

Abstract
A second scaffold of 1,4-thienodiazepine-2,5-diones was discovered and is synthetically accessible from Gewald 2-aminothiophenes via Ugi-Deprotection-Cyclization (UDC) strategy. This approach yielded hybrid peptidomimetic diazepine structures with six points of diversity introduced from readily available starting materials. A virtual compound library (N = 50,000) was generated and evaluated for chemical space distribution and drug-like properties.
Keywords: multicomponent reaction; Ugi reaction; Gewald reaction; 1,4-thienodiazepine-2,5-dione; 2-aminothiophene; peptidomimetic
The synthesis and evaluation of new scaffolds based on privileged structures play an important role in the drug discovery process. Privileged structures are often preferentially chosen by medicinal chemists, since there is the believe that the “spirit” of an approved and successful compound based on such a scaffold can be transferred into a new compound for a different indication (1). Over 40 medications highlight 1,4-benzodiazepine as a classic privileged structure with a broad range of therapeutic treatment (2). Since the discovery of benzodiazepine family, many synthetic derivatives with a wide pharmacological spectrum have been extensively developed (3,4). For example, farnesyltransferase (FTase) inhibitor BMS-214662 has undergone evaluation as an anti-cancer drug in phase I and II clinical trials (5). Moreover, 1,4-benzodiazepines may act as mimetics of peptide secondary structures such as α-helix and β-turn, due to their unique structural motifs and physicochemical properties (6-9). It is noteworthy that 1,4-benzodiazepine-2,5-diones (BDZ) as a sub-family of benzodiazepine scaffolds have been synthesized as cyclic peptide structures with diversely substituted moieties (10-12). Recently, hybrid peptidomimetic BDZs have been investigated during the lead generation process, such as tripeptide compounds 1-3 (Figure 1) (13-17). Therefore, the development of new synthetic scaffolds based on 1,4-diazepines has attracted considerable attention in the design of biologically active compounds (18,19).
Figure 1.

Hybrid peptidomimetic 1,4-benzodiazepine-2,5-diones
The chemistry developed based on 1,4-benzodiazepine scaffolds has contributed to the emergence of thiophene as a useful isostere of a benzene ring (20). Bioisosterism successfully led to the discovery of a series of 1,4-thienodiazepine drugs (such as olanzapine, clotiazepam, and brotizolam), which are heterocyclic compounds containing a 1,4-diazepine ring fused to a thiophene ring. It’s of great interest to develop the synthesis of new 1,4-thienodiazepine scaffolds, which are much less investigated compared to 1,4-benzodiazepines. Recently, compound 4 has been identified as an inhibitor of MAPKAP Kinase-2 (MK-2) (21). The co-crystal structure of compound 4 and MK-2 complex reveals the unique properties of a 1,4-thienodiazepine scaffold (Figure 2) (21). However, chemical space and the structural biology of 1,4-thienodiazepines is still largely unexplored. We are particularly interested in the synthesis of new 1,4-thienodiazepine-2,5-dione (TDZ) scaffolds, which have been not investigated as potential pharmacophore so far.
Figure 2.

X-ray crystal structure of compound 4 bound to MAPKAP kinase-2 (MK-2, PDB code: 3FYK). Compound 4 is shown in yellow sticks. The receptor binding pocket is shown, and some amino acids are labeled. The hydrogen bonding network is shown in red dash lines.
Multicomponent reaction (MCR) chemistry is particularly useful for the fast and efficient discovery of diverse scaffolds (22). Isocyanide-based MCRs provide powerful tools for producing diverse arrays of compounds with high atom economy (23). In search of a novel synthetic route for the construction of TDZ scaffolds, 2-aminothiophene has been considered as the most appropriate precursor. Gewald three-component reaction (G-3CR) is a unique method using elemental sulfur to yield 2-aminothiophenes 5, which builds a platform for the synthesis of new thiophene scaffolds (24,25). We found that Gewald 2-aminothiophene is clearly an isostere of anthranilic acids (26). Ugi four-component reaction (U-4CR) is known to be one of the most versatile tools for the construction of α-aminoacylcarbonamides and related backbones (27). For example, orthogonally protected anthranilic acids were used as a key synthon for the synthesis of 1,4-benzodiazepin-2,5-diones via the Ugi-Deprotection-Cyclization (UDC) approach (28-31). In our recent study, we developed Ugi-Deprotection-Cyclization (UDC) strategy using 2-aminothiophene derivatives 6 and ethyl glyoxalate to access 1,4-thienodiazepine-2,5-dione derivatives as potential p53-Mdm2 antagonists (32). Herein, we utilized the UDC strategy (Figure 3) to synthesize another new scaffold of 1,4-thienodiazepine-2,5-diones starting from 2-aminothiophenes, which were prepared by Gewald reactions.
Figure 3.

UDC approach for the synthesis of 1,4-thienodiazepine-2,5-dione scaffolds (R1, R2, R3, R4, R5, R6: points of diversity)
Materials and Methods
General
All reagents were purchased from commercial sources and used without further purification. The reactions were conducted under air atmosphere unless otherwise indicated. Analytical thin-layer chromatography (TLC) was performed on SiO2 plates on Alumina available from Whatman. Visualization was accomplished by UV irradiation at 254 nm. Chromatography was conducted using Preparative Silica gel TLC plates (1000 μm, 20cm×20cm). Proton and carbon NMR spectra were determined on Bruker Avance™ 600 MHz NMR spectrometer. Chemical shifts are reported as δ values in parts per million (ppm) as referenced to residual solvent. 1H NMR spectra are tabulated as follows: chemical shift, number of protons, multiplicity (s = singlet, br.s = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet), and coupling constant. High Resolution Mass spectra were obtained at the University of Pittsburgh Mass Spectrometry facility. LC-MS analysis was performed on an SHIMADZU instrument, using an analytical C18 column (Dionex Acclaim 120 Å, 2.1 × 50 mm, 3.0 μm, 0.2 mL/min). Acetonitrile/water mixtures were used as mobile phase for reverse-phase HPLC coupled to electrospray ionization-mass spectrometry (ESI-MS).
N-(tert-butyl)-2-(2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno[2,3-e][1,4] diazepin-4-yl)acetamide (7a, Method A)
The mixture of 6a (59.4 mg, 0.2 mmol), glycine methyl ester hydrochloride (0.2 mmol, 25.0 mg), triethylamine (0.2 mmol, 27.9 μL), aqueous formaldehyde (0.2 mmol, 14.9 μL), tert-butyl isocyanide (0.2 mmol, 22.6 μL) in 0.5 mL of methanol was stirring under RT for 2 days. The reaction was quenched by water, and extracted by DCM. The organic layer was washed by saturated potassium carbonate (aq), and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by 0.5 mL of TFA, stirring under RT for 24 h. The reaction was added 10 mL of DCM, then neutralized by saturated potassium carbonate (aq). The mixture was extracted by DCM, the organic layer was combined and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by triethylamine (50 μL) and TBD (10 mg) in 0.5 mL of THF, stirring overnight under 40°C. 7a was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (32 mg, yield: 46% over three steps). HPLC/MS: tR = 9.42 min; m/z = 350.1 [M+H]+ HRMS: 349.145979 (found); C17H23N3O3S, 349.14601 (calcd.). 1H NMR (600 MHz, CDCl3): 1.33 (9H, s), 1.77 (2H, m), 1.83 (2H, m), 2.62 (2H, m), 2.76 (2H, m), 4.08 (2H, s), 4.12 (2H, s), 6.21 (1H, s). 13C NMR (150 MHz, CDCl3): 22.3, 23.0, 24.5, 25.8, 28.7, 51.5, 52.5, 52.7, 121.7, 129.2, 135.0, 141.4, 164.4, 167.4, 169.0.
N-(cyclopropylmethyl)-2-(2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno [2,3-e][1,4]diazepin-4-yl)acetamide (7b, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (12 mg, yield: 17% over three steps). HPLC/MS: tR = 9.12 min; m/z = 348.1 [M+H]+ HRMS: 347.131963 (found); C17H21N3O3S, 347.13036 (calcd.). 1H NMR (600 MHz, CDCl3): 0.19 (2H, m), 0.48-0.50 (2H, m), 0.94 (1H, m), 1.77-1.84 (4H, m), 2.64 (2H, m), 2.77 (2H, m), 3.11 (2H, m), 4.10 (2H, s), 4.21 (2H, s), 6.49 (1H, s). 13C NMR (150 MHz, CDCl3): 3.4, 10.5, 22.3, 23.0, 24.5, 25.8, 44.3, 52.0, 52.5, 121.7, 129.2, 135.1, 141.3, 164.5, 168.0, 168.8.
N-(2-phenylethyl)-2-(2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno[2,3-e] [1,4]diazepin-4-yl)acetamide (7c, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (10 mg, yield: 13% over three steps). HPLC/MS: tR = 9.81 min; m/z = 398.1 [M+H]+ HRMS: 397.145798 (found); C21H23N3O3S, 397.14601 (calcd.). 1H NMR (600 MHz, CDCl3): 1.77-1.83 (4H, m), 2.63 (2H, m), 2.73 (2H, m), 2.80 (2H, m), 3.51 (2H, m), 3.99 (1H, s), 4.15 (2H, s), 6.48 (1H, m), 7.17-7.21 (3H, m), 7.25-7.28 (2H, m). 13C NMR (150 MHz, CDCl3): 22.3, 23.0, 24.5, 25.8, 35.5, 40.6, 52.1, 52.4, 121.5, 126.5, 128.6, 128.8, 129.1, 135.0, 138.7, 141.6, 164.5, 168.2, 168.7.
N-(tert-butyl)-2-(2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno[2,3-e][1,4] diazepin-4-yl)-3-methylbutanamide (7d, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (12 mg, yield: 15% over three steps). HPLC/MS: tR = 10.69 min; m/z = 392.2 [M+H]+ HRMS: 391.192703 (found); C20H29N3O3S, 391.19296 (calcd.). 1H NMR (600 MHz, d6-DMSO): 0.72 (3H, d, J = 6.0 Hz), 0.89 (3H, d, J = 6.6 Hz), 1.23 (9H, s), 1.61-1.78 (5H, m), 2.10 (1H, m), 2.58 (2H, m), 3.16 (1H, m), 3.86 (1H, d, J = 14.4 Hz), 4.63 (1H, d, J = 10.8 Hz), 7.72 (1H, s). 13C NMR (150 MHz, d6-DMSO): 19.2, 19.6, 22.5, 23.1, 24.4, 26.1, 28.0, 28.8, 47.2, 50.7, 50.8, 121.0, 127.2, 134.5, 159.9, 164.2, 169.9.
N-(tert-butyl)-1-(2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno[2,3-e][1,4] diazepin-4-yl)cyclohexanecarboxamide (7e, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (8 mg, yield: 10% over three steps). HPLC/MS: tR = 11.00 min; m/z = 418.3 [M+H]+ HRMS: 440.1981 (found); C22H31N3NaO3S, 440.19783 (calcd.). 1H NMR (600 MHz, CD3OD): 1.34 (9H, s), 1.45-1.51 (3H, m), 1.61-1.65 (2H, m), 1.81-1.92 (6H, m), 2.03 (2H, m), 2.23 (1H, m), 2.45 (1H, m), 2.67 (2H, m), 2.97 (1H, m), 4.11 (2H, s), 6.55 (1H, s). 13C NMR (150 MHz, CD3OD): 22.2, 22.8, 23.9, 25.1, 25.7, 27.48, 27.50, 46.8, 50.7, 50.8, 66.69, 66.71, 122.6, 128.4, 134.4, 142.1, 165.4, 170.7, 174.3.
N-(tert-butyl)-2-(4-chlorophenyl)-2-(2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1] benzothieno[2,3-e][1,4]diazepin-4-yl)acetamide (7f, Method B)
The mixture of 6a (59.4 mg, 0.2 mmol), glycine methyl ester hydrochloride (0.2 mmol, 25.0 mg), triethylamine (0.2 mmol, 27.9 μL), o-chloro-benzylaldehyde (0.2 mmol, 28.0 mg), tert-butyl isocyanide (0.2 mmol, 22.6 μL) in 0.5 mL of methanol was stirring under RT for 2 days. The reaction was quenched by water, and extracted by DCM. The organic layer was washed by saturated potassium carbonate (aq), and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by 0.5 mL of TFA, stirring under RT for 24 h. The reaction was added 10 mL of DCM, then neutralized by saturated potassium carbonate (aq). The mixture was extracted by DCM, the organic layer was combined and dried over anhydrous sodium sulfate. After the evaporation of the solvent, 7f was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (34 mg, yield: 37% over two steps). HPLC/MS: tR = 11.08 min; m/z = 460.1 [M+H]+ HRMS: 459.137649 (found); C23H26ClN3O3S, 459.13834 (calcd.). 1H NMR (600 MHz, CD3OD): 1.31-1.34 (2H, m), 1.38 (9H, s), 1.74-1.90 (4H, m), 2.67 (2H, m), 3.23 (1H, m), 3.87 (1H, m), 6.22 (1H, s), 7.33 (2H, d, J = 7.8 Hz), 7.43 (2H, d, J = 7.8 Hz), 7.94 (1H, s). 13C NMR (150 MHz, CD3OD): 7.8, 22.2, 22.8, 23.9, 25.5, 27.4, 46.5, 51.2, 120.7, 128.2, 128.8, 130.8, 134.1, 134.2, 134.4, 142.8, 165.1, 169.3, 169.6.
N-(tert-butyl)-2-(2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno[2,3-e][1,4] diazepin-4-yl)-3-phenylpropanamide (7g, Method B)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (18 mg, yield: 21% over two steps). HPLC/MS: tR = 11.03 min; m/z = 440.3 [M+H]+ HRMS: 439.193597 (found); C24H29N3O3S, 439.19296 (calcd.). 1H NMR (600 MHz, d6-DMSO): 0.86 (1H, s), 1.22 (9H, s), 1.61-1.69 (4H, m), 2.55 (2H, s), 2.88 (1H, m), 3.14 (1H, m), 3.96 (1H, m), 5.36 (1H, s), 7.22 (1H, m), 7.26 (3H, m), 7.29 (1H, m), 7.44 (1H, m), 7.51 (1H, s). 13C NMR (150 MHz, d6-DMSO): 22.4, 23.1, 24.3, 26.0, 28.2, 28.8, 35.8, 48.0, 50.9, 57.6, 120.7, 126.6, 127.1, 128.4, 129.4, 134.6, 137.9, 164.0, 169.7.
Methyl 2-(2-(tert-butoxycarbonylamino)-N-(2-(tert-butylamino)-2-oxoethyl)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamido)-3-methylbutanoate (8)
The mixture of 6a (74.3 mg, 0.25 mmol), valine methyl ester hydrochloride (0.25 mmol, 41.8 mg), triethylamine (0.25 mmol, 34.8 μL), aqueous formaldehyde (0.25 mmol, 18.6 μL), tert-butyl isocyanide (0.25 mmol, 28.3 μL) in 0.5 mL of methanol was stirring under RT for 1 day. The reaction was quenched by water, and extracted by DCM. The organic layer was washed by saturated potassium carbonate (aq), and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 3:1) as yellowish solids (90 mg, yield: 69%). HRMS: 523.272509 (found); C26H41N3O6S, 523.27161 (calcd.). 1H NMR (600 MHz, CDCl3): 0.82 (1H, d, J = 6.0 Hz), 0.85 (1H, d, J = 6.6 Hz), 1.36 (9H, s), 1.50 (9H, s), 1.74-1.82 (4H, m), 2.05 (1H, m), 2.24 (1H, m), 2.62-2.65 (3H, m), 3.68 (3H, s), 3.86 (1H, d, J = 15.0 Hz), 4.02 (1H, d, J = 10.2 Hz), 4.62 (1H, d, J = 15.6 Hz), 5.53 (1H, s), 10.03 (1H, s). 13C NMR (150 MHz, CDCl3): 18.9, 19.4, 22.6, 23.4, 24.0, 28.2, 28.3, 28.6, 29.6, 45.0, 51.8, 52.1, 66.3, 80.3, 126.6, 129.7, 137.5, 153.0, 167.6, 168.8, 170.6.
N-(tert-butyl)-2-(3-isopropyl-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno [2,3-e][1,4]diazepin-4-yl)acetamide (9a, Method C)
The mixture of 6a (74.3 mg, 0.25 mmol), valine methyl ester hydrochloride (0.25 mmol, 41.8 mg), triethylamine (0.25 mmol, 34.8 μL), aqueous formaldehyde (0.25 mmol, 18.6 μL), tert-butyl isocyanide (0.25 mmol, 28.3 μL) in 0.5 mL of methanol was stirring under RT for 2 days. The reaction was quenched by water, and extracted by DCM. The organic layer was washed by saturated potassium carbonate (aq), and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by 0.5 mL of TFA, stirring under 40 °C overnight. The reaction was added 10 mL of DCM, then neutralized by saturated potassium carbonate (aq). The mixture was extracted by DCM, the organic layer was combined and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by triethylamine (50 μL) and TBD (10 mg) in 0.5 mL of THF, stirring overnight under 40 °C. 9a was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (22 mg, yield: 23% over three steps). HPLC/MS: tR = 10.44 min, m/z = 392.3 [M+H]+ HRMS: 391.192778 (found); C20H29N3O3S, 391.19296 (calcd.). 1H NMR (600 MHz, CDCl3, major rotamer): 0.92 (1H, d, J = 6.6 Hz), 0.96 (1H, d, J = 6.6 Hz), 1.32 (9H, s), 1.69 (1H, m), 1.77 (1H, m), 1.89-1.96 (3H, m), 2.43 (1H, m), 2.60-2.68 (2H, m), 3.07 (1H, m), 3.70 (1H, d, J = Hz), 4.07-4.16 (2H, ABd, J = Hz), 6.66 (1H, s). 13C NMR (150 MHz, CDCl3, major rotamer): 19.4, 20.0, 23.0, 24.5, 25.8, 26.7, 28.6, 51.3, 57.3, 73.8, 122.0, 128.8, 134.7, 140.1, 163.6, 167.6, 170.0.
N-(cyclopropylmethyl)-2-(3-isopropyl-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1] benzothieno[2,3-e][1,4]diazepin-4-yl)acetamide (9b, Method C)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (16 mg, yield: 21% over three steps). HPLC/MS: tR = 10.10 min; m/z = 490.3 [M+H]+ HRMS: 389.176195 (found); C20H27N3O3S, 389.17731 (calcd.). 1H NMR (600 MHz, CDCl3, major rotamer): 0.16-0.19 (2H, m), 0.46- 0.48 (2H, m), 0.88 (3H, d, J = 6.6 Hz), 0.92-0.94 (1H, m), 0.95 (3H, d, J = 6.6 Hz), 1.66-1.76 (2H, m), 1.88-1.96 (3H, m), 2.43-2.46 (1H, m), 2.59-2.66 (2H, m), 3.02-3.08 (2H, m), 3.11-3.14 (1H, m), 3.71 (1H, d, J = 11.4 Hz), 4.17 (1H, ABd, J = 15.0 Hz), 4.25 (1H, ABd, J = 15.0 Hz), 6.91 (1H, s). 13C NMR (150 MHz, CDCl3, major rotamer): 3.4, 3.5, 10.6, 19.1, 20.0, 22.3, 23.0, 24.5, 25.9, 26.7, 44.3, 56.1, 73.8, 121.8, 128.6, 134.7, 140.7, 163.7, 168.4, 169.9.
N-benzyl-2-(3-isopropyl-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno[2,3-e][1,4]diazepin-4-yl)acetamide (9c, Method C)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (20 mg, yield: 24% over three steps). HPLC/MS: tR = 10.50 min; m/z = 426.3 [M+H]+ HRMS: 425.177499 (found); C23H27N3O3S, 425.17731 (calcd.). 1H NMR (600 MHz, CDCl3, major rotamer): 0.79 (3H, d, J = 6.6 Hz), 0.90 (3H, d, J = 6.0 Hz), 1.65-1.72 (2H, m), 1.83-1.93 (3H, m), 2.35-2.38 (1H, m), 2.57-2.61 (1H, m), 2.62-2.63 (1H, m), 2.98-3.00 (1H, m), 3.68 (1H, d, J = 11.4 Hz), 4.11 (1H, d, J = 15.0 Hz), 4.33-4.45 (3H, m), 7.22-7.31 (6H, m). 13C NMR (150 MHz, CDCl3, major rotamer): 19.2, 19.9, 22.3, 22.9, 24.5, 25.8, 26.7, 43.5, 56.2, 74.0, 121.7, 127.3, 127.8, 128.6, 128.8, 134.6, 137.9, 140.8, 163.8, 168.5, 169.8.
N-(tert-butyl)-2-(3-benzyl-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno [2,3-e][1,4]diazepin-4-yl)acetamide (9d, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (18 mg, yield: 21% over three steps). HPLC/MS: tR = 10.89 min; m/z = 440.2 [M+H]+ HRMS: 439.194543 (found); C24H29N3O3S, 439.19296 (calcd.). 1H NMR (600 MHz, CDCl3, 1:1 mixture of rotamers): 1.30 (9H, s), 1.34 (9H, s), 1.71-1.81 (4H, m), 1.91-1.95 (4H, m), 2.52-2.66 (6H, m), 2.97 (2H, m), 3.10 (2H, m), 3.20 (1H, m), 3.62 (2H, m), 3.88 (1H, m), 4.09 (1H, m), 4.19 (1H, m), 4.35 (1H, m), 4.49 (1H, m), 5.99 (1H, s), 6.47 (1H, s), 7.05-7.06 (2H, m), 7.21-7.28 (8H, m). 13C NMR (150 MHz, CDCl3, 1:1 mixture of rotamers): 22.3, 22.4, 22.99, 23.03, 24.58, 24.60, 25.5, 26.2, 28.59, 28.63, 32.6, 34.6, 48.3, 51.3, 51.4, 55.4, 57.7, 68.1, 122.3, 122.5, 126.9, 127.3, 128.5, 128.6, 128.9, 129.0, 129.1, 129.45, 129.50, 134.7, 135.0, 135.6, 136.6, 140.2, 141.4, 163.2, 165.5, 167.4, 168.3, 168.4, 169.6.
N-(cyclopropylmethyl)-2-(3-benzyl-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1] benzothieno[2,3-e][1,4]diazepin-4-yl)acetamide (9e, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (32 mg, yield: 37% over three steps). HPLC/MS: tR = 10.53 min; m/z = 438.2 [M+H]+ HRMS: 460.1687 (found); C24H27N3NaO3S, 460.16708 (calcd.). 1H NMR (600 MHz, CDCl3, 1:1 mixture of rotamers): 0.12 (2H, m), 0.16-0.18 (2H, m), 0.40-0.42 (2H, m), 0.45-0.46 (2H, m), 0.84-0.95 (2H, m), 1.75-1.77 (4H, m), 1.90-1.93 (4H, m), 2.55-2.63 (6H, m), 2.92-2.96 (2H, m), 2.98-3.02 (2H, m), 3.06-3.08 (4H, m), 3.25 (1H, m), 3.56 (1H, m), 3.67-3.75 (2H, m), 4.02 (1H, m), 4.13 (1H, m), 4.22 (1H, m), 4.38 (1H, m), 4.51 (1H, m), 6.44 (1H, m), 6.85 (1H, m), 7.04-7.05 (2H, m), 7.19-7.28 (8H, m). 13C NMR (150 MHz, CDCl3, 1:1 mixture of rotamers): 3.3, 3.4, 10.5, 10.6, 22.3, 22.4, 23.00, 23.04, 24.6, 25.5, 26.2, 32.6, 34.3, 41.0, 44.3, 47.2, 52.1, 54.8, 55.7, 57.8, 68.3, 122.2, 122.3, 126.9, 127.3, 128.57, 128.60, 128.8, 128.9, 129.1, 129.3, 129.4, 129.5, 134.6, 134.8, 135.7, 136.6, 140.7, 141.8, 163.4, 165.7, 168.1, 168.5, 168.8, 169.6.
N-(tert-butyl)-2-(3-isobutyl-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1]benzothieno [2,3-e][1,4]diazepin-4-yl)acetamide (9f, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (11 mg, yield: 14% over three steps). HPLC/MS: tR = 10.87 min; m/z = 406.2 [M+H]+ HRMS: 405.209380 (found); C21H31N3O3S, 405.20861 (calcd.). 1H NMR (600 MHz, CDCl3, a mixture of rotamers): 0.88-0.91 (6H, m), 0.93-0.98 (6H, m), 1.34 (9H, s), 1.37 (5H, s), 1.47-1.50 (2H, m), 1.57-1.65 (3H, m), 1.71-1.75 (3H, m), 1.80-1.82 (2H, m), 1.90-1.93 (4H, m), 2.09 (1H, m), 2.45-2.54 (2H, m), 2.63-2.71 (3H, m), 2.92 (1H, m), 3.07-3.11 (2H, m), 3.22 (1H, m), 3.32 (1H, m), 3.79 (1H, d, J = 15.6 Hz), 3.94 (1H, d, J = 15.0 Hz), 4.12 (1H, d, J = 15.6 Hz), 4.18 (1H, m), 4.22 (1H, m), 4.31 (1H, d, J = 15.0 Hz), 6.35 (1H, s), 6.46 (1H, s). 13C NMR (150 MHz, CDCl3, a mixture of rotamers): 22.0, 22.26, 22.28, 22.32, 22.4, 22.7, 22.8, 22.96, 23.02, 24.55, 24.59, 25.0, 25.1, 25.5, 25.7, 26.0, 28.6, 28.7, 35.0, 37.9, 42.7, 48.3, 50.5, 51.2, 51.3, 51.7, 52.0, 54.8, 56.1, 60.1, 65.1, 122.4, 122.7, 128.7, 129.5, 134.8, 135.0, 139.7, 141.0, 163.5, 165.9, 167.6, 168.2, 168.8, 170.4.
N-(tert-butyl)-2-[3-(4-hydroxyphenyl)-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1] benzothieno[2,3-e][1,4]diazepin-4-yl]acetamide (9g, Method A)
Sodium bicarbonate was used for the workup instead of saturated potassium carbonate (aq). The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (20 mg, yield: 23% over three steps). HPLC/MS: tR = 9.53 min; m/z = 442.2 [M+H]+ HRMS: 441.173542 (found); C23H27N3O4S, 441.17223 (calcd.). 1H NMR (600 MHz, CD3OD): 1.36 (9H, s), 1.50 (1H, m), 1.67-1.77 (3H, m), 2.22 (1H, m), 2.46 (2H, m), 2.70 (1H, m), 2.85 (1H, m), 3.22 (1H, m), 4.13 (1H, m), 4.64 (1H, m), 5.22 (1H, m), 6.62 (2H, m), 6.97 (2H, m), 7.62 (1H, m). 13C NMR (150 MHz, CD3OD): 22.0, 22.7, 23.7, 25.2, 27.6, 34.1, 43.3, 51.0, 53.3, 63.5, 68.5, 114.8, 123.2, 124.2, 125.2, 128.2, 133.4, 140.3, 156.8, 164.7, 167.7, 170.6.
N-(tert-butyl)-2-[3-(1H-indol-3-ylmethyl)-2,5-dioxo-1,2,3,5,6,7,8,9-octahydro-4H-[1] benzothieno [2,3-e][1,4]diazepin-4-yl]acetamide (9h, Method A)
The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (15 mg, yield: 16% over three steps). HPLC/MS: tR = 10.68 min; m/z = 479.3 [M+H]+ HRMS: 478.203784 (found); C26H30N4O3S, 478.20386 (calcd.). 1H NMR (600 MHz, CDCl3, 1:1 mixture of rotamers): 1.32 (9H, s), 1.38 (9H, s), 1.68-1.96 (8H, m), 2.53-2.65 (6H, m), 3.11-3.20 (4H, m), 3.36 (1H, m), 3.68 (1H, m), 3.86 (1H, m), 3.99-4.03 (2H, m), 4.18 (1H, m), 4.48-4.50 (2H, m), 6.01 (1H, s), 6.38 (1H, s), 6.89 (1H, s), 7.05 (1H, s), 7.09-7.13 (3H, m), 7.18 (1H, m), 7.25 (1H, d, J = 7.8 Hz), 7.34 (1H, d, J = 7.8 Hz), 7.53 (1H, d, J = 7.8 Hz), 7.58 (1H, d, J = 7.8 Hz), 8.27 (1H, s), 8.52 (1H, s). 13C NMR (150 MHz, CDCl3, 1:1 mixture of rotamers): 22.2, 22.3, 22.4, 23.0, 24.3, 24.5, 24.6, 25.6, 26.3, 28.6, 28.7, 48.0, 51.36, 51.44, 55.6, 56.9, 67.1, 109.3, 110.2, 111.3, 111.4, 118.17, 118.23, 119.6, 122.00, 122.04, 122.1, 122.5, 123.4, 123.8, 126.8, 127.0, 128.8, 129.5, 134.6, 135.1, 135.9, 136.1, 140.0, 141.1, 163.3, 165.7, 167.6, 168.2, 168.7, 170.0.
N-(tert-butyl)-2-(3-isopropyl-2,5-dioxo-1,2,3,5-tetrahydro-4H-thieno[2,3-e][1,4] diazepin-4-yl)acetamide (10a)
The mixture of 6b (48.6 mg, 0.2 mmol), valine methyl ester hydrochloride (0.2 mmol, 33.5 mg), triethylamine (0.2 mmol, 27.9 μL), aqueous formaldehyde (0.2 mmol, 14.9 μL), tert-butyl isocyanide (0.2 mmol, 22.6 μL) in 0.5 mL of methanol was stirring under RT for 2 days. The Ugi product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 5:1), then treated by 0.5 mL of DCM (10% TFA). The reaction mixture was stirring under RT for 24 hours. After the evaporation of the solvent, the residue was treated by triethylamine (100 μL) and TBD (10 mg) in 0.5 mL of THF, stirring overnight under 40 °C. The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 1:1) as yellowish solids (5 mg, yield: 7% over three steps). HPLC/MS: tR = 8.99 min; m/z = 338.3 [M+H]+ HRMS:337.145879 (found); C16H23N3O3S, 337.14601 (calcd.). 1H NMR (600 MHz, CDCl3, major rotamer): 0.94 (1H, d, J = 6.6 Hz), 0.98 (1H, d, J = 6.6 Hz), 1.34 (9H, s), 1.95 (1H, m), 3.75 (1H, d, J = 9.6 Hz), 4.03 (1H, d, J = 15.0 Hz), 4.27 (1H, d, J = 15.0 Hz), 6.44 (1H, m), 6.87 (1H, d, J = 6.0 Hz), 8.60 (1H, br.s). 13C NMR (150 MHz, CDCl3, major rotamer): 19.4, 19.9, 27.2, 28.6, 57.2, 73.7, 117.0, 123.8, 128.1, 142.3, 163.2, 167.0, 169.0.
N-(tert-butyl)-2-(2,5-dioxo-7-phenyl-1,2,3,5-tetrahydro-4H-thieno[2,3-e][1,4] diazepin-4-yl)acetamide (10b)
The mixture of 6c (63.8 mg, 0.2 mmol), glycine methyl ester hydrochloride (0.2 mmol, 25.0 mg), triethylamine (0.2 mmol, 27.9 μL), aqueous formaldehyde (0.2 mmol, 14.9 μL), tert-butyl isocyanide (0.2 mmol, 22.6 μL) in 0.5 mL of methanol was stirring under RT for 2 days. The reaction was quenched by water, and extracted by DCM. The organic layer was washed by saturated potassium carbonate (aq), and dried over anhydrous sodium sulfate. After the evaporation of the solvent, the residue was treated by 1.0 mL of DCM (10% TFA), stirring under RT for 24 h. After the evaporation of the solvent, the residue was treated by triethylamine (200 μL) and TBD (10 mg) in 0.5 mL of THF, stirring overnight under 40 °C. The product was isolated by silica gel chromatography (petroleum ether/ ethyl acetate, 2:1) as yellowish solids (8 mg, yield: 11% over three steps). HPLC/MS: tR = 9.66 min; m/z = 372.1 [M+H]+ HRMS: 371.128759 (found); C19H21N3O3S, 371.13036 (calcd.). 1H NMR (600 MHz, CD3OD): 1.37 (9H, s), 4.09 (2H, s), 4.21 (2H, s), 7.31-7.33 (1H, m), 7.40-7.42 (2H, m), 7.47 (1H, s), 7.58-7.59 (2H, m). 13C NMR (150 MHz, CD3OD): 27.5, 50.9, 51.1, 52.9, 122.3, 123.3, 124.9, 127.7, 128.8, 133.0, 135.3, 165.0, 167.7, 168.7.
Results and Discussion
Synthesis of 1,4-thienodiazepine-2,5-dione scaffold
Since 2-aminothiophenes are optimal starting materials for the synthesis of thienodiazepine backbone, we utilized the versatile Gewald multicomponent reactions to prepare compounds 5a-c. We recently developed a general synthetic protocol to synthesize Boc protected thiophene carboxylic acids 6a-c (Scheme 1) (32). In the first step, 2-aminothiophenes 5a-c were obtained by the Gewald reaction of cyclohexanone, 1,4-dithiane-2,5-diol, phenylacetaldehyde, respectively. In the second step, N-Boc thiophene carboxylic acids 6 were prepared by Boc protection of 5 and following hydrolysis transformation. Hence, we intended to employ the bifunctional orthogonally protected intermediates 6 and amino acid derived methyl esters for the synthesis of new TDZ scaffold via UDC approach.
Scheme 1.

Synthesis of 6a-c
The synthetic method was designed to allow rapid access to 1,4-thienodiazepine-2,5-diones in just three steps from the variable precursor building blocks. Initially, we tried the Ugi reaction of 6a, tert-butyl isocyanide, formaldehyde, with glycine methyl ester hydrochloride in the presence of triethylamine under room temperature for 48 hours. After simple extraction workup, the intermediate Ugi product was subject to deprotection with TFA and subsequent cyclization using a catalytic amount of TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene). The product 7a was isolated by chromatography in 46% yield over three steps. The ‘three-step, one-separation’ procedure was applied for the synthesis of 7a-e with variable isocyanides and oxo components (Table 1). Interestingly, compounds 7f and 7g were obtained in two steps after the treatment with TFA. It’s possible that the intramolecular cyclization is favorable even without the treatment of TBD (29).
Table 1.
UDC approach for the synthesis of 1,4-thienodiazepine-2,5-diones from glycine methyl ester
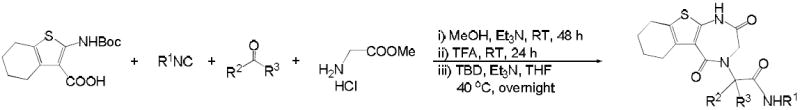
| entry | R1 | oxo component | product | yield |
|---|---|---|---|---|
| 7a | t-Bu | formaldehyde |  |
46%a |
| 7b | cyclopropyl methyl | formaldehyde |  |
17%a |
| 7c | phenyl ethyl | formaldehyde |  |
13%a |
| 7d | t-Bu | isobutylaldehyde |  |
15%a |
| 7e | t-Bu | cyclohexanone | 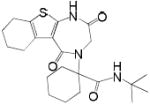 |
10%a |
| 7f | t-Bu | o-chloro-benzaldehyde |  |
37%b |
| 7g | t-Bu | benzylaldehyde |  |
21%b |
Method A, isolated yields (over three steps);
Method B, isolated yields (over two steps).
Next, we investigated a series of amino acid methyl esters for the synthesis of new TDZ scaffolds. We tried the Ugi reaction of 6a, tert-butyl isocyanide, formaldehyde, with valine methyl ester hydrochloride in the presence of triethylamine under room temperature for 24 hours (Scheme 2). The Ugi product 8 was isolated by chromatography in 69% yield. The deprotection of 8 and the following cyclization afford 9a in 59% yield over two steps (41% over three steps). Running the sequence without any isolation yielded compound 9a 23%. Although the overall yield is lower, the ‘three-step, one-separation’ procedure avoids the additional separation step.
Scheme 2.

Synthesis of 9a
Hence, 6a was applied to UDC approach for the synthesis of 1,4-thienodiazepine-2,5-diones with variable amino acid derivatives (Table 2). Valine, phenylalanine, leucine, 4-hydroxy phenylglycine, trptophan methyl esters as well as several isocyanides are tolerant to this procedure. Compounds 9a-h were isolated by chromatography in 14-37% yield over three steps. Since TBD serves as the catalyst, racemization of chiral center at amino acid nucleus is unclear (33).
Table 2.
1,4-Thienodiazepine-2,5-diones from the variation of amino acids
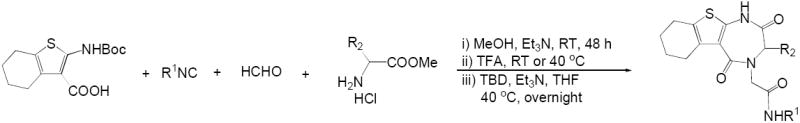
| entry | R1 | amino acid | product | yield |
|---|---|---|---|---|
| 9a | t-Bu | valine |  |
23%b |
| 9b | cyclopropyl methyl | valine |  |
21%b |
| 9c | benzyl | valine |  |
24%b |
| 9d | t-Bu | phenylalanine | 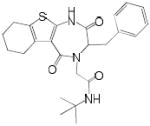 |
21%a |
| 9e | cyclopropyl methyl | phenylalanine |  |
37%a |
| 9f | t-Bu | leucine |  |
14%a |
| 9g | t-Bu | 4-hydroxy phenylglycine |  |
23%a |
| 9h | t-Bu | trptophan |  |
16%a |
Method A, isolated yields (over three steps)
Method C, isolated yields (over three steps).
We also investigated other aminothiophene backbones 6b and 6c for the synthesis of 1,4-thienodiazepine-2,5-diones (Table 3). The corresponding Ugi product of 6b was isolated by chromatography in 46% yield. Compound 10a was obtained in 16% yield by the further transformation of the Ugi product. A 10% solution of TFA in dichloromethane was used for the deprotection step under a mild condition. Similarly, compound 10b was isolated by chromatography in 11% yield over three steps.
Table 3.
1,4-Thienodiazepine-2,5-diones from the variation of aminothiophenes
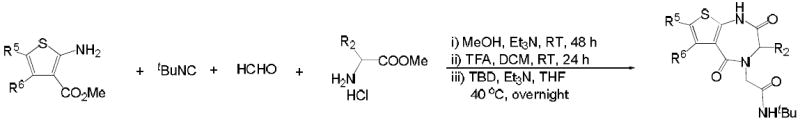
| entry | R5 | R6 | amino acid | product | yielda |
|---|---|---|---|---|---|
| 10a | H | H | valine |  |
7% |
| 10b | Ph | H | glycine |  |
11% |
isolated yields, over three steps.
Conformation analysis of TDZ scaffold
NMR spectra of some compounds substituted with amino acid side chains show clearly the presence of two rotamers, which are not chromatographically separable. For instance, compounds 9d, 9e, 9h show two rotamers at a ratio of 1:1 in CDCl3. The population of rotamers was found related to the deuterium solvent used. For example, the ratio of 9d shows roughly 2:3 in CD3OD (Figure S1 in Supporting Information). This observation suggests that seven-membered diazepine nucleus of thienodiazepinedione is quite rigid, similar to the scaffold of benzodiazepinediones (34,35). The energy barrier for the interconversion of benzodiazepinedione conformers (pseudo-axial and pseudo-equatorial conformers) was calculated up to 14.8 kcal/mol (11). We also speculate that 1,4-thienodiazepine ring inversion is slow enough, therefore conformers can be detected by NMR under room temperature.
Recently, we used MCR methods to develop α-helix mimetics, which could become very important lead structures to (ant-)agonize protein-protein interactions (36). In our ongoing interest for the application of peptidomimetic structures generated from MCRs, the design and synthesis of peptide β-turn mimetic scaffolds and libraries are also desirable (37). A β-turn is most often defined as any tetrapeptide unit occurring in a nonhelical region that causes a reversal of the direction of the peptide chain (Figure 4A) (38). Since 1,4-benzodiazepines were found to act as β-turn mimetics (7-9), we speculate that our 1,4-thienodiazepine-2,5-dione scaffold could also have β-turn mimetic moiety. Thus, the tripeptide fragment of TDZ scaffold was compared with known protein β-turns (Figure 4). The core scaffold was investigated as a model superimposed onto a type II β-turn backbone (PDB code: 1H2C, turn region Ile142-Pro146).
Figure 4.
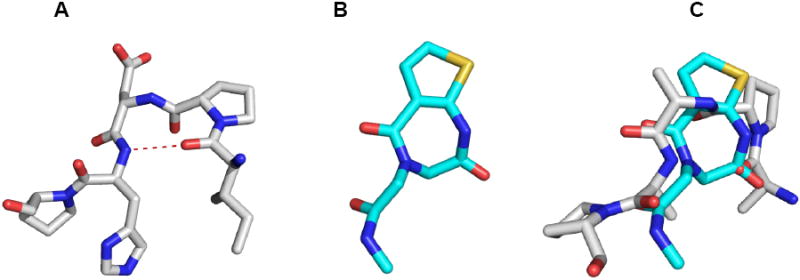
(A) Structure of a typical β-turn. The PDB code for the protein is 1H2C (chain A, turn region Ile142-Pro146). (B) The core scaffold was chosen as a model of the investigated β-turn mimetic. (C) Minimized conformation of core scaffold as a β-turn mimetic superimposed onto a type II β-turn. For clarity, only the backbone and α atoms are shown.
Cheminformatics study of TDZ scaffold
Due to in silico and computational advances, chemoinformatics would help to identify promising scaffolds of greater importance in lead discovery (39). We generated a virtual compound library (N = 50,000) from a random sample of starting materials to evaluate the chemical space of the 1,4-thienodiazepine-2,5-diones (Supplemental Information). These compounds generated from the virtual library were introduced into Instant JChem (Instant JChem 2.5.1, 2009, www.chemaxon.com) for calculating their physical properties. The distributions of this random virtual library (N = 50,000) and commercial available benzodiazepines (N = 2,498) were presented in Figure 5. This new scaffold covers unexplored chemical space of benzodiazepine family. Due to the diversity of chemical space, this scaffold is potentially useful for virtual screening and lead discovery.
Figure 5.

Comparison of chemical space distribution: (A) benzodiazepine library of a substructure search from eMolecules (N = 2,498); (B) a random virtual library of 1,4-thienodiazepine-2,5-diones (N = 50,000).
In order to evaluate the potential of combinatorial library design, the physicochemical properties of the virtual compound library (N = 50,000) were calculated (40). The data was analyzed statistically by frequency distributions in PASW Statistics 18 (Figure S2 in Supplemental Information). Due to the diversity of reactant components, the range of molecular weight is between 267.3 Da. and 580.7 Da. The mean of LogP is 2.46 with standard deviation of 1.84, which indicates acceptable permeability of most compounds. Total polar surface area (TPSA) of the majority of compounds is between 122.3 Å2 and 176.9 Å2. The mean number of rotatable bonds (NRB), hydrogen bond acceptors (HBA), and hydrogen bond donors (HBD) are shown in Table 4. In terms of drug likeness, 79.7% of 50,000 compounds obey Lipinski’s rule. And 93.7% of them are predicted to be bioavailable (mass ≤ 500, LogP ≤ 5, HBD ≤ 5, HBA ≤ 10, PSA ≤ 200, NRB ≤ 10, fused aromatic rings ≤ 5). For comparison, the physicochemical properties of synthesized compounds (N = 17) and a random virtual library (N = 50,000) were summarized in Table 4. The structure-property relationship indicates the rationale of diversity-oriented library design with drug-like properties. Moreover, 3D structures of a random virtual library were generated by the software Omega (Supplemental Information). The compound library of this new thienodiazepine scaffold (N = 5,000) could be used for docking program and other virtual analysis software to discover possible hits of suitable receptors.
Table 4.
Physicochemical properties of synthesized compounds (N = 17) and a random virtual library (N = 50,000)a
| Mean | MW (Da.) | LogP | TPSA (Å2) | NRB | HBA | HBD |
|---|---|---|---|---|---|---|
| synthesized compounds | 407.1 ± 40.7 | 3.73 ± 0.94 | 108.9 ± 6.0 | 4 ± 1 | 4 ± 0 | 2 ± 0 |
| virtual library | 453.7 ± 44.1 | 2.46 ± 1.84 | 149.6 ± 27.3 | 7 ± 2 | 6 ± 1 | 3 ± 1 |
All values listed are mean value plus/minus standard deviation.
Scaffold Hopping
Isofunctional molecules based on different chemical scaffolds are key to the early drug development process. Leads based on different scaffolds can be found by a process called scaffold hopping (41). This process can rely on known or intuitive bioisosteres or on advanced chemoinformatic strategies. Early development of several leads based on different scaffolds has the advantage of reducing the very high attrition rate in preclinical and early clinical development. Additionally, scaffold hopping has great implications for the maintenance of intellectual property. For example, for the benzodiazepine scaffold 4672 structures are registered in SciFinder, whereas only 381 thienodiazepines are known (Figure 6A). We described here two thienodiazepine scaffolds with different 2D and 3D distribution of hydrogen bond donors and acceptors. They are clearly related to their benzodiazepine scaffolds amenable by the UDC method (Figure 6B,C). The chemical space behind the four related scaffolds is however very much different, since the thiophene building block allows for many more simple variations based on the versatile Gewald MCR.
Figure 6.

(A) Known structures; (B) MCR acessible isosteric azepine scaffolds; (C) Other MCR accessible benzodiazepines. The scaffold space of benzodiazepines accessible by MCR is very rich. Six different scaffolds can be synthesized using different isocyanide-based MCR strategies. Of the bioisosteric thienodiazepines currently only two scaffolds are generally amenable by MCR.
Conclusions and Future Directions
In summary, we have synthesized a series of 1,4-thienodiazepine-2,5-diones by using the union of Gewald reaction and Ugi-Deprotection-Cyclization strategy. This approach possesses novel hybrid peptidomimetic 1,4-diazepines with well-defined diversity, which can be achieved from readily available starting materials. UDC strategy allows convenient preparation of 1,4-thienodiazepine-2,5-diones without using the traditional peptide coupling methods. Similar to the compound libraries of benzodiazepines, the TDZ scaffold could also be suitable for high-dimensional combinatorial synthesis to meet the screening purpose. The conformation analysis and chemical space of this novel scaffold was studied. Based on the commonly accepted descriptors, it’s potentially useful to obtain lead-like compounds based on this scaffold.
Supplementary Material
Acknowledgments
This work was supported the NIH grant 1R21GM087617-01 (to AD), and is part of a NCI-RAND program.
Abbreviations
- BDZ
1,4-benzodiazepine-2,5-dione
- G-3CR
Gewald three-component reaction
- MCR
multicomponent reaction
- TBD
1,5,7-triazabicyclo[4,4,0]dec-5-ene
- TDZ
1,4-thienodiazepine-2,5-dione
- UDC
Ugi-Deprotection-Cyclization
- U-4CR
Ugi four-component reaction
Footnotes
Supporting Information Additional supporting information may be found in the online version of this article.
Figure S1: NMR spectra of compound 9d in CDCl3 and CD3OD.
Figure S2: Statistical distributions of physical properties of a random virtual library (N = 50,000).
References
- 1.Duarte CD, Barreiro EJ, Fraga CAM. Privileged structures: A useful concept for the rational design of new lead drug candidates. Mini-Rev Med Chem. 2007;7:1108–1119. doi: 10.2174/138955707782331722. [DOI] [PubMed] [Google Scholar]
- 2.Essman WB. Subcellular actions of benzodiazepines. In: Garattini S, Mussini E, Randall LO, editors. The Benzodiazepines. Raven Press; New York: 1973. pp. 177–190. [Google Scholar]
- 3.Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 4.Kamal A, Reddy KL, Devaiah V, Shankaraiah N, Reddy DR. Recent advances in the solid-phase combinatorial synthetic strategies for the benzodiazepine based privileged structures. Mini-Rev Med Chem. 2006;6:53–69. doi: 10.2174/138955706775197875. [DOI] [PubMed] [Google Scholar]
- 5.Reid TS, Beese LS. Crystal structures of the anticancer clinical candidates R1 15777 (Tipifarnib) and BMS-214662 complexed with protein farnesyltransferase suggest a mechanism of FTI selectivity. Biochemistry. 2004;43:6877–6884. doi: 10.1021/bi049723b. [DOI] [PubMed] [Google Scholar]
- 6.Leonard K, Marugan JJ, Raboisson P, Calvo R, Gushue JM, Koblish HK, et al. Novel 1,4-benzodiazepine-2,5-diones as Hdm2 antagonists with improved cellular activity. Bioorg Med Chem Lett. 2006;16:3463–3468. doi: 10.1016/j.bmcl.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Verdie P, Subra G, Averland-Petit M-C, Amblard M, Martinez J. Solid-phase synthesis of 4-methylcarboxy-1,4-benzodiazepine-2,5-diones. J Comb Chem. 2008;10:869–874. doi: 10.1021/cc800085d. [DOI] [PubMed] [Google Scholar]
- 8.Saudo M, Garcia-Valverde M, Marcaccini S, Delgado JJ, Rojo J, Torroba T. Synthesis of benzodiazepine beta-turn mimetics by an Ugi 4CC/Staudinger/aza-Wittig sequence. Solving the conformational behavior of the Ugi 4CC adducts. J Org Chem. 2009;74:2189–2192. doi: 10.1021/jo8025862. [DOI] [PubMed] [Google Scholar]
- 9.Rosenstrom U, Skold C, Lindeberg G, Botros M, Nyberg F, Karlen A, et al. Design, synthesis, and incorporation of a beta-turn mimetic in angiotensin II forming novel pseudopeptides with affinity for AT1 and AT2 receptors. J Med Chem. 2006;49:6133–6137. doi: 10.1021/jm051222g. [DOI] [PubMed] [Google Scholar]
- 10.McDowell RS, Blackburn BK, Gadek TR, McGee LR, Rawson T, Reynolds ME, et al. From peptide to non-peptide. 2. The de novo design of potent, non-peptidal inhibitors of platelet aggregation based on a benzodiazepinedione scaffold. J Am Chem Soc. 2002;116:5077–5083. [Google Scholar]
- 11.Loudni L, Roche J, Potiron V, Clarhaut J, Bachmann C, Gesson J-P, et al. Design, synthesis and biological evaluation of 1,4-benzodiazepine-2,5-dione-based HDAC inhibitors. Bioorg Med Chem Lett. 2007;17:4819–4823. doi: 10.1016/j.bmcl.2007.06.067. [DOI] [PubMed] [Google Scholar]
- 12.Carlier PR, Zhao H, MacQuarrie-Hunter SL, DeGuzman JC, Hsu DC. Enantioselective synthesis of diversely substituted quaternary 1,4-benzodiazepin-2-ones and 1,4-benzodiazepine-2,5-diones. J Am Chem Soc. 2006;128:15215–20. doi: 10.1021/ja0640142. [DOI] [PubMed] [Google Scholar]
- 13.Goff DA, Zuckermann RN. Solid-phase synthesis of defined 1,4-benzodiazepine-2,5-dione mixtures. J Org Chem. 1995;60:5744–5745. [Google Scholar]
- 14.Alig L, Chucholowski A, Weller T. Benzazepinone and quinazoline derivatives inhibiting the binding of adhesive proteins to vitronectin receptors. 2001 WO2001004103. [Google Scholar]
- 15.Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Marugan JJ, Raboisson P, et al. Enhanced pharmacokinetic properties of 1,4-benzodiazepine-2,5-dione antagonists of the HDM2-p53 protein-protein interaction through structure-based drug design. Bioorg Med Chem Lett. 2006;16:3310–3314. doi: 10.1016/j.bmcl.2006.03.055. [DOI] [PubMed] [Google Scholar]
- 16.Verdie P, Subra G, Feliu L, Sanchez P, Berge G, Garcin G, et al. Online synthesis of pseudopeptide library incorporating a benzodiazepinone turn mimic: Biological evaluation on MC1 receptors. J Comb Chem. 2007;9:254–262. doi: 10.1021/cc060054q. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Chen X, Connors RV, Dai K, Fu Y, Jaen JC, et al. Preparation of benzo-fused heterocycles for treatment of obesity and eating disorders. 2006 WO2006020959. [Google Scholar]
- 18.Costantino L, Barlocco D. Privileged structures as leads in medicinal chemistry. Curr Med Chem. 2006;13:65–85. [PubMed] [Google Scholar]
- 19.DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Privileged structures: Applications in drug discovery. Comb Chem High Throughput Screen. 2004;7:473–493. doi: 10.2174/1386207043328544. [DOI] [PubMed] [Google Scholar]
- 20.Burger A. Isosterism and bioisosterism in drug design. Prog Drug Res. 1991;37:287–371. doi: 10.1007/978-3-0348-7139-6_7. [DOI] [PubMed] [Google Scholar]
- 21.Anderson DR, Meyers MJ, Kurumbail RG, Caspers N, Poda GI, Long SA, et al. Benzothiophene inhibitors of MK2. Part 1: Structure-activity relationships, assessments of selectivity and cellular potency. Bioorg Med Chem Lett. 2009;19:4878–4881. doi: 10.1016/j.bmcl.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J, Bienaymé H, editors. Multicomponent Reactions. Wiley-VCH: Weinheim; 2005. [Google Scholar]
- 23.Dömling A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- 24.Sabnis RW, Rangnekar DW, Sonawane ND. 2-Aminothiophenes by the Gewald reaction. J Heterocycl Chem. 1999;36:333–345. [Google Scholar]
- 25.Wang K, Kim D, Dömling A. Cyanoacetamide MCR (III): Three-component Gewald reactions revisited. J Comb Chem. 2010;12:111–118. doi: 10.1021/cc9001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Dömling A. The Gewald multicomponent reaction. Mol Divers. 2010;14 doi: 10.1007/s11030-010-9229-6. in press. [DOI] [PubMed] [Google Scholar]
- 27.Dömling A, Ugi I. Multicomponent reactions with isocyanides. Angew Chem Int Ed. 2000;39:3169–3210. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 28.Keating TA, Armstrong RW. A remarkable two-Step synthesis of diverse 1,4-benzodiazepine-2,5-diones using the Ugi four-component condensation. J Org Chem. 1996;61:8935–8939. doi: 10.1021/jo961517p. [DOI] [PubMed] [Google Scholar]
- 29.Hulme C, Ma L, Kumar NV, Krolikowski PH, Allen AC, Labaudiniere R. Novel applications of resin bound alpha-amino acids for the synthesis of benzodiazepines (via Wang resin) and ketopiperazines (via hydroxymethyl resin) Tetrahedron Lett. 2000;41:1509–1514. [Google Scholar]
- 30.Faggi C, Marcaccini S, Pepino R, Pozo MC. Studies on isocyanides and related compounds; Synthesis of 1,4-benzodiazepine-2,5-diones via Ugi four-component condensation. Synthesis. 2002:2756–2760. [Google Scholar]
- 31.Liu A, Zhou H, Su G, Zhang W, Yan B. Microwave-assisted fluorous synthesis of a 1,4-benzodiazepine-2,5-dione library. J Comb Chem. 2009;11:1083–1093. doi: 10.1021/cc900109e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Wolf S, Bista M, Meireles L, Holak TA, Camacho C, et al. 1,4-Thienodiazepine-2,5-diones via MCR (I): Synthesis, virtual space and p53-Mdm2 activity. Chem Biol Drug Des. 2010 doi: 10.1111/j.1747-0285.2010.00989.x. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Joyner S, Khoury K, Dömling A. (-)-Bacillamide C: the convergent approach. Org Biomol Chem. 2010;8:529–532. doi: 10.1039/b918214d. [DOI] [PubMed] [Google Scholar]
- 34.Jadidi K, Aryan R, Mehrdad M, Lugger T, Hahn FE, Ng SW. Simple synthesis, structure and ab initio study of 1,4-benzodiazepine-2,5-diones. J Mol Struct. 2004;692:37–42. [Google Scholar]
- 35.Araujo AC, Nicotra F, Airoldi C, Costa B, Giagnoni G, Fumagalli P, et al. Synthesis and biological evaluation of novel rigid 1,4-benzodiazepine-2,5-dione chimeric scaffolds. Eur J Org Chem. 2008:635–639. [Google Scholar]
- 36.Antuch W, Menon S, Chen Q-Z, Lu Y, Sakamuri S, Beck B, et al. Design and modular parallel synthesis of a MCR derived alpha-helix mimetic protein-protein interaction inhibitor scaffold. Bioorg Med Chem Lett. 2006;16:1740–1743. doi: 10.1016/j.bmcl.2005.11.102. [DOI] [PubMed] [Google Scholar]
- 37.Souers AJ, Ellman JA. Beta-turn mimetic library synthesis: Scaffolds and applications. Tetrahedron. 2001;57:7431–7448. [Google Scholar]
- 38.Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 39.Oprea TI. Chemical space navigation in lead discovery. Curr Opin Chem Biol. 2002;6:384–389. doi: 10.1016/s1367-5931(02)00329-0. [DOI] [PubMed] [Google Scholar]
- 40.Oprea TI. Property distribution of drug-related chemical databases. J Comput Aided Mol Des. 2000;14:251–264. doi: 10.1023/a:1008130001697. [DOI] [PubMed] [Google Scholar]
- 41.Schneider G, Schneider P, Renner S. Scaffold-hopping: How far can you jump? QSAR Comb Sci. 2006;25:1162–1171. [Google Scholar]
- 42.Keating TA, Armstrong RW. Postcondensation modifications of Ugi four-component condensation products: 1-isocyanocyclohexene as a convertible isocyanide. Mechanism of conversion, synthesis of diverse structures, and demonstration of resin capture. J Am Chem Soc. 1996;118:2574–2583. [Google Scholar]
- 43.Hulme C, Peng J, Tang S-Y, Burns CJ, Morize I, Labaudiniere R. Improved procedure for the solution phase preparation of 1,4-benzodiazepine-2,5-dione libraries via Armstrong’s convertible isonitrile and the Ugi reaction. J Org Chem. 1998;63:8021–8023. [Google Scholar]
- 44.Tempest P, Ma V, Kelly MG, Jones W, Hulme C. MCC/SNAr methodology. Part 1: Novel access to a range of heterocyclic cores. Tetrahedron Lett. 2001;42:4963–4968. [Google Scholar]
- 45.Marcaccini S, Miliciani M, Pepino R. A facile synthesis of 1,4-benzodiazepine derivatives via Ugi four-component condensation. Tetrahedron Lett. 2005;46:711–713. [Google Scholar]
- 46.Cuny G, Bois-Choussy M, Zhu J. Palladium- and copper-catalyzed synthesis of medium- and large-sized ring-fused dihydroazaphenanthrenes and 1,4-benzodiazepine-2,5-diones Control of reaction pathway by metal-switching. J Am Chem Soc. 2004;126:14475–14484. doi: 10.1021/ja047472o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.