigen presentation, they secrete cytotoxic granules that induce apoptosis in neighboring DCs (35), a process which is thought to be an important immunoregulatory mechanism. The amount of the antiapoptotic protein Bcl-2 present in DCs determines the longevity of these cells in response to the death stimuli (36, 37). As a result, raising the amount of Bcl-2 in DCs provides a way to prolong their lifespan and hence sustain antigen presentation. Kim et al. have previously shown that in vivo transfection of DCs with Bcl-2 together with DNA encoding the E7 onco-protein of human papillomavirus type-16 could increase DC survival, which correlated with an enhanced frequency of E7-specific CD8+ T cells as well as improved therapeutic effects against E7-expressing tumors (38). Because Bcl-2 functions through inhibition of the proapoptotic proteins Bax and Bak, the silencing of these two molecules in DCs by RNAi may directly increase the duration of antigen presentation in the context of a vaccine (39, 40). The delivery of RNAi would circumvent concerns of oncogenicity associated with administration of DNA plasmids. It was shown that siRNA targeting Bax/Bak could be efficiently delivered to silence expression of these molecules in vivo, and co-administration of this siRNA with E7 DNA elicited strong E7-specific T cell-mediated immunity and antitumor effects in a preclinical model (40).
Collectively, these studies demonstrate that RNAi technology directed against proapoptotic molecules can be incorporated into vaccines to produce potent therapeutic immune responses against cancer.
3. Cancer Immunotherapy by Genetic Modification of T Cells
Even the most potent degree of antigen presentation by DCs would not generate significant control of cancer if activated T cells do not efficiently home to, proliferate, and function in the tumor microenvironment. In this regard, the genetic modification of antigen-specific T cells provides a direct and effective route to generate antitumor immunity. For example, the biology of T cells could be altered such that they exhibit rapid and high frequency accumulation in tumors, enhanced clonal expansion, as well as improved helper and cytotoxic function. While development of the technology for the in vitro and in vivo transfection of naïve or activated T cells is still in its infancy, there are many areas in which RNAi may be used to modulate the properties of these cells for therapeutic purpose. In this section, we review one of these areas: suppression of immunosuppressive pathways.
3.1. Suppression of Immunosuppressive Pathways in T Cells
Just as the antigen processing and presentation functions of DCs are regulated by a variety of immunosuppressive pathways, the helper and cytolytic functions of T cells are subject to many negative feedback mechanisms as well. The attenuation of these mechanisms through RNAi technology thus represents a way to unleash the full functional capacity of these T cells, as they are primed to recognize and eliminate cancers. Here, we discuss three such relevant and classical mechanisms: the TGF-β, CTLA-4, and PD-1 signaling pathways.
Many cancers, such as neuroblastoma and Hodgkin Reed–Sternberg tumors, secrete abundant amounts of the immunosuppressive cytokine TGF-β (41–43). When TGF-β binds to and signals through the TGF-β receptor (TGF-βR) on T cells, the phosphorylation and nuclear translocation of the transcription factors Smad 2 and 3 is triggered (44, 45), resulting in growth arrest, terminal differentiation (46, 47), and induction of tolerance (48, 49). This signaling cascade is a major route through which tumors routinely escape immunological surveillance and therefore provides an excellent molecular target for clinical intervention. The use of siRNA to silence the expression of TGF-β receptor in tumor-specific T cells could protect them from tumor-mediated tolerance and thus generate a highly functional therapeutic immune response.
In addition to the TGF-β pathway, the cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) pathway also imposes strong inhibitory control over T cells. CTLA-4 is expressed highly on T cells following antigen exposure and activation. It is a homolog of the CD28, the major receptor for the costimulatory protein B7. However, CTLA-4 binds to B7 with several thousand times the affinity of CD28 and, as a result, has an overwhelmingly negative influence on T cell priming. CTLA-4 signaling also suppresses IL-2 synthesis and release by T cells, thereby inhibiting their proliferation. It has been shown that blockade of CTLA-4 on T cells with monoclonal antibodies could lead to successful rejection of tumors as well as protection from subsequent challenge (50). Therefore, knockdown of CTLA-4 with siRNA in T cells may be a potentially promising avenue for exploration as a cancer immunotherapy.
PD-1 is another member of the CD28 receptor family and, like CTLA-4, strongly inhibits T cell proliferation and cytokine secretion. The pathway of this molecule is described above. It has been shown in mouse models that blockade of PD-1 signaling improved the accumulation of effector T cells in the tumor, enhanced the cytolytic function of these cells, and inhibited the spread of both B16 melanoma and CT26 colon cancer cells (51). Thus, siRNA-mediated silencing of PD-1 in T cells may also represent a fruitful strategy for cancer immunotherapy.
As the role of effector T cells in the antitumor immune response becomes increasingly apparent, it has too become clear that an effective response would require the presence of functional T cells. Thus, the genetic modification of T cells in this regard has emerged as a distinctly attractive strategy for cancer immunotherapy. Hopefully, as more becomes understood about the molecular pathways governing T cell activation and tolerance, RNAi technology can be exploited for this purpose.
4. Translation of RNAi Technology into the Clinic as a Form of Immunotherapy
Recent advances in our understanding of the RNAi process, as well as how the immune system is regulated, have created great opportunities in the field of cancer immunotherapy. The development of synthetic siRNA over the past few years has made it feasible to apply this technology for clinical purposes. However, the efficient in vivo delivery of RNAi remains a significant challenge.
The efficacy of in vivo siRNA-mediated gene silencing is limited by the transient nature of RNAi as well as the inherent instability of the RNA molecule. It has been shown that on average dsRNA is degraded about 36–48 h after introduction into cells (for review, see (52)). Furthermore, the efficiency of gene knockdown varies by tissue depending on the readiness with which siRNA is uptaken into cells. Although the effects of RNAi may persist for several weeks in terminally differentiated or senescent cells, they typically disappear within one week in rapidly proliferating cells.
To overcome the issues associated with siRNA half-life, investigators have created chemically modified, synthetic dsRNAs that have increased stability and resist degradation in blood or tissue. In addition, the dsRNAs can be designed to be protected from serum RNase, linked with fusogenic peptides, encased in lipid complexes, or conjugated to membrane protein-specific antibodies for cell-specific delivery (for review, see (3)). To enhance this specificity, conditionally replicating viral vectors harboring the siRNA have been developed (53). The incorporation of tissue-specific RNA polymerase II promoters to drive expression of the siRNA (54), or conjugation to nano-complexes (55), has been exploited as a strategy to further enhance preferential uptake of the molecules into a particular type of tissue. With these novel developments, it is likely that delivery of siRNA will become much more practical in the clinic in the near future.
To achieve potency in addition to specificity, siRNA may be incorporated into viral vectors, which have the ability to integrate into the host genome and thereby induce long-term gene silencing. Previous studies have investigated this approach using lentiviral (56–58), retroviral (58–60), or adenoviral vectors (57, 61, 62). However, in general, these vectors lack tissue specificity and raise concerns of toxicity associated with the viral structural proteins.
Although much progress has been made in the enhancement of RNAi administration, numerous hurdles remain. Intracellular stability, tissue-specific delivery, and sustained gene silencing are issues that must be addressed before this technology can become widely applicable in the clinic. Additionally, questions related to the so-called non-specific or “off-target” effects, as well as cellular resistance to siRNA or miRNA, must be answered before the therapeutic use of RNAi technology may be considered.
Nonetheless, as scientists continue to unravel the complexities of the RNAi machinery, optimism is generated that what originated as the discovery of a remarkable cellular phenomenon at the turn of the century may soon evolve into a technology that is safe and effective to administer in humans. At this point, it would be possible to use this technology to augment the potency of conventional vaccines against cancer. Specifically, the down-regulation of immunosuppressive molecules in DCs and T cells (Fig. 2), key components of the immune system, are promising strategies that could abrogate the ability of tumors to escape immunological control. At the rapid pace of our understanding of the intricate regulation of the innate and adaptive immune systems, it is hopeful that the use of RNAi in cancer immunotherapy may soon become a reality that saves the lives of many people worldwide.
Fig. 2.

Immunosuppressive molecules expressed by DCs that can be targeted by RNAi for cancer immunotherapy. A20, SOCS1, and Tyro3/Axl/Mer suppress the antigen presentation and T cell activation capacity of DCs. IDO, PD-L1, DIgR2, Notch ligands, and FasL directly inhibit T cell proliferation, function, or survival. CCL17 and CCL22 mediate the recruitment of Treg cells to the tumor. miR-155 and miR-146a are predicted to be suppressive miRNAs which inhibit DC function
Acknowledgments
This review is not intended to be an encyclopedic one, and we apologize to any authors not cited. We would like to thank Ms. Archana Monie for help with preparation of the manuscript and Ms. Lucy Wangaruro for excellent secretarial support. This work is funded by the National Cancer Institute SPORE (P50CA098252) and the NCDDG program (U19 CA113341).
References
- 1.Caplen NJ. Gene therapy progress and prospects. Downregulating gene expression: the impact of RNA interference. Gene Ther. 2004;11:1241–1248. doi: 10.1038/sj.gt.3302324. [DOI] [PubMed] [Google Scholar]
- 2.Leung RK, Whittaker PA. RNA interference: from gene silencing to gene-specific therapeutics. Pharmacol Ther. 2005;107:222–239. doi: 10.1016/j.pharmthera.2005.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shankar P, Manjunath N, Lieberman J. The prospect of silencing disease using RNA interference. JAMA. 2005;293:1367–1373. doi: 10.1001/jama.293.11.1367. [DOI] [PubMed] [Google Scholar]
- 4.Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF, Wu TC. Prospects of RNA interference therapy for cancer. Gene Ther. 2006;13:464–477. doi: 10.1038/sj.gt.3302694. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22:1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 9.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–7116. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 10.Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14:258–265. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou H, Zhang D, Wang Y, Dai M, Zhang L, Liu W, et al. Induction of CML28-specific cytotoxic T cell responses using co-transfected dendritic cells with CML28 DNA vaccine and SOCS1 small interfering RNA expression vector. Biochem Biophys Res Commun. 2006;347:200–207. doi: 10.1016/j.bbrc.2006.06.093. [DOI] [PubMed] [Google Scholar]
- 12.Yang R, Yang X, Zhang Z, Zhang Y, Wang S, Cai Z, et al. Single-walled carbon nanotubes-mediated in vivo and in vitro delivery of siRNA into antigen-presenting cells. Gene Ther. 2006;13:1714–1723. doi: 10.1038/sj.gt.3302808. [DOI] [PubMed] [Google Scholar]
- 13.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 14.Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- 15.Wallet MA, Sen P, Flores RR, Wang Y, Yi Z, Huang Y, et al. MerTK is required for apoptotic cell-induced T cell tolerance. J Exp Med. 2008;205:219–232. doi: 10.1084/jem.20062293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 17.Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–290. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–643. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 19.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 20.Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 21.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 22.Shi L, Luo K, Xia D, Chen T, Chen G, Jiang Y, et al. DIgR2, dendritic cell-derived immunoglobulin receptor 2, is one representative of a family of IgSF inhibitory receptors and mediates negative regulation of dendritic cell-initiated antigen-specific T-cell responses. Blood. 2006;108:2678–2686. doi: 10.1182/blood-2006-04-015404. [DOI] [PubMed] [Google Scholar]
- 23.Hoyne GF, Le Roux I, Corsin-Jimenez M, Tan K, Dunne J, Forsyth LM, et al. Serrate1-induced notch signalling regulates the decision between immunity and tolerance made by peripheral CD4(+) T cells. Int Immunol. 2000;12:177–185. doi: 10.1093/intimm/12.2.177. [DOI] [PubMed] [Google Scholar]
- 24.Wong KK, Carpenter MJ, Young LL, Walker SJ, McKenzie G, Rust AJ, et al. Notch ligation by Delta1 inhibits peripheral immune responses to transplantation antigens by a CD8+ cell-dependent mechanism. J Clin Invest. 2003;112:1741–1750. doi: 10.1172/JCI18020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–526. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- 26.Stallwood Y, Briend E, Ray KM, Ward GA, Smith BJ, Nye E, et al. Small interfering RNA-mediated knockdown of notch ligands in primary CD4+ T cells and dendritic cells enhances cytokine production. J Immunol. 2006;177:885–895. doi: 10.4049/jimmunol.177.2.885. [DOI] [PubMed] [Google Scholar]
- 27.Huang B, Mao CP, Peng S, Hung CF, Wu TC. RNA interference-mediated in vivo silencing of fas ligand as a strategy for the enhancement of DNA vaccine potency. Hum Gene Ther. 2008;19:763–773. doi: 10.1089/hum.2007.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194:847–853. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 32.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu G, Ng H, Akasaki Y, Yuan X, Ehtesham M, Yin D, et al. Small interference RNA modulation of IL-10 in human monocyte-derived dendritic cells enhances the Th1 response. Eur J Immunol. 2004;34:1680–1687. doi: 10.1002/eji.200425081. [DOI] [PubMed] [Google Scholar]
- 34.Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39:531–536. doi: 10.1016/s0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 35.Ingulli E, Mondino A, Khoruts A, Jenkins MK. In vivo detection of dendritic cell antigen presentation to CD4(+) T cells. J Exp Med. 1997;185:2133–2141. doi: 10.1084/jem.185.12.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hou WS, Van Parijs L. A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat Immunol. 2004;5:583–589. doi: 10.1038/ni1071. [DOI] [PubMed] [Google Scholar]
- 37.Nopora A, Brocker T. Bcl-2 controls dendritic cell longevity in vivo. J Immunol. 2002;169:3006–3014. doi: 10.4049/jimmunol.169.6.3006. [DOI] [PubMed] [Google Scholar]
- 38.Kim TW, Hung CF, Ling M, Juang J, He L, Hardwick JM, et al. Enhancing DNA vaccine potency by coad-ministration of DNA encoding antiapoptotic proteins. J Clin Invest. 2003;112:109–117. doi: 10.1172/JCI17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng S, Kim TW, Lee JH, Yang M, He L, Hung CF, et al. Vaccination with dendritic cells transfected with BAK and BAX siRNA enhances antigen-specific immune responses by prolonging dendritic cell life. Hum Gene Ther. 2005;16:584–593. doi: 10.1089/hum.2005.16.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim TW, Lee JH, He L, Boyd DA, Hardwick JM, Hung CF, et al. Modification of professional antigen-presenting cells with small interfering RNA in vivo to enhance cancer vaccine potency. Cancer Res. 2005;65:309–316. [PubMed] [Google Scholar]
- 41.Hsieh CL, Chen DS, Hwang LH. Tumor-induced immunosuppression: a barrier to immunotherapy of large tumors by cytokine-secreting tumor vaccine. Hum Gene Ther. 2000;11:681–692. doi: 10.1089/10430340050015581. [DOI] [PubMed] [Google Scholar]
- 42.Poppema S, Potters M, Visser L, van den Berg AM. Immune escape mechanisms in Hodgkin's disease. Ann Oncol. 1998;9 5:S21–S24. doi: 10.1093/annonc/9.suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- 43.Scarpa S, Coppa A, Ragano-Caracciolo M, Mincione G, Giuffrida A, Modesti A, et al. Transforming growth factor beta regulates differentiation and proliferation of human neuroblastoma. Exp Cell Res. 1996;229:147–154. doi: 10.1006/excr.1996.0352. [DOI] [PubMed] [Google Scholar]
- 44.Jayaraman L, Massague J. Distinct oligomeric states of SMAD proteins in the transforming growth factor-beta pathway. J Biol Chem. 2000;275:40710–40717. doi: 10.1074/jbc.M005799200. [DOI] [PubMed] [Google Scholar]
- 45.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 46.Depoortere F, Pirson I, Bartek J, Dumont JE, Roger PP. Transforming growth factor beta(1) selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-cdk4 with nuclear p27(kip1) Mol Biol Cell. 2000;11:1061–1076. doi: 10.1091/mbc.11.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sandhu C, Garbe J, Bhattacharya N, Bhattacharya N, Daksis J, Pan CH, et al. Transforming growth factor beta stabilizes p15INK4B protein, increases p15INK4B-cdk4 complexes, and inhibits cyclin D1-cdk4 association in human mammary epithelial cells. Mol Cell Biol. 1997;17:2458–2467. doi: 10.1128/mcb.17.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fargeas C, Wu CY, Nakajima T, Cox D, Nutman T, Delespesse G. Differential effect of transforming growth factor beta on the synthesis of Th1- and Th2-like lymphokines by human T lymphocytes. Eur J Immunol. 1992;22:2173–2176. doi: 10.1002/eji.1830220833. [DOI] [PubMed] [Google Scholar]
- 49.Palladino MA, Morris RE, Starnes HF, Levinson AD. The transforming growth factor-betas. A new family of immunoregulatory molecules. Ann NY Acad Sci. 1990;593:181–187. doi: 10.1111/j.1749-6632.1990.tb16110.x. [DOI] [PubMed] [Google Scholar]
- 50.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 51.Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–144. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- 52.Ryther RC, Flynt AS, Phillips JA, 3rd, Patton JG. siRNA therapeutics: big potential from small RNAs. Gene Ther. 2005;12:5–11. doi: 10.1038/sj.gt.3302356. [DOI] [PubMed] [Google Scholar]
- 53.Carette JE, Overmeer RM, Schagen FH, Alemany R, Barski OA, Gerritsen WR, et al. Conditionally replicating adeno-viruses expressing short hairpin RNAs silence the expression of a target gene in cancer cells. Cancer Res. 2004;64:2663–2667. doi: 10.1158/0008-5472.can-03-3530. [DOI] [PubMed] [Google Scholar]
- 54.Song J, Pang S, Lu Y, Yokoyama KK, Zheng JY, Chiu R. Gene silencing in androgen-responsive prostate cancer cells from the tissue-specific prostate-specific antigen promoter. Cancer Res. 2004;64:7661–7663. doi: 10.1158/0008-5472.CAN-04-1751. [DOI] [PubMed] [Google Scholar]
- 55.Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G, et al. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nano-particle. Nucleic Acids Res. 2004;32:e149. doi: 10.1093/nar/gnh140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sumimoto H, Miyagishi M, Miyoshi H, Yamagata S, Shimizu A, Taira K, et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004;23:6031–6039. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 57.Sumimoto H, Yamagata S, Shimizu A, Miyoshi H, Mizuguchi H, Hayakawa T, et al. Gene therapy for human small-cell lung carcinoma by inactivation of Skp-2 with virally mediated RNA interference. Gene Ther. 2005;12:95–100. doi: 10.1038/sj.gt.3302391. [DOI] [PubMed] [Google Scholar]
- 58.Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. Retrovirally mediated RNA interference targeting the M2 subunit of ribonucleotide reductase: A novel therapeutic strategy in pancreatic cancer. Surgery. 2004;136:261–269. doi: 10.1016/j.surg.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 59.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–247. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 60.Chen J, Wall NR, Kocher K, Duclos N, Fabbro D, Neuberg D, et al. Stable expression of small interfering RNA sensitizes TEL-PDGFbetaR to inhibition with imatinib or rapamycin. J Clin Invest. 2004;113:1784–1791. doi: 10.1172/JCI20673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen LM, Le HY, Qin RY, Kumar M, Du ZY, Xia RJ, et al. Reversal of the phenotype by K-rasval12 silencing mediated by adenovirus-delivered siRNA in human pancreatic cancer cell line Panc-1. World J Gastroenterol. 2005;11:831–838. doi: 10.3748/wjg.v11.i6.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uchida H, Tanaka T, Sasaki K, Kato K, Dehari H, Ito Y, et al. Adenovirus-mediated transfer of siRNA against survivin induced apoptosis and attenuated tumor cell growth in vitro and in vivo. Mol Ther. 2004;10:162–171. doi: 10.1016/j.ymthe.2004.05.006. [DOI] [PubMed] [Google Scholar]