

Abstract
The pathogenesis of several neuropsychiatric disorders, including anxiety and depression, has been linked to oxidative stress, in part via alterations in cyclic nucleotide signaling. Phosphodiesterase-2 (PDE2), which regulates cGMP and cAMP signaling, may affect anxiety-related behavior through reduction of oxidative stress. The present study evaluated the effects of oxidative stress on behavior and assessed the anxiolytic effects of the PDE2 inhibitor Bay 60-7550 [(2-(3,4-dimethoxybenzyl)-7-{(1R)-1-[(1R)-1-hydroxyethyl]-4-phenylbutyl}-5-methyl imidazo-[5,1-f][1,2,4]triazin-4(3H)-one)]. Treatment of mice with L-buthionine-(S,R)-sulfoximine (300 mg/kg), an inducer of oxidative stress, caused anxiety-like behavioral effects in elevated plus-maze, open-field, and hole-board tests through the NADPH oxidase pathway; these effects were antagonized by Bay 60-7550 (3 mg/kg) and apocynin (3 mg/kg), an inhibitor of NADPH oxidase. The Bay 60-7550-mediated decrease in oxidative stress (i.e., superoxide anion and reactive oxygen species generation in cultured neurons and total antioxidant capacity and lipid peroxides in amygdala and hypothalamus) and expression of NADPH oxidase subunits (i.e., p47 phox and gp91 phox expression in amygdala, hypothalamus, and cultured neurons) was associated with increased cGMP and phosphorylation of vasodilator-stimulated phosphoprotein at Ser239, suggesting an important role of cGMP-protein kinase G signaling in reduction of anxiety. Overall, the present results indicate that oxidative stress induces anxiety-like behavior in mice and that PDE2 inhibition reverses it through an increase in cGMP signaling. Thus, PDE2 may be a novel pharmacological target for treatment of anxiety in neuropsychiatric and neurodegenerative disorders that involve oxidative stress.
Anxiety and fear are normal emotional responses to threatening stimuli. These responses are abnormal in human anxiety disorders, such as panic disorder, post-traumatic stress disorder, social phobias, and generalized anxiety disorder. The development of anxiety/stress-related disorders involves complex interactions among various body mechanisms involving the limbic system and the hypothalamic-pituitary-adrenal axis; their interactions play a significant role in the manifestation of disease pathology (Chrousos and Gold, 1992; Ray et al., 1993). Exposure to stressful stimuli produces widespread physiological and behavioral effects in animals. In recent studies, oxidative stress has been shown to be associated with anxiety in different behavioral models (Gingrich, 2005; Hovatta et al., 2005; Berry et al., 2007).
The nervous system, due to enriched concentrations of polyunsaturated fatty acids, is particularly susceptible to the deleterious effects of oxidative stress; this can lead to loss of membrane integrity, protein damage, and neuronal dysfunction. Recent studies have shown that social phobia, depression, anxiety, and other neuropsychiatric disorders result in signs of oxidative stress such as increased reactive oxygen generation and decreased antioxidant capacity (Arranz et al., 2007; Bouayed et al., 2007). There is increasing evidence that oxidative stress in neurons is involved in pathological manifestations of many neurological disorders. Thus, there is a need to evaluate the direct effects of oxidative stress on anxiety-related behavior.
Phosphodiesterase-2 (PDE2) belongs to a family of proteins that regulate the intracellular levels of both cGMP and cAMP. cGMP/cAMP signaling, being generally anti-inflammatory in nature, could play an important role in the reduction of oxidative stress. Increased cGMP/cAMP signaling in many systems, including the nervous system, has been shown to suppress reactive oxygen species (ROS) generation and oxidative stress (Urushitani et al., 2000). However, the role of cGMP/cAMP signaling has not been studied in relation to oxidative stress and anxiety.
PDE2 expression is high in many regions of the brain (Boess et al., 2004; Reyes-Irisarri et al., 2007) and in the adrenal gland (Nikolaev et al., 2005). Inhibition of PDE2 results in increased cGMP levels that could influence anxiety/stress-related events (Werner et al., 2004). Several lines of evidence also indicate that targeting PDE2 with selective inhibitors may offer novel strategies in the treatment of age-related and Alzheimer’s disease-associated impairments in memory and behavior (Boess et al., 2004), which are thought to involve oxidative stress (de la Monte and Wands, 2006).
The present study was conducted to evaluate the effects of oxidative stress on anxiety-like behavior in mice and its modulation by cGMP-protein kinase G (PKG) signaling through PDE2 inhibition. It was found that oxidative stress leads to anxiogenic behavior in mice, which is reversed by PDE2 inhibition, probably through an increase in cGMP-PKG signaling.
Materials and Methods
Animals
Male ICR mice, 25 to 30 g, were used (Harlan, Indianapolis, IN). Rodent chow and tap water were freely available. Mice were kept in a temperature-controlled room under standard laboratory conditions, with a 12-h light/dark cycle (lights on at 7:00 AM). All experiments were carried out according to the Institute of Laboratory Animal Resources (1996) and were approved by the Institutional Animal Care and Use Committee of West Virginia University.
Drugs and Chemicals
Bay 60-7550 (Bayer AG, Wuppertal, Germany), KT-5823, H89, diphenyliodonium, and 8-Br-cGMP (Sigma-Aldrich, St. Louis, MO) were dissolved in 50% dimethyl sulfoxide (Fisher Scientific Co., Pittsburgh, PA), whereas L-buthionine-(S,R)-sulfoximine (BSO) and apocynin (Sigma-Aldrich) were dissolved in normal saline. Dulbecco’s modified Eagle’s medium (DMEM), penicillin G, streptomycin, and amphotericin B (Fungizone) were purchased from Invitrogen (Carlsbad, CA). Poly-L-lysine, DNase, and plasma-derived horse serum were purchased from Sigma-Aldrich. Trypsin was purchased from Worthington Biochemicals (Freehold, NJ), and bicinchoninic acid protein assay kits were purchased from Pierce Chemical (Rockford, IL). Tetrodotoxin was purchased from Alexis Laboratories (San Diego, CA).
Behavioral Tests
Elevated Plus-Maze Test
Behavior in the elevated plus-maze test was assessed as described previously (Masood et al., 2003). The elevated plus-maze (San Diego Instruments, San Diego, CA) consisted of two open arms (30 × 5 cm) and two enclosed arms (30 × 5 × 15 cm) that extended from a central platform (5 × 5 cm). The entire maze was elevated 40 cm above the floor. During the first 5 min of free exploration, the number of entries into and the time spent in open and closed arms were recorded. An entry was defined as all four paws in an arm.
Hole-Board Test
Behavior in the hole-board test was assessed as described previously (Bhattacharya et al., 1997). The hole-board apparatus consisted of a Perspex box (60 × 60 × 35 cm) with four equidistant holes 4 cm in diameter in the floor. Each animal was placed in one corner of the hole board and was observed for 5 min for the number of head dips and total time spent head dipping.
Open-Field Test
Behavior in the open-field test was assessed as described previously (Masood et al., 2003). The open field was made of white acrylic (50 × 50 cm) with 22-cm-high walls. The floor was divided into 16 squares by black parallel and intersecting lines. Mice were placed singly in one corner of the open field, and entry latency (i.e., time to enter the first adjacent square), ambulation, and rearing were observed during a 5-min test.
The experimental room for behavioral testing was an isolated quiet room with dimly lit environment. All behavioral experiments were conducted by a human observer in a blind fashion.
Drug Treatments
BSO (300 mg/kg i.p.) was administered 48 and 24 h before behavioral testing (Liu et al., 1996). Apocynin (3 mg/kg i.p.), Bay 60-7550 (3 mg/kg i.p.), and diazepam (1 mg/kg i.p.) were given 30 min before each BSO treatment. Control mice also were treated with apocynin (3 mg/kg i.p.), Bay 60-7550 (3 mg/kg i.p.), or diazepam (1 mg/kg i.p.), in the absence of BSO, to assess the effects of these drugs alone, 48 and 24 h before behavioral testing. Bay 60-7550 shows 50-fold selectivity for PDE2 compared with PDE1, 100-fold compared with PDE5, and greater than 200-fold compared with the other PDE families (Boess et al., 2004). All drug solutions were prepared fresh and administered in a volume of 5 ml/kg. BSO has been reported earlier to enhance the generation of reactive oxygen species (ROS) and increase oxidative stress in the nervous system (de Bernardo et al., 2004). After behavioral testing, mice were killed by decapitation, brains were removed, and hypothalamus and amygdala dissected and stored at −80°C until biochemical and molecular analyses were carried out as shown in Fig. 1.
Fig. 1.

Schematic presentation of the protocol used in the present study for behavioral, neurochemical, and molecular experiments in mice (A) and parallel neurochemical and molecular experiments in vitro using cultured neurons (B).
Primary Cultures of Rat Cerebral Cortical Neurons
Primary cultures of rat cerebral cortical neurons were prepared as described previously (Suvarna and O’Donnell, 2002). The dishes were then incubated at 37°C in a 5% CO2, 95% O2 atmosphere. On day 3, the medium was replaced with fresh DMEM containing 10 μM β-cytosine arabinoside to diminish glial cell proliferation. On day 5, the β-cytosine arabinoside-containing media was aspirated, and regular DMEM was added to the cells. The cells were grown for 7 more days before they were used in experiments on day 12.
The cells were rinsed and incubated for 50 min at 37°C in HEPES buffer (140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 100 nM glycine, 15 mM glucose, and 25 mM HEPES, pH 7.4) containing 1 μM tetrodotoxin. Activators and inhibitors [8-Br-cGMP (250 nM), diphenyliodonium (10 μM), Bay 60-7550 (1 μM), apocynin (10 μM), KT-5823 (20 μM), H89 (10 μM)] were added 30 min before the addition of BSO (1 mM). Four hours after the addition of BSO, the cells were used for biochemical (superoxide anion, ROS, and cGMP production) and immunocytochemical (gp91 phox and p47 phox immunoreactivity) analyses as shown in Fig. 1.
Biochemical and Molecular Assays
Total Antioxidant Capacity
Total antioxidant capacity of non-enzymatic antioxidants in amygdala and hypothalamus was measured by the method of Benzie and Strain (1996) as described previously (Nadeem et al., 2006). Ferric tripyridyltriazine complex is reduced by an antioxidant present in the sample, which can be monitored by measuring the change in absorption at 593 nm, which is directly related to the total reducing power of electron-donating nonenzymatic antioxidants present in the reaction mixture. The components that contribute to total antioxidant capacity are ascorbate, α-tocopherol, uric acid, bilirubin, and other, less abundant antioxidants. Results are expressed as micromoles per milligram of protein.
Lipid Peroxides
Lipid peroxides in amygdala and hypothalamus were measured as malondialdehyde (MDA)-thiobarbituric acid (TBA) adducts by the method of Jentzsch et al. (1996). The sample was incubated with 5 mM butylated hydroxyl toluene, 0.2 M ortho-phosphoric acid, and 0.11 M TBA in a total volume of 500 μl at 90°C for 45 min, followed by ice-cooling and extraction of MDA-TBA adducts in n-butanol. Absorption was read at 535 and 572 nm for baseline correction in a multititer plate reader. MDA-TBA adducts were calculated using the difference in absorption at the two wavelengths compared with the standard curve generated by the use of tetraethoxypropane. Results are expressed in micromoles per milligram of protein.
Real-Time RT-PCR for NADPH Oxidase Subunits
Total RNA was isolated from amygdala and hypothalamus using the TRIzol reagent (Invitrogen) followed by DNase treatment to eliminate potential genomic DNA contamination as described by Nadeem et al. (2007). This was followed by conversion of 0.5 μg of total RNA into cDNA using the High Capacity cDNA archive kit (Applied Biosystems, Foster City, CA) in a total volume of 100 μl. Real-time PCR was performed on an ABI PRISM 7300 Detection System (Applied Biosystems) using TaqMan Universal Mastermix (Applied Biosystems). The reaction volume (25 μl) included 12.5 μl of 2× TaqMan Universal Mastermix, 1 μl of cDNA, and 1.25 μl of 20× 5-carboxy-fluorescein-labeled TaqMan gene expression assay master mix solution. For the real-time PCR for NADPH oxidase subunit genes (p40 phox, p47 phox, p67 phox, and gp91 phox), the TaqMan inventoried assays-on-demand gene expression products were purchased from Applied Biosystems. 18S rRNA was used as an endogenous control. The fold difference in expression of target cDNA was determined using the comparative threshold cycle method described by Livak and Schmittgen (2001).
Western Blot for p-VASPSer239 and gp91 Phox
Aliquots of the supernatant from amygdala and hypothalamus (40 μg protein/well) were separated using 10% SDS-polyacrylamide gel electrophoresis; prestained protein molecular markers (20- to 112-KDa low range) were run in parallel. Proteins were then transferred to nitrocellulose membranes and probed with antibodies for the detection of vasodilator-stimulated phosphoprotein (VASP) phosphorylated at Ser239 (anti-p-VASPSer239 rabbit polyclonal IgG; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and gp91 phox (anti-gp91 phox mouse monoclonal IgG; BD Biosciences, San Jose, CA), both diluted 1:1000. This was followed by incubation with the respective secondary horseradish peroxidase-conjugated antibodies (GE Healthcare, Chalfont St. Giles, UK) for 1 h at room temperature. For detection of bands, the membranes were treated with enhanced chemiluminescence reagent (GE Healthcare) for 1 min and exposed to enhanced chemiluminescence Hyperfilm; relative band intensities were quantified by densitometry. Labeled protein bands were compared within individual gels/blots and expressed as percentage of control density value.
Extracellular Superoxide Anion Generation
Superoxide anion generation by neuronal cultures after the indicated treatments (see above) was measured as the superoxide dismutase-inhibitable reduction of WST-1 by the method of Tan and Berridge (2000). Release of superoxide anion was monitored for 60 min at 37°C in the presence of 500 μM WST-1. The difference in absorption of culture medium, in the presence or absence of superoxide dismutase, was determined by spectrophotometry at 450 nm. The amount of superoxide produced was calculated on the basis of molar extinction coefficient of 37 mM/cm for WST-1. Results are expressed as percentage of superoxide production in controls.
Intracellular ROS Generation
For intracellular ROS generation in neuronal cultures, cells were grown in 96-well microplates; after the indicated treatment schedule (see above), the cells were incubated in 100 μM 6-carboxy-2′,7′-dichlorofluorescein diacetate with 5% CO2/95% air at 37°C for 30 min. 6-Carboxy-2′,7′-dichlorofluorescein diacetate forms a fluorescent product, dichlorofluorescein, upon oxidation with ROS. Fluorescence caused by dichlorofluorescein in each well was measured and recorded for 15 min using an HD Synergy multiwell fluorescence plate reader (PerSeptive Biosystems Inc., Framingham, MA) with temperature maintained at 37°C (Wang and Joseph, 1999). The excitation filter was set at 485 nm and the emission filter at 530 nm. The final values are expressed as percentage of fluorescence in control wells.
Measurement of cGMP
After the indicated treatments (see above), neurons were scraped into 100 μl of 0.1 N ice-cold hydrochloric acid, sonicated, and centrifuged at 13,000g for 10 min at 4°C. The supernatant was separated for the cGMP assay (Suvarna and O’Donnell, 2002), and the pellet was used for protein estimation by the Bradford method (Bio-Rad, Hercules, CA). Levels of cGMP were measured by enzyme-based immunoassay (Assay Designs, Ann Arbor, MI); cGMP was normalized to the protein content for each sample.
Immunocytochemistry for gp91 phox and p47 phox Subunits of NADPH Oxidase
For immunocytochemistry, cells were grown on coverslips and, after the indicated treatments (see above), fixed in acetone/methanol (ratio of 6:4). For detection of NADPH oxidase subunits, gp91 phox and p47 phox, defrosted coverslips were incubated with an anti-gp91 phox mouse monoclonal IgG (1:100 dilution; BD Biosciences) or anti-p47 phox rabbit polyclonal IgG (1:100 dilution; Santa Cruz Biotechnology, Inc.) diluted in phosphate-buffered saline-Triton X-bovine serum albumin, pH 7.8, in a humid chamber at 4°C overnight followed by incubation with fluorescein isothiocyanate-labeled chicken anti-mouse IgG (Zymed Laboratories, South San Francisco, CA) for gp91 phox and rhodamine-conjugated goat anti-rabbit IgG (Millipore, Billerica, MA) for p47 phox at 37°C for 45 min. The coverslips were mounted with Fluoro-mount and observed with an Olympus AX70 fluorescence microscope (Melville, NY) equipped for fluorescein (excitation wavelengths, 455–500 nm; emission wavelengths, >510 nm) and rhodamine (excitation, 540–504 nm; emission, >580 nm). Nonspecific background labeling was determined by omission of primary antisera.
Statistical Analysis
Data are expressed as means ± S.E.M. Data were analyzed by analysis of variance followed by Tukey’s multiple comparison test. A p value less than 0.05 was considered statistically significant.
Results
Effect of Oxidative Stress on Behavior and Its Reversal by Bay 60-7550 and Apocynin
Elevated Plus Maze
Induction of oxidative stress with BSO (300 mg/kg i.p.) produced a decrease in both percentage of open arm entries and percentage of open arm time compared with control; the difference was significant for percentage of open arm entries. The PDE2 inhibitor Bay 60-7550 (3 mg/kg i.p.) reversed the oxidative stress-induced decrease in both elevated plus-maze test parameters significantly. Apocynin (3 mg/kg i.p.), a specific NADPH oxidase inhibitor, had effects similar to Bay 60-7550, increasing both elevated plus-maze test parameters, but significance was achieved only for percentage of open arm entries (Fig. 2A). The benzodiazepine anxiolytic diazepam (1 mg/kg i.p.) did not reverse the BSO-induced changes in behavior in the elevated plus-maze test. Mice treated with apocynin or Bay 60-7550 alone showed a trend toward an anxiolytic effect with increased percentage of open arm time, but this effect was not significant. Likewise, diazepam alone also produced a slight, albeit nonsignificant, anxiolytic effect.
Fig. 2.

The PDE2 inhibitor Bay 60-7550 (Bay) and the NADPH oxidase inhibitor apocynin, but not diazepam (DZP), reversed the anxiogenic effect of BSO, an inducer of oxidative stress. This was evidenced by increased open arm entries and open arm time in the elevated plus-maze (A), increased number of and time spent head dipping in the hole-board test (B), and reduced entry latency in the open-field test (C). Values are expressed as means ± S.E.M., n = 6–7 per group. #, p < 0.05 compared with control; *, p < 0.05 compared with BSO-treated group.
Hole-Board Test
Induction of oxidative stress with BSO (300 mg/kg i.p.) produced a decrease in the number of head dips and the time spent in dipping, but only the latter was significantly decreased compared with control. The PDE2 inhibitor Bay 60-7550 (3 mg/kg i.p.) reversed the decrease in the hole-board parameters caused by oxidative stress, with significantly increased number of head dips and time spent head dipping. Apocynin (3 mg/kg i.p.) also reversed the oxidative stress-induced effect, increasing both hole-board test parameters (Fig. 2B). Diazepam (1 mg/kg i.p.) did not reverse any of the changes induced by BSO in the hole-board test. Behavior of mice was not affected significantly by Bay 60-7550 or apocynin administered alone. Diazepam alone produced a small increase in head-dipping behavior, but this effect was not significant.
Open-Field Test
Induction of oxidative stress with BSO (300 mg/kg i.p.) resulted in an increase in entry latency (Fig. 2C) and a decrease in both ambulation and rearing (Fig. 2D) compared with control; however, only entry latency and rearing response were significant. The PDE2 inhibitor Bay 60-7550 (3 mg/kg i.p.) attenuated oxidative stress-induced effects by reversing the effects of BSO on all three parameters significantly. Apocynin (3 mg/kg i.p.) also reversed oxidative stress-induced effects on all three parameters, but significance was achieved only for entry latency (Fig. 2C) and rearing (Fig. 2D). Diazepam (1 mg/kg i.p.) did not reverse any of the BSO-induced changes in open field parameters significantly but tended to reduce entry latency. Treatment of mice with apocynin, Bay 60-7550, or diazepam alone resulted in a tendency toward reduced entry latency.
Induction of Oxidative Stress in the Hypothalamus and Amygdala with BSO Treatment and Its Reversal by Bay 60-7550 and Apocynin
Because the amygdala and hypothalamus are involved in anxiety-related behaviors, the role of PDE2 inhibition in reversal of oxidative stress-induced changes in these brain regions was assessed. Treatment with BSO (300 mg/kg i.p.) led to increased lipid peroxides in both hypothalamus (Fig. 3A) and amygdala (Fig. 4A), but the effect was significant only in the hypothalamus. The PDE2 inhibitor Bay 60-7550 (3 mg/kg i.p.) reversed BSO-induced lipid peroxidation, as did the NADPH oxidase inhibitor apocynin (3 mg/kg i.p.), as shown in Figs. 3A and 4A. Bay 60-7550 and apocynin alone had no effect on the level of lipid peroxides in hypothalamus and amygdala.
Fig. 3.

The PDE2 inhibitor Bay 60-7550 (Bay) and NADPH oxidase inhibitor apocynin reversed the BSO-induced increase in oxidative stress in the hypothalamus. This was evidenced by a decrease in lipid peroxides (A) and an increase in total antioxidant capacity (B). Values are expressed as means ± S.E.M., n = 4/group. #, p < 0.05 compared with control; *, p < 0.05 compared with BSO-treated group.
Fig. 4.

The PDE2 inhibitor Bay 60-7550 (Bay) and NADPH oxidase inhibitor apocynin reversed the BSO-induced increase in oxidative stress in the amygdala. This was evidenced by a decrease in lipid peroxides (A) and an increase in total antioxidant capacity (B). Values are expressed as means ± S.E.M., n = 4/group. #, p < 0.05 compared with control; *, p < 0.05 compared with BSO-treated group.
Treatment with BSO (300 mg/kg i.p.) also reduced total antioxidant capacity in the hypothalamus (Fig. 3B) and amygdala (Fig. 4B). The PDE2 inhibitor Bay 60-7550 (3 mg/kg i.p.) significantly attenuated the BSO-induced decrease in total antioxidant capacity, both in hypothalamus and amygdala. Apocynin (3 mg/kg i.p.) also reversed the BSO-induced decrease in total antioxidant capacity, but the difference was significant only for the hypothalamus, as shown in Figs. 3B and 4B. Bay 60-7550 and apocynin alone each increased total antioxidant capacity slightly, both in hypothalamus and amygdala, but these changes were not significant.
Induction of NADPH Oxidase Expression in the Hypothalamus and Amygdala with BSO Treatment and Its Reversal by Bay 60-7550 and Apocynin
To further explore whether BSO-induced oxidative stress and anxiety resulted from activation of the NADPH oxidase pathway, the expression of NADPH oxidase subunits in both amygdala and hypothalamus was measured. Treatment of mice with BSO (300 mg/kg i.p.) resulted in increased gene expression of subunits of NADPH oxidase in both hypothalamus [p47 phox (Fig. 5B), gp91 phox (Fig. 5A), and p40 phox (data not shown)] and amygdala [p67 phox (Fig. 6B) and gp91 phox (Fig. 6A)]. Western blotting was done to confirm the real-time PCR data; this showed increased gp91 phox protein expression after BSO treatment in both hypothalamus (Fig. 5C) and amygdala (Fig. 6C). As shown in Figs. 5 and 6, the PDE2 inhibitor Bay 60-7550 (3 mg/kg i.p.) attenuated the BSO-induced increase in the expression of NADPH oxidase subunits in hypothalamus and amygdala, respectively, as did the NADPH oxidase inhibitor apocynin (3 mg/kg i.p.). Bay 60-7550 and apocynin alone produced slight decreases in the expression of NADPH oxidase subunits both in hypothalamus and amygdala, but these effects were not significant.
Fig. 5.

BSO caused oxidative stress by increasing the expression of the NADPH oxidase subunits gp91 phox (A) and p47 phox (B) as shown by real-time PCR (n = 7–10) and gp91 phox protein (C) as shown by Western blotting (n = 3–4) in the hypothalamus. The PDE2 inhibitor Bay 60-7550 (Bay) and the NADPH oxidase inhibitor apocynin reversed the BSO-induced increase in the expression of NADPH oxidase subunits. Values are expressed as means ± S.E.M. #, p < 0.05 compared with control; *, p < 0.05 compared with BSO-treated group.
Fig. 6.

BSO caused oxidative stress by increasing the expression of NADPH oxidase subunits gp91 phox (A) and p47 phox (B) as shown by real-time PCR (n = 7–10) and gp91 phox protein (C) as shown by Western blotting (n = 3–4) in the amygdala. The PDE2 inhibitor Bay 60-7550 (Bay) and the NADPH oxidase inhibitor apocynin reversed the BSO-induced increase in the expression of NADPH oxidase subunits. Values are expressed as means ± S.E.M. #, p < 0.05 compared with control; *, p < 0.05 compared with BSO-treated group.
Effects of BSO, Bay 60-7550, and Apocynin on p-VASPSer239 Expression in the Hypothalamus and Amygdala of Mice
Induction of oxidative stress by treatment with BSO (300 mg/kg i.p.) had little effect on the expression p-VASPSer239, whereas the PDE2 inhibitor Bay 60-7550 increased phosphorylation of VASPSer239, both in hypothalamus and amygdala (Fig. 7). Phosphorylation of VASP at Ser239 has been used as a specific marker of cGMP-mediated signaling through PKG activation (Smolenski et al., 1998; Deguchi et al., 2002). Apocynin, a specific inhibitor of NADPH oxidase, had no effect on the expression of p-VASPSer239.
Fig. 7.

The PDE2 inhibitor Bay 60-7550 (Bay) increased p-VASPSer239 phosphoprotein expression as measured by Western blotting (n = 3–4) in the hypothalamus (A) and amygdala (B) of mice treated with BSO, an inducer of oxidative stress. BSO and apocynin did not change p-VASPSer239 expression. Values are expressed as means ± S.E.M. *, p < 0.05 compared with control.
Effects of BSO, Bay 60-7550, and Apocynin on NADPH Oxidase, Superoxide/ROS Generation, and cGMP Levels in Primary Cultures of Cerebral Cortical Neurons
Immunofluorescent photomicrographs show NADPH oxidase subunits, i.e., gp91 phox (Fig. 8, A–F, green fluorescence) and p47 phox (Fig. 8, G–L, red fluorescence) in cultured neurons. These show increased immunoreactivity of the NADPH oxidase subunits gp91 phox (Fig. 8B) and p47 phox (Fig. 8H) in BSO-treated cells compared with control. Bay 60-7550 (Fig. 8, C and I) or apocynin (Fig. 8, F and L) treatment before BSO resulted in a decrease in gp91 phox and p47 phox immunoreactivity, respectively, compared with BSO-treated cells. The PKG inhibitor KT-5823 (20 μM) given before Bay 60-7550 (1 μM) + BSO (1 mM) reversed the Bay 60-7550-induced decrease in immunoreactivity of gp91 phox and p47 phox subunits (Fig. 8, D and J), respectively, whereas the PKA inhibitor H89 (10 μM) had little effect (Fig. 8, E and K).
Fig. 8.

Immunoreactivity of gp91 phox and p47 phox, subunits of NADPH oxidase in the neuronal cultures treated with BSO, an inducer of oxidative stress; the PDE2 inhibitor Bay 60-7550 (Bay) and the NADPH oxidase inhibitor apocynin reversed the BSO-induced increase in the immunoreactivity of gp91 phox and p47 phox subunits. The PKG inhibitor KT-5823 markedly blocked the Bay 60-7550-induced effect, increasing gp91 phox and p47 phox immunoreactivity. The PKA inhibitor H89 had little effect. Representative immunofluorescent photomicrographs of gp91 phox (A–F; green staining) and p47 phox immunostaining (G–L; red staining) in neuronal cultures are shown (magnification, 200×).
End products of NADPH oxidase activation, i.e., superoxide and ROS generation, were measured to confirm immunocytochemical observations. BSO treatment (1 mM) of cultured neurons resulted in increased superoxide anion (Fig. 9A) and ROS generation (Fig. 9B). Bay 60-7550 (1 μM), apocynin (10 μM), and the flavoprotein inhibitor diphenyliodonium (10 μM, data not shown) significantly reversed the BSO-induced increase in superoxide and ROS generation. The PKG inhibitor KT-5823 (20 μM) reversed the Bay 60-7550-induced decrease in superoxide and ROS generation, whereas the PKA inhibitor H89 (10 μM) had little effect. The cGMP analog 8-Br-cGMP (250 nM) mimicked the effects produced by Bay 60-7550. Figure 9C shows the effect of the PDE2 inhibitor Bay 60-7550 on cGMP levels in cultured neurons. Bay 60-7550 increased cGMP levels, whereas BSO and apocynin had no significant effect on cGMP levels in cultured neurons. The studies in neuronal cultures confirm the findings observed in hypothalamus and amygdala that NADPH oxidase is primarily responsible for oxidative stress, leading to anxiety-like behavior in mice. These data further show that increased cGMP-PKG signaling due to inhibition of PDE2 is responsible for the inhibition of NADPH oxidase and subsequent oxidative stress.
Fig. 9.
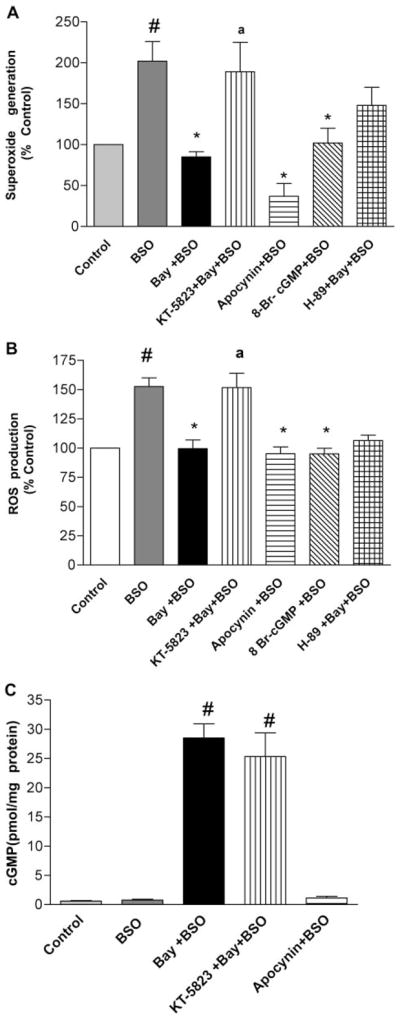
The PDE2 inhibitor Bay 60-7550 (Bay) reversed the BSO-induced increase in superoxide (A) and ROS (B) generation (end products of NADPH oxidase activation) while increasing cGMP levels (C). The PKG inhibitor KT-5823 reversed the Bay 60-7550-induced changes, increasing superoxide and ROS generation. The PKA inhibitor H89 had minimal effects on these parameters. Values are expressed as means ± S.E.M., n = 8 to 10/group. #, p < 0.05 compared with control; *, p < 0.05 compared with BSO-treated group; a, p < 0.05 compared with Bay + BSO-treated group.
Discussion
The present study demonstrates for the first time that PDE2 inhibition, by increasing cGMP signaling, is able to inhibit oxidative stress-induced anxiety in mice. The hypothalamus and amygdala, which are implicated in anxiety-related disorders, showed oxidative stress after treatment with BSO. There was an increase in expression of NADPH oxidase subunits and subsequent production of superoxide/ROS and lipid peroxides and a decrease in total antioxidant capacity after treatment with BSO. This was associated with increased anxiety-related behaviors in the mice. Pretreatment with the PDE2 inhibitor Bay 60-7550 increased cGMP-PKG signaling and reversed both BSO-induced oxidative stress and anxiogenic effects on behavior. Pretreatment with apocynin, a specific NADPH oxidase inhibitor, produced effects similar to the PDE2 inhibitor. Although PDE2 inhibition and increased cGMP production reversed the neurochemical and behavioral effects of oxidative stress, this treatment showed a trend toward anxiolytic effects in the absence of oxidative stress; larger doses of the PDE2 inhibitor may produce anxiolytic effects on its own.
Organisms exposed to stressful stimuli generate survival responses that counteract departures from homeostasis. Previous studies have shown that anxiolytic drugs attenuate stress-induced changes in autonomic, visceral, and immunological responses, as well as behavioral changes, such as those observed in the elevated plus-maze, open-field, and hole-board tests. These tests provide quantitative indices of the neurobehavioral profile of animals under the influence of stressful conditions or anxiogenic/anxiolytic drugs (Carli et al., 1989; Bhattacharya and Satyan, 1997). Recently, a new insight emerged from the studies of Hovatta et al. (2005) and Berry et al. (2007), which suggests a link between genes involved in oxidative stress metabolism and anxiety using these behavioral models in mice.
ROS production is maintained in equilibrium by the presence of antioxidant compounds such as vitamin E, vitamin C, bilirubin, and glutathione. Increased ROS generation by depletion of these antioxidants can upset the oxidant-antioxidant equilibrium in brain, moving these cells into a pro-oxidative state. If not effectively removed, ROS may cause oxidative cell injury (Gutteridge and Halliwell, 2000), protein damage, and lipid peroxidation. Energy failure caused by oxidative stress is reported to alter neuronal function and overall brain activity (Halliwell and Gutteridge, 2000; Calabrese et al., 2006). In the present study, oxidative stress induced by BSO led to decreased antioxidant capacity and increased lipid peroxides in hypothalamus and amygdala, regions linked to anxiety (Charney, 2003). BSO-mediated oxidative stress resulted from activation of the NADPH oxidase pathway as amygdala and hypothalamus showed increased expression of NADPH oxidase subunits. In addition, cultured neurons showed increased NADPH oxidase activation and superoxide/ROS generation in response to BSO treatment.
Most research on anxiety focuses on unraveling abnormalities in the GABA and serotonin systems; oxidative stress has not been considered to be major mediator of anxiety disorders. However, recent work has begun to suggest that increased oxidative stress in hypothalamus and amygdala can alter the function of fear-related circuits in the brain; this may play an important role in behavioral regulation and development of anxiety (Gingrich, 2005; Hovatta et al., 2005). The present study lends support to this notion because oxidative stress in hypothalamus and amygdala paralleled the anxiety-like behavioral pattern in the elevated plus-maze, hole-board, and open-field tests.
Oxidative stress has been reported to be associated with anxiety and cognitive defects in both human and animal models. Desrumaux et al. (2005) showed that vitamin E deficiency in the brain increases ROS generation, which results in anxiety-like behavior in mice. A recent study also suggests that a highly palatable diet leads to oxidative stress and anxiety-like behavior (Souza et al., 2007). Liu et al. (2003) have shown that oxidative stress is associated with age-related cognitive deficits, which are reversed by treatment with ROS scavengers. Sklan et al. (2004) have suggested that lower paraoxonase activity, which is involved in reduction of oxidative stress, may be responsible for the anxiety in aged people. However, these studies did not examine the direct effects of oxidative stress on anxiety-like behavior.
PDE2 is linked to cGMP/cAMP signaling pathways; inhibition of this enzyme increases concentrations of these second messengers. Several studies have shown that increased cGMP is associated with a decrease in oxidative stress/ROS generation (Mattson et al., 1999; Urushitani et al., 2000), but no study to date has explored its role in relation to oxidative stress-related anxiety. The present study shows for the first time that PDE2 inhibition leads to reversal of BSO-induced oxidative stress and anxiety-like behavioral effects in mice through inhibition of the NADPH oxidase pathway. This effect is probably mediated by cGMP-PKG signaling because Bay 60-7550 increased p-VASPSer239 levels in the brain; this was accompanied by decreased expression of NADPH oxidase subunits and anxiety-related behavioral measures. These findings were further confirmed in neuronal cultures where BSO-induced activation of NADPH oxidase and subsequent superoxide/ROS production were markedly inhibited by the PDE2 inhibitor Bay 60-7550. Furthermore, consistent with the effect of Bay 60-7550 on cGMP signaling and anxiety-related behavior, it has been reported that mice deficient in cGMP-dependent protein kinase II, a mediator of the intracellular actions of cGMP, exhibit an anxiogenic-like behavioral phenotype (Werner et al., 2004). Moreover, PDE5, which is a cGMP-specific PDE, has been reported to inhibit NADPH oxidase via an increase in cGMP levels (Muzaffar et al., 2005).
Apocynin, like Bay 60-7550, also reduced BSO-induced anxiety, but this effect was mediated via direct inhibition of NADPH oxidase. This was confirmed by using neuronal cultures that did not show any increase in cGMP levels after apocynin treatment but concurrently showed decreased ROS generation and immunoreactivity for NADPH oxidase subunits. Apocynin pretreatment also did not cause any changes in the expression of p-VASPSer239 in amygdala or hypothalamus, confirming that it did not alter cGMP signaling. On the other hand, the inhibitory role of cGMP on NADPH oxidase pathway was supported by the finding that the Bay 60-7550-induced decrease in superoxide/ROS production was paralleled by an increase in cGMP levels in cultured neurons. Moreover, the effects of Bay 60-7550 were antagonized by the PKG inhibitor KT-5823 but not by the PKA inhibitor H89, further suggesting that Bay 60-7550 exerts its effects via altered cGMP-PKG signaling. Some earlier studies also have shown inhibitory effects on oxidative stress via the cGMP-PKG pathway (Lee et al., 2003; Nakamizo et al., 2003). Because many neuropsychiatric/neurodegenerative disorders are associated with dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis, as well as with oxidative stress, it is possible that oxidative stress disturbs the HPA-axis function, altering the level of corticosterone with subsequent effects on hypothalamus and amygdala. Therefore, effects observed with PDE2 or NADPH oxidase inhibition could be through direct or indirect effects on HPA axis function.
Increased oxidative stress through activation of the NADPH oxidase pathway is an important underlying factor for the pathogenesis of a number of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (de la Monte and Wands, 2006; Wu et al., 2006; Sun et al., 2007). Patients with these illnesses develop anxiety and depression and also are susceptible to other neurobehavioral disturbances, which could be a result of ongoing oxidative injury to the central nervous system (Kurt et al., 2007; Starkstein et al., 2007). Moreover, the aged, who are more likely to have memory impairment and cognitive deficits, also are more likely to develop anxiety-related disorders due to enhanced oxidative stress (Liu et al., 2003; Berry et al., 2007). These observations, along with the present findings, suggest that PDE2 inhibitors could be uniquely beneficial in reduction of oxidative stress-induced anxiety in these disorders. This is supported by the finding that the benzodiazepine anxiolytic diazepam, at a dose of 1 mg/kg, did not fully reverse the oxidative stress-induced behavioral effects related to anxiety; previous work has shown that this dose of diazepam reverses restraint stress-induced anxiety (Masood et al., 2003). Thus, activation of GABA receptor-mediated signaling might not be as effective against anxiety caused by oxidative stress compared with that resulting from physical stress. It will be of interest to determine whether pharmacologically induced changes in benzodiazepine/GABA receptor function, including the assessment of the actions of inverse agonists, alters oxidant-antioxidant balance with corresponding neurochemical changes. This would begin to show whether the effects of PDE2 inhibitors on oxidative stress-induced anxiety are distinct from those of anxiolytics that act via benzodiazepine receptors.
Overall, the present study shows that oxidative stress leads to anxiety-like behavior in mice; this is reversed by PDE2 inhibition through increased cGMP-PKG signaling. Therefore, PDE2 may be a novel pharmacological target for treatment of anxiety in neuropsychiatric and neurodegenerative disorders that involve oxidative stress.
Acknowledgments
J.M.O. received research support from Memory Pharmaceuticals and Lund-beck Pharmaceuticals. This study was supported by the National Institute of Mental Health (Grants MH040964 and MH051175) and the National Heart, Lung, and Blood Institute (Grant HL027339).
ABBREVIATIONS
- PDE2
phosphodiesterase-2
- PKG
protein kinase G
- Bay 60-7550
(2-(3,4-dimethoxybenzyl)-7-{(1R)-1-[(1R)-1-hydroxyethyl]-4-phenylbutyl}-5-methyl imidazo[5,1-f][1,2,4]triazin-4(3H)-one)
- H89
N-[2-(4-bromocinnamylamino)ethyl]-5-isoquinoline
- BSO
L-buthionine-(S,R)-sulfoximine
- ROS
reactive oxygen species
- MDA
malondialdehyde
- TBA
thiobarbituric acid
- PCR
polymerase chain reaction
- VASP
vasodilator-stimulated phosphoprotein
- HPA
hypothalamic-pituitary-adrenal
- WST-1
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt
- KT-5823
(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, methyl ester
References
- Arranz L, Guayerbas N, De la Fuente M. Impairment of several immune functions in anxious women. J Psychosom Res. 2007;62:1–8. doi: 10.1016/j.jpsychores.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Chem. 1996;239:70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- Berry A, Capone F, Giorgio M, Pelicci PG, de Kloet ER, Alleva E, Minghetti L, Cirulli F. Deletion of the life span determinant p66Shc prevents age-dependent increases in emotionality and pain sensitivity in mice. Exp Gerontol. 2007;42:37–45. doi: 10.1016/j.exger.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Bhattacharya SK, Satyan KS. Experimental methods for the evaluation of psychotropic agents: I–Anti-anxiety agents. Indian J Exp Biol. 1997;35:565–575. [PubMed] [Google Scholar]
- Boess FG, Hendrix M, van der Staay FJ, Erb C, Schreiber R, van Staveren W, de Vente J, Prickaerts J, Blokland A, Koenig G. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology. 2004;47:1081–1092. doi: 10.1016/j.neuropharm.2004.07.040. [DOI] [PubMed] [Google Scholar]
- Bouayed J, Rammal H, Younos C, Soulimani R. Positive correlation between peripheral blood granulocyte oxidative status and level of anxiety in mice. Eur J Pharmacol. 2007;564:146–149. doi: 10.1016/j.ejphar.2007.02.055. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Guagliano E, Sapienza M, Mancuso C, Butterfield DA, Stella AM. Redox regulation of cellular stress response in neurodegenerative disorders. Ital J Biochem. 2006;55:263–282. [PubMed] [Google Scholar]
- Carli M, Prontera C, Samanin R. Effects of 5 HT-1A agonist in stress-induced deficit in the open field locomotor activity of rats, evidence that this model identifies anxiolytic activity. Neuropharmacology. 1989;28:471–476. doi: 10.1016/0028-3908(89)90081-6. [DOI] [PubMed] [Google Scholar]
- Charney DS. Neuroanatomical circuits modulating fear and anxiety behaviors. Acta Psychiatr Scand Suppl. 2003;417:38–50. doi: 10.1034/j.1600-0447.108.s417.3.x. [DOI] [PubMed] [Google Scholar]
- Chrousos GP, Gold PW. The concepts of stress and stress system disorders. JAMA. 1992;26:1244–1252. [PubMed] [Google Scholar]
- de Bernardo S, Canals S, Casarejos MJ, Solano RM, Menendez J, Mena MA. Role of extracellular signal-regulated protein kinase in neuronal cell death induced by glutathione depletion in neuron/glia mesencephalic cultures. J Neurochem. 2004;91:667–682. doi: 10.1111/j.1471-4159.2004.02744.x. [DOI] [PubMed] [Google Scholar]
- Deguchi A, Soh JW, Li H, Pamukcu R, Thompson WJ, Weinstein IB. Vasodilator-stimulated phosphoprotein (VASP) phosphorylation provides a biomarker for the action of exisulind and related agents that activate protein kinase G. Mol Cancer Ther. 2002;1:803–809. [PubMed] [Google Scholar]
- de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis. 2006;9:167–181. doi: 10.3233/jad-2006-9209. [DOI] [PubMed] [Google Scholar]
- Desrumaux C, Risold PY, Schroeder H, Deckert V, Masson D, Athias A, Laplanche H, Le Guern N, Blache D, Jiang XC, et al. Phospholipid transfer protein (PLTP) deficiency reduces brain vitamin E content and increases anxiety in mice. FASEB J. 2005;19:296–297. doi: 10.1096/fj.04-2400fje. [DOI] [PubMed] [Google Scholar]
- Gingrich JA. Oxidative stress is the new stress. Nat Med. 2005;12:1281–1282. doi: 10.1038/nm1205-1281. [DOI] [PubMed] [Google Scholar]
- Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000: a historical look to the future. Ann N Y Acad Sci. 2000;899:136–147. doi: 10.1111/j.1749-6632.2000.tb06182.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Free Radicals in Biology and Medicine. 3. Clarendon; Oxford, UK: 2000. [Google Scholar]
- Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, Ellison JA, Schadt EE, Verma IM, Lockhart DJ, et al. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005;438:662–666. doi: 10.1038/nature04250. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources. Guide for the Care and Use of Laboratory Animals. 7. Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council; Washington, DC: 1996. [Google Scholar]
- Jentzsch AM, Bachmann H, Furst P, Biesalski HK. Improved analysis of malondialdehyde in human body fluids. Free Radic Biol Med. 1996;20:251–256. doi: 10.1016/0891-5849(95)02043-8. [DOI] [PubMed] [Google Scholar]
- Kurt A, Nijboer F, Matuz T, Kübler A. Depression and anxiety in individuals with amyotrophic lateral sclerosis: epidemiology and management. CNS Drugs. 2007;21:279–291. doi: 10.2165/00023210-200721040-00003. [DOI] [PubMed] [Google Scholar]
- Lee SY, Andoh T, Murphy DL, Chiueh CC. 17beta-estradiol activates ICI 182,780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. FASEB J. 2003;17:947–948. doi: 10.1096/fj.02-0807fje. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang X, Shigenaga MK, Yeo HC, Mori A, Ames BN. Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J. 1996;10:1532–1538. [PubMed] [Google Scholar]
- Liu R, Lui IY, Bi X, Thompson RE, Doctrow SR, Malfry B, Baudry M. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc Natl Acad Sci U S A. 2003;100:8526–8531. doi: 10.1073/pnas.1332809100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Masood A, Banerjee B, Vijayan VK, Ray A. Modulation of stress induced neurobehavioral changes by nitric oxide in rats. Eur J Pharmacol. 2003;458:135–139. doi: 10.1016/s0014-2999(02)02688-2. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Guo ZH, Geiger JD. Secreted form of amyloid precursor protein enhances basal glucose and glutamate transport and protects against oxidative impairment of glucose and glutamate transport in synaptosomes by a cyclic GMP-mediated mechanism. J Neurochem. 1999;73:532–537. doi: 10.1046/j.1471-4159.1999.0730532.x. [DOI] [PubMed] [Google Scholar]
- Muzaffar S, Shukla N, Srivastava A, Angelini GD, Jeremy JY. Sildenafil citrate and sildenafil nitrate (NCX 911) are potent inhibitors of superoxide formation and gp91phox expression in porcine pulmonary artery endothelial cells. Br J Pharmacol. 2005;146:109–117. doi: 10.1038/sj.bjp.0706305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeem A, Fan M, Ansari HR, Ledent C, Mustafa SJ. Enhanced airway reactivity and airway inflammation in A2A adenosine receptor deficient allergic mice. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1335–L1344. doi: 10.1152/ajplung.00416.2006. [DOI] [PubMed] [Google Scholar]
- Nadeem A, Masood A, Masood N, Gilani RA, Shah ZA. Immobilization stress causes extra-cellular oxidant-antioxidant imbalance in rats: restoration by L-NAME and vitamin E. Eur Neuropsychopharmacol. 2006;16:260–267. doi: 10.1016/j.euroneuro.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Nakamizo T, Kawamata J, Yoshida K, Kawai Y, Kanki R, Sawada H, Kihara T, Yamashita H, Shibasaki H, Akaike A, et al. Phosphodiesterase inhibitors are neuroprotective to cultured spinal motor neurons. J Neurosci Res. 2003;71:485–495. doi: 10.1002/jnr.10483. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Gambaryan S, Engelhardt S, Walter U, Lohse MJ. Real-time monitoring of the PDE2 activity of live cells: hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. J Biol Chem. 2005;280:1716–1719. doi: 10.1074/jbc.C400505200. [DOI] [PubMed] [Google Scholar]
- Ray A, Henke PG, Gulati K, Sen P. The amygdaloid complex, corticotropin releasing factor and stress induced gastric ulcerogenesis in rats. Brain Res. 1993;624:286–290. doi: 10.1016/0006-8993(93)90089-6. [DOI] [PubMed] [Google Scholar]
- Reyes-Irisarri E, Markerink-Van Ittersum M, Mengod G, de Vente J. Expression of the cGMP-specific phosphodiesterases 2 and 9 in normal and Alzheimer’s disease human brains. Eur J Neurosci. 2007;25:3332–3338. doi: 10.1111/j.1460-9568.2007.05589.x. [DOI] [PubMed] [Google Scholar]
- Sklan EH, Lowenthal A, Korner M, Ritov Y, Landers DM, Rankinen T, Bouchard C, Leon AS, Rice T, Rao DC, et al. Acetylcholinesterase/paraoxonase genotype and expression predict anxiety scores in health, risk factors, exercise training, and genetics study. Proc Natl Acad Sci U S A. 2004;101:5512–5517. doi: 10.1073/pnas.0307659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolenski A, Bachmann C, Reinhard K, Honig-Liedl P, Jarchau T, Hoschuetzky H, Walter U. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J Biol Chem. 1998;273:20029–20035. doi: 10.1074/jbc.273.32.20029. [DOI] [PubMed] [Google Scholar]
- Souza CG, Moreira JD, Siqueira IR, Pereira AG, Rieger DK, Souza DO, Souza TM, Portela LV, Perry ML. Highly palatable diet consumption increases protein oxidation in rat frontal cortex and anxiety-like behavior. Life Sci. 2007;81:198–203. doi: 10.1016/j.lfs.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Starkstein SE, Jorge R, Petracca G, Robinson RG. The construct of generalized anxiety disorder in Alzheimer disease. Am J Geriatr Psychiatry. 2007;15:42–49. doi: 10.1097/01.JGP.0000229664.11306.b9. [DOI] [PubMed] [Google Scholar]
- Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A(2) in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem. 2007;103:1–16. doi: 10.1111/j.1471-4159.2007.04670.x. [DOI] [PubMed] [Google Scholar]
- Suvarna NU, O’Donnell JM. Hydrolysis of N-methyl-D-aspartate receptor-stimulated cAMP and cGMP by PDE4 and PDE2 phosphodiesterases in primary neuronal cultures of rat cerebral cortex and hippocampus. J Pharmacol Exp Ther. 2002;302:249–256. doi: 10.1124/jpet.302.1.249. [DOI] [PubMed] [Google Scholar]
- Tan AS, Berridge MV. Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: a simple colorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents. J Immunol Methods. 2000;238:59–68. doi: 10.1016/s0022-1759(00)00156-3. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Inoue R, Nakamizo T, Sawada H, Shibasaki H, Shimohama S. Neuroprotective effect of cGMP against radical-induced toxicity in cultured spinal motor neurons. J Neurosci Res. 2000;61:443–448. doi: 10.1002/1097-4547(20000815)61:4<443::AID-JNR11>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med. 1999;27:612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- Werner C, Raivich G, Cowen M, Strekalova T, Sillaber I, Buters JT, Spanagel R, Hofmann F. Importance of NO/cGMP signalling via cGMP-dependent protein kinase II for controlling emotionality and neurobehavioural effects of alcohol. Eur J Neurosci. 2004;20:3498–3506. doi: 10.1111/j.1460-9568.2004.03793.x. [DOI] [PubMed] [Google Scholar]
- Wu DC, Ré DB, Nagai M, Ischiropoulos H, Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A. 2006;103:12132–12137. doi: 10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]