

SUMMARY
The lipolytic processing of triglyceride-rich lipoproteins by lipoprotein lipase (LPL) is the central event in plasma lipid metabolism, providing lipids for storage in adipose tissue and fuel for vital organs such as the heart. LPL is synthesized and secreted by myocytes and adipocytes but then finds its way into the lumen of capillaries, where it hydrolyzes lipoprotein triglycerides. The mechanism by which LPL reaches the lumen of capillaries represents one of the most persistent mysteries of plasma lipid metabolism. Here, we show that GPIHBP1 is responsible for the transport of LPL into capillaries. In Gpihbp1-deficient mice, LPL is mislocalized to the interstitial spaces surrounding myocytes and adipocytes. Also, we show that GPIHBP1 is located at the basolateral surface of capillary endothelial cells and actively transports LPL across endothelial cells. Our experiments define the function of GPIHBP1 in triglyceride metabolism and provide a mechanism for the transport of LPL into capillaries.
INTRODUCTION
The lipolytic processing of triglyceride-rich lipoproteins by lipoprotein lipase (LPL) provides fuel to vital tissues such as the heart and lipids for storage in adipose tissue (Olivecrona and Olivecrona, 2009; Wang and Eckel, 2009). Lipolysis also leads to the production of atherogenic remnant lipoproteins (e.g., low-density lipoproteins) and provides lipids for the formation of high-density lipoprotein particles. LPL is a homodimer and its activity requires a protein cofactor, apolipoprotein (apo-) CII, which is carried within lipoproteins. A deficiency of LPL or apo-CII causes familial chylomicronemia, a severe form of hypertriglyceridemia (Brunzell and Deeb, 2001).
The lipolysis of lipoproteins by LPL occurs at the surface of capillaries, mainly in skeletal muscle, heart, and adipose tissue (Havel and Kane, 2001; Olivecrona and Olivecrona, 2009; Wang and Eckel, 2009). Remarkably, however, LPL is not expressed by capillary endothelial cells. Instead, LPL is produced by the parenchymal cells, myocytes and adipocytes, and secreted into the surrounding interstitial spaces. From there, LPL finds its way into the lumen of capillaries. An older study suggested that the VLDL receptor (VLDLR) might be involved in the transport of LPL across capillaries (Obunike et al., 2001), but the significance of that finding is uncertain because VLDLR deficiency does not lead to hypertriglyceridemia (Frykman et al., 1995). No further progress has been made on this issue, and the mechanism for the entry of LPL into capillaries remains unknown.
Recently, Beigneux et al. (Beigneux et al., 2007) identified a critical role for GPIHBP1 in lipolysis. GPIHBP1 is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein of the Lymphocyte Antigen 6 (Ly6) family and is expressed in endothelial cells. Cell transfection studies revealed that GPIHBP1 binds LPL and that this binding could be blocked with heparin. GPIHBP1-deficient mice develop severe hypertriglyceridemia, even on a chow diet, with plasma triglyceride levels >3000 mg/dl. These findings prompted Beigneux et al. (Beigneux et al., 2007) to speculate that GPIHBP1 could serve as a binding site for LPL in capillaries; however, the precise role of this molecule in lipolysis was not elucidated.
In this study, we examined the role of GPIHBP1 in proper localization of LPL within capillaries and tested whether GPIHBP1 is the critical molecule in shuttling LPL from the interstitial spaces to the luminal surface of capillary endothelial cells.
RESULTS
GPIHBP1 Is Present on the Luminal Face of Capillaries
Early immunohistochemical studies with a rabbit antiserum against mouse GPIHBP1 revealed that GPIHBP1 is found on capillaries of heart, skeletal muscle, and adipose tissue (Beigneux et al., 2007). In this study, to determine if GPIHBP1 is present on the luminal surface of capillaries, wild-type and Gpihbp1-deficient mice (Gpihbp1−/−) were given an intravenous injection of an Alexa555-labeled GPIHBP1-specific monoclonal antibody (antibody 11A12) and an antibody against CD31 (an endothelial cell marker). Five minutes later, the mice were euthanized and their tissues were perfused extensively with PBS. In the adipose tissue of wild-type mice, antibody 11A12 binding was restricted to capillaries, colocalizing with antibodies to CD31. In the adipose tissue of Gpihbp1−/− mice, CD31 was found in capillaries but GPIHBP1 was absent (Figure 1A). Similarly, GPIHBP1 and CD31 colocalized in the capillaries of the heart in wild-type mice, but only CD31 was detected in Gpihbp1−/− mice (Figure 1B). Interestingly, GPIHBP1 was confined to capillaries surrounding myocytes, whereas CD31 was found in both capillaries and larger vessels (Figure 1B). GPIHBP1 was not detectable in the capillaries of the brain (Figure S1).
Figure 1. GPIHBP1 is located within the lumen of capillaries.
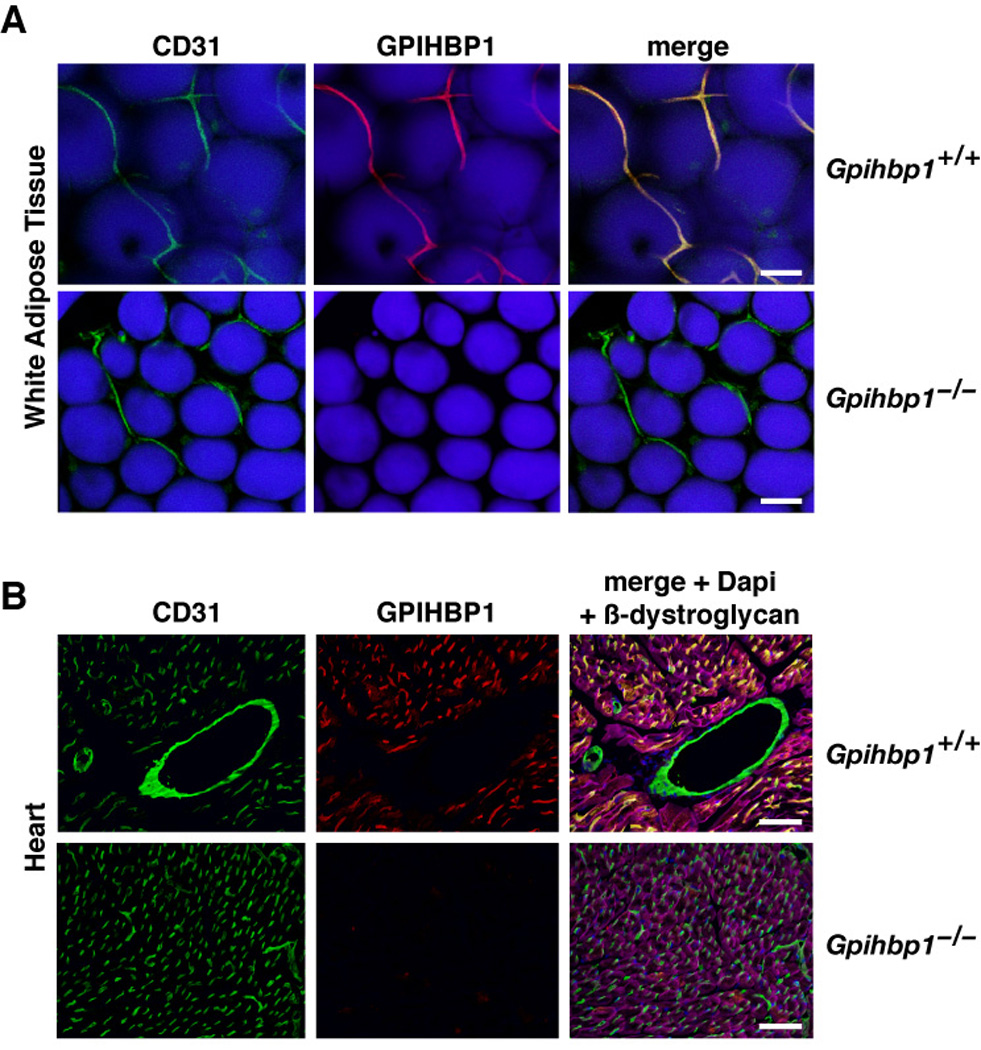
(A) Confocal images of white adipose tissue revealing GPIHBP1 in the capillaries of wild-type but not Gpihbp1−/− mice. Mice were injected intravenously with Alexa555-labeled anti-GPIHBP1 (red) and hamster anti-CD31 antibodies and then perfused extensively with PBS. Fixed adipose explants were stained with FITC-labeled anti-hamster antibodies (green) and BODIPY to label adipocytes (blue). The scale bar represents a distance of 100 µm.
(B) Confocal images of heart tissue from the same antibody-injected wild-type and Gpihbp1−/− mice showing GPIHBP1 staining in the capillaries of wild-type but not Gpihbp1−/− mice. Tissues were stained with an antibody against β-dystroglycan (magenta), a plasma membrane protein of myocytes, to outline myocytes. The scale bar represents a distance of 100 µm. (See also Figure S1.)
GPIHBP1 and LPL Normally Colocalize in Capillaries
Because expression of GPIHBP1 in CHO cells confers the ability to bind LPL (Beigneux et al., 2007; Beigneux et al., 2009a; Beigneux et al., 2009b; Gin et al., 2008), we hypothesized that at least some of the LPL in mouse tissues might colocalize with GPIHBP1 in capillaries. To test this hypothesis, we performed immunohistochemistry with a chicken antibody against bovine LPL and a goat antibody against a recombinant mouse LPL fragment (Olivecrona and Bengtsson, 1983; Weinstein et al., 2008). Both antibodies identified a single 52-kDa protein in CHO cells expressing V5-tagged mouse LPL (Figure S2A–B). By immunofluorescence, both antibodies bound mouse LPL–transfected CHO cells (Figure S2C). When LPL-transfected and GPIHBP1-transfected CHO cells were mixed, immunofluorescence microscopy revealed that LPL and GPIHBP1 colocalized on the surface of cells (i.e., the LPL secreted by LPL-expressing cells bound avidly to GPIHBP1-transfected cells) (Figure S2D), showing that both antibodies bind LPL when it is also bound to GPIHBP1. Both antibodies detected mouse LPL in the heart of wild-type mice, but neither detected LPL (human or mouse) in the hearts of Lpl−/− mice that had been rescued with a muscle-specific human LPL transgene (Weinstock et al., 1997) (Figure S2E–F).
By immunofluorescence microscopy, most of the LPL in brown adipose tissue was located in capillary endothelial cells, colocalizing with GPIHBP1 (Figure 2A–B). Both LPL and GPIHBP1 were identified in capillaries; neither was found in larger blood vessels (Figure 2B). The capillaries of the heart, all positive for GPIHBP1, also stained for LPL; this capillary staining was intense and was visible above the less-intense LPL staining in myocytes (Figure S2E–F, Figure 2C).
Figure 2. GPIHBP1 and LPL colocalize within capillaries.
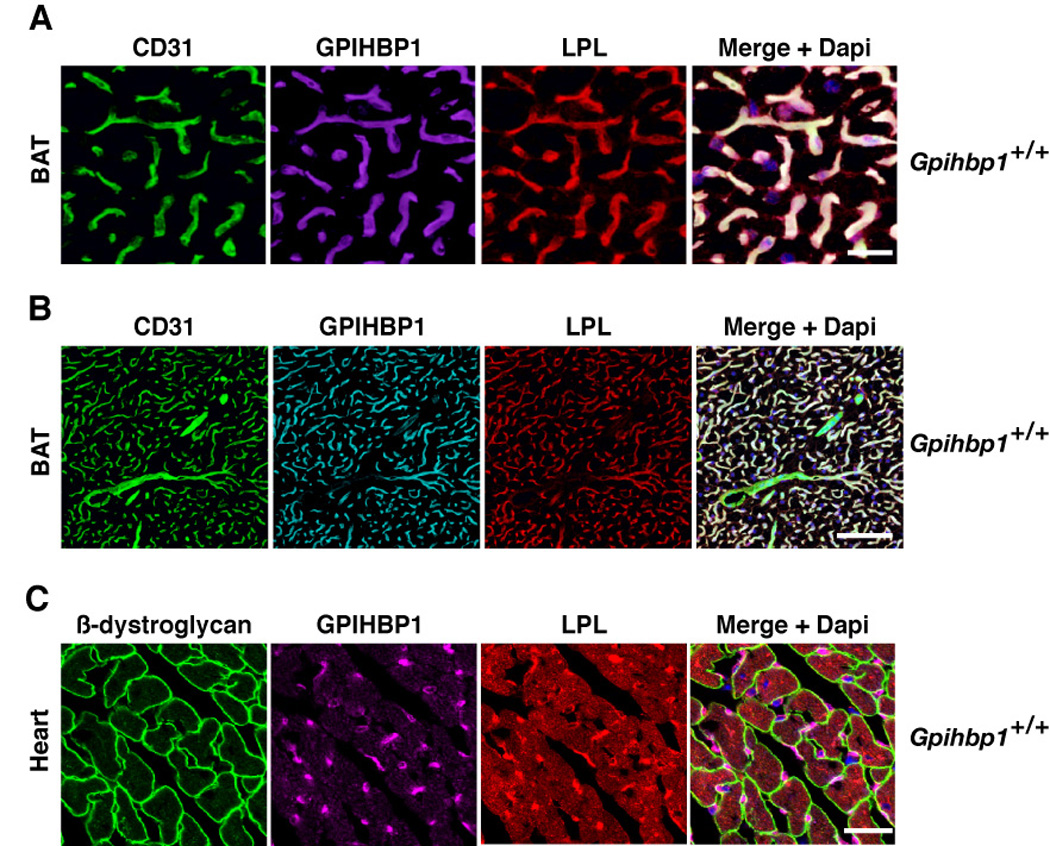
(A and B) Confocal images showing the binding of GPIHBP1-, LPL-, and CD31-specific antibodies to brown adipose tissue (BAT) from a wild-type mouse. Images were taken with a 63× objective; (A) close-up (scale bar represents distance of 50 µm) and (B) wide-field (scale bar represents distance of 100 µm).
(C) Confocal images showing the binding of GPIHBP1-, LPL-, and β-dystroglycan-specific antibodies to heart tissue from a wild-type mouse. The scale bar represents a distance of 25 µm. (See also Figure S2.)
LPL Is Mislocalized Away from Capillaries in Gpihbp1−/− Mice
To determine if GPIHBP1 plays a role in the targeting of LPL to capillaries, we examined the localization of LPL in the tissues of Gpihbp1−/− mice. The LPL in brown adipose tissue of Gpihbp1−/− mice surrounded adipocytes, colocalizing with the extracellular matrix protein collagen IV (Figure 3A). Similar mislocalization was observed in both heart and skeletal muscle of Gpihbp1−/− mice; the LPL surrounded myocytes, colocalizing with β-dystroglycan (a plasma membrane protein of myocytes) (Figure 3B–C, Figure S3A) or laminin, a basement membrane protein surrounding myocytes (Figure S3B–C).
Figure 3. Mislocalization of LPL in Gpihbp1−/− mice.
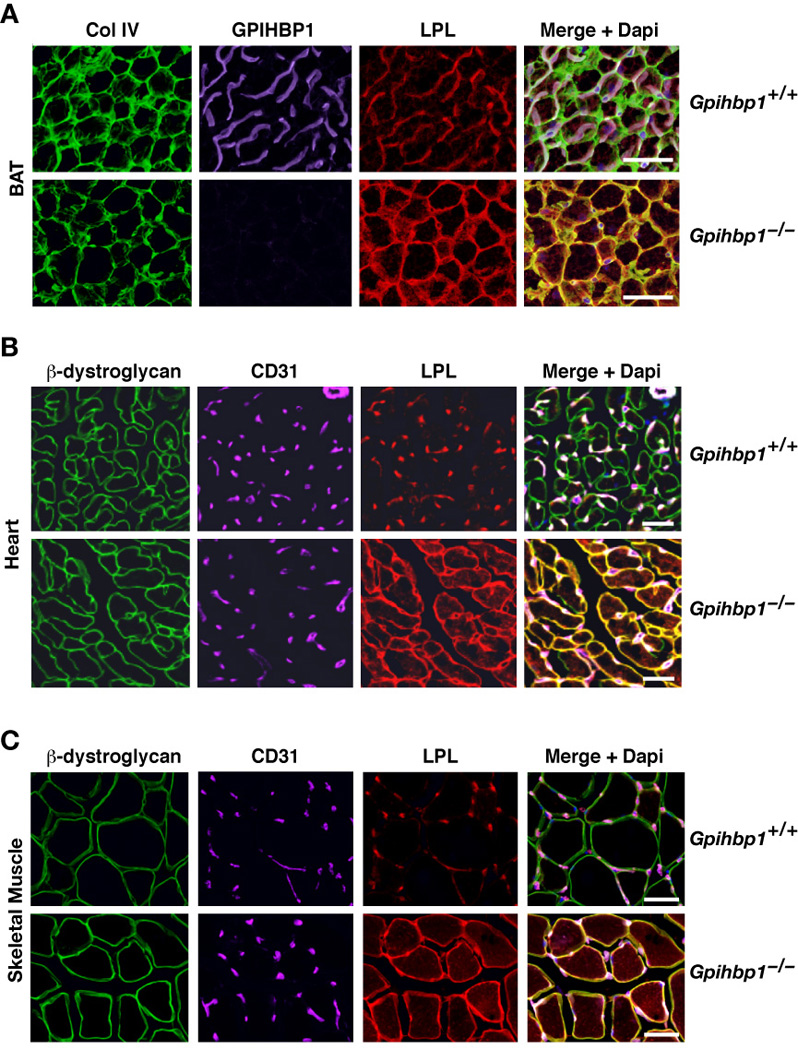
(A) Confocal microscopy showing the binding of GPIHBP1-, LPL-, and collagen IV–specific antibodies to brown adipose tissue (BAT) from wild-type and Gpihbp1−/− mice. The scale bars represent a distance of 100 µm.
(B and C) Confocal microscopy showing the binding of CD31-, LPL-, and β-dystroglycan-specific antibodies to heart (B) and skeletal muscle (C) of wild-type and Gpihbp1−/− mice. The scale bars represent a distance of 100 µm (skeletal muscle) or 50 µm (heart).
GPIHBP1-Mediated Transport Across Endothelial Cells
The mislocalization of LPL in Gpihbp1−/− mice prompted us to consider the possibility that GPIHBP1 plays a role in the transport of LPL across capillary endothelial cells. We reasoned that the involvement of GPIHBP1 in transport would require that GPIHBP1 be present on the basolateral surface of cells (in proximity to the LPL secreted by adipocytes and myocytes). To determine if some GPIHBP1 is present on the basolateral surface of cells, we examined rat heart microvessel endothelial cells (RHMVECs) that expressed an S-protein–tagged version of GPIHBP1. We used RHMVECs that had been transduced with a mouse GPIHBP1 lentivirus because the expression of Gpihbp1 from the endogenous gene is nearly undetectable within a day after plating primary cells (Davies et al., 2008). We compared GPIHBP1 expression levels in transduced RHMVECs with GPIHBP1 expression levels in endothelial cells of mouse tissues by qRT-PCR, normalizing the data to Cd31 (expressed only in endothelial cells). GPIHBP1 expression in lentivirus-transduced RHMVECs was about ~20% of endothelial cells in brown adipose tissue and ~40% of endothelial cells in the heart (Figure 4A).
Figure 4. GPIHBP1 is present at the basolateral and apical surfaces of cultured endothelial cells and transports a GPIHBP1-specific monoclonal antibody across cells.
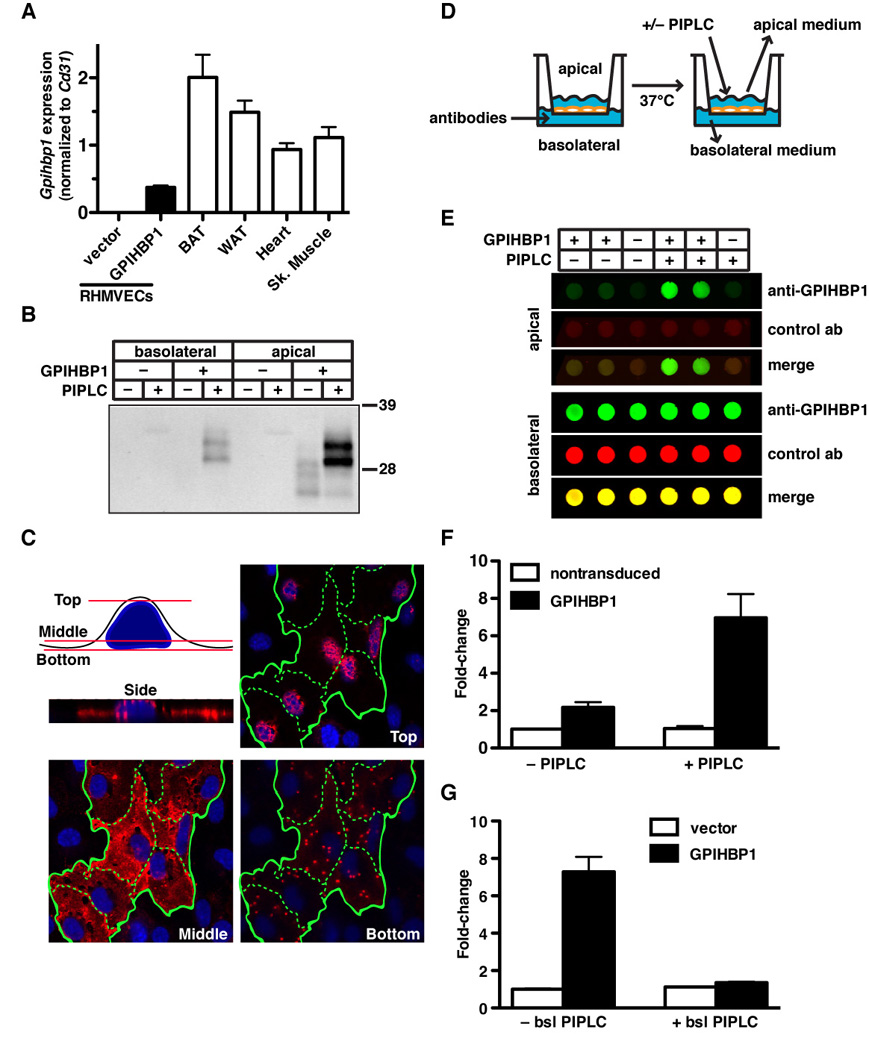
(A) Gpihbp1 expression levels in lentivirus-transduced RHMVECs and mouse heart, skeletal muscle, brown adipose tissue (BAT), and white adipose tissue (WAT). Gpihbp1 expression levels were measured by quantitative RT-PCR and normalized to Cd31 (mean ± SEM).
(B) Release of GPIHBP1 by PIPLC from apical and basolateral surfaces of GPIHBP1-transduced RHMVEC monolayers grown on filters. After incubating the cells with PIPLC (5 U/ml) for 1.5 h, the medium was harvested from the apical and basolateral chambers, precipitated with 10% TCA, and size-fractionated by SDS-PAGE. Western blots were performed with antibody 11A12.
(C) Confocal microscopy showing the binding of antibody 11A12 to GPIHBP1-transduced RHMVECs. Alexa555-labeled antibody 11A12 was added to both the apical and basolateral chambers, and images of optical sections were obtained with a confocal microscope (63× objective) near the top, middle, and bottom of the cells. Green lines outline individual cells.
(D) Schematic of the transport assay. RHMVECs were grown on filters (1-µm pore size) until they formed a tight monolayer. Antibodies were added to the basolateral chamber and cells were incubated at 37°C. After the incubation, PIPLC was added to the apical chamber to release GPIHBP1; medium from the apical and basolateral chambers was collected and analyzed.
(E and F) Dot blots demonstrating the transport of antibody 11A12 from the basolateral to the apical surface of GPIHBP1-transduced RHMVEC monolayers. IRDye800-labeled antibody 11A12 (green) and an IRDye680-labeled anti-goat IgG control antibody (red) were added to the basolateral chamber and incubated for 3 h at 37°C. The apical surface was treated with PIPLC (1 U/ml; 30 min) or PBS, and both apical and basolateral media were dot blotted, scanned, and quantified with the Odyssey scanner. Bar graph in Panel F shows the fold change (mean ± SEM) compared to untreated vector-transduced cells from four independent experiments.
(G) Analysis of the ability of basolateral PIPLC to block the transport of antibody 11A12 from the basolateral to the apical chamber. IRDye800-labeled antibody 11A12 and IRDye680-labeled anti-goat IgG antibody were added to the basolateral chamber in the presence or absence of PIPLC (1 U/ml) and incubated for 1.5 h at 37°C. The apical surface was treated with PIPLC (1 U/ml; 30 min) and the apical medium was dot blotted, scanned, and quantified with the Odyssey scanner. The bar graph shows the fold change (mean ± SEM) compared to untreated vector-transduced cells.
The GPIHBP1- and empty vector–transduced RHMVECs were grown on polyethylene terephthalate filters (1-µm pore size), which makes it possible to expose the basolateral surface of cells to one medium and the apical surface to another. The RHMVECs formed tight monolayers, as judged by electrical resistance measurements and the ability of the monolayer to prevent the flow of medium from one chamber to the other. To determine if GPIHBP1 is present at both the apical and basolateral surfaces of endothelial cells, phosphatidylinositol-specific phospholipase C (PIPLC) was added to the apical or the basolateral chamber, and the release of GPIHBP1 into the medium was monitored with western blots. PIPLC released GPIHBP1 from both the basolateral and apical surface of cells (Figure 4B). One possible explanation for the lower amount of GPIHBP1 release from the basolateral surface was that the access of PIPLC to the basolateral surface of cells may have been limited (pores represent only ~4% of the surface area of the filters).
GPIHBP1 localization in endothelial cell monolayers was also examined by microscopy. For these experiments, Alexa555-labeled antibody 11A12 was added to both the apical and basolateral chambers. By confocal microscopy, GPIHBP1 was detected in the uppermost and middle optical slices of endothelial cells. When the most inferior slices of the cells were examined, small patches of intense GPIHBP1 staining were observed, corresponding to regions of the plasma membrane adjacent to the filter’s pores (i.e., the regions in direct contact with the antibody in the medium) (Figure 4C).
To test whether GPIHBP1 could traverse endothelial cells, we measured its ability to transport a specific anti-GPIHBP1 monoclonal antibody (11A12) from the basolateral media to the apical media. RHMVECs were grown on filters (Figure 4D) and IRDye800-conjugated antibody 11A12 and an IRDye680-conjugated irrelevant antibody (a donkey anti–goat IgG) were added to the basolateral chamber. After a 3-h incubation at 37°C, the apical surface of the cells was incubated with PIPLC, releasing GPIHBP1 along with any bound antibody into the medium. The amount of each antibody in the apical medium was assessed by dot blot with an Odyssey infrared scanner. Neither the GPIHBP1- nor the vector-transduced cells transported the irrelevant antibody into the upper chamber efficiently, but the GPIHBP1-transduced cells transported antibody 11A12 into the upper chamber (Figure 4E–F). The transport of antibody 11A12 required the presence of GPIHBP1 at the basolateral surface of cells; when basolateral GPIHBP1 was clipped off with PIPLC, transport of the antibody to the apical chamber was abolished (Figure 4G).
GPIHBP1 Transports LPL Across Endothelial Cells
To test whether GPIHBP1 could transport LPL across endothelial cell monolayers, human LPL (hLPL) was added to the basolateral side of GPIHBP1-transduced RHMVECs. After a 1-h incubation at 37°C, the LPL content of the apical medium was assessed by dot blot. In some studies, cells were treated with heparin to release any surface-bound LPL into the medium. Even in the absence of heparin, more LPL was present in the apical medium of GPIHBP1-transduced cells than in vector-transduced cells (Figure 5A). When heparin was added to release cell-surface LPL, the differences in the content of LPL in the apical medium were more pronounced (Figure 5A). In control experiments, we found that bovine serum albumin (BSA) transport was not affected by the expression of GPIHBP1 or by the addition of heparin to the apical surface of cells (Figure S4A). We also found that the amount of LPL transported to the apical surface of cells was dependent on the amount of LPL in the basolateral medium (Figure S4B–C). Finally, LPL transport occurred when GPIHBP1-transduced bovine aortic endothelial cells were used instead of RHMVECs (Figure S4D–E).
Figure 5. Transport of LPL across endothelial cell monolayers.
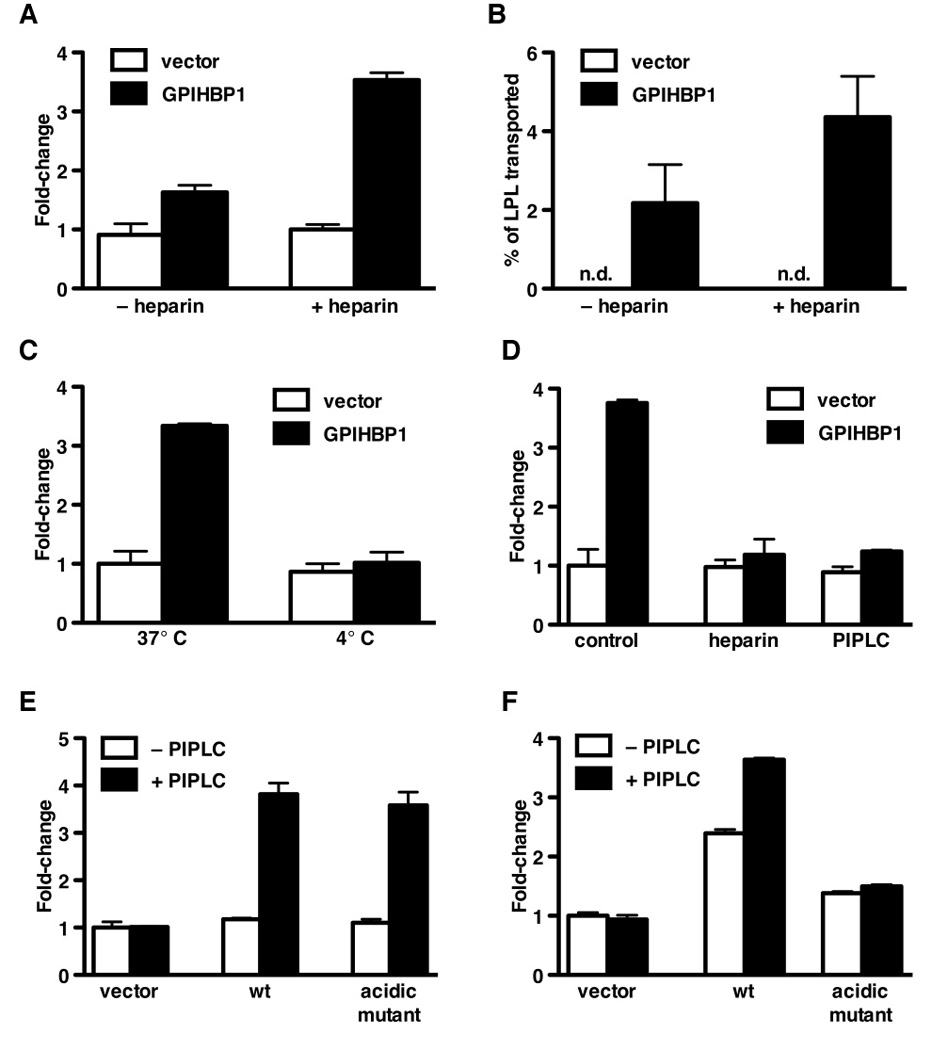
(A) Dot blot analysis demonstrating the transport of V5-tagged human LPL (hLPL) from the basolateral to the apical chamber by GPIHBP1-transduced RHMVECs. After adding LPL to the basolateral chamber and incubating for 1 h at 37°C, either heparin (100 U/ml) or PBS was added to the apical chamber and the apical medium was dot blotted with an antibody against the V5 tag and quantified with the Odyssey scanner. Bar graph shows the fold change (mean ± SEM) compared to vector-transduced cells from four independent experiments.
(B) Transport of bovine LPL from the basolateral to the apical chamber by GPIHBP1- or vector-transduced RHMVECs grown on filters. After adding bovine LPL to the basolateral chamber and incubating for 1 h at 37°C, the apical surface of cells was treated with heparin (100 U/ml) or PBS for 15 min at room temperature. The apical medium was collected, and the LPL concentration was determined with an ELISA.
(C) GPIHBP1-mediated LPL transport across endothelial cells at 4° and 37°C. After adding V5-tagged hLPL to the basolateral chamber, GPIHBP1-transduced RHMVECs were incubated for 1 h at 4° or 37°C. Heparin was then added to the apical chamber (100 U/ml; 15 min at 4°C), and the amount of LPL in the apical medium was assessed by dot blot with an antibody against the V5 tag. The blot was scanned and quantified with the Odyssey scanner. Bar graph shows the fold change (mean ± SEM) compared to vector-transduced cells incubated at 37°C.
(D) Ability of PIPLC or heparin in the basolateral medium to block GPIHBP1-mediated transport of the V5-tagged hLPL from the basolateral to the apical medium of RHMVECs. After adding LPL and either heparin (100 U/ml) or PIPLC (2.5 U/ml) to the basolateral chamber, the cells were incubated for 1 h at 37°C. Heparin was then added to the apical chamber (100 U/ml; 15 min at room temperature), and the amount of LPL in the apical medium was then assessed by dot blotting with an antibody against the V5 tag. The blot was scanned and quantified with the Odyssey scanner. Bar graph shows the fold change (mean ± SEM) compared to vector-transduced cells.
(E) Dot blot analysis of antibody 11A12 transport in RHMVECs transduced with wild-type or mutant (D,E(24–48)A) GPIHBP1. IRDye800-labeled antibody 11A12 was added to the basolateral chamber and incubated for 1 h at 37°C. The apical surface was then treated with PIPLC (1 U/ml; 30 min) or PBS; the apical medium was dot blotted, scanned, and analyzed with the Odyssey scanner. Bar graph shows fold change (mean ± SEM) compared to vector-transduced control cells.
(F) Dot blot analysis of LPL transport in RHMVECs transduced with either wild-type or mutant (D,E(24–48)A) GPIHBP1. After adding LPL to the basolateral chamber and incubating for 1 h at 37°C, the apical surface was treated with PIPLC (2.5 U/ml) or PBS (30 min at 37°C). The apical medium was then dot blotted with an antibody against the V5 tag, and the signal was quantified with the Odyssey scanner. Bar graph shows the fold change (mean ± SEM) compared to vector-transduced control cells. (See also Figure S4.)
We also assessed transport of a highly purified bovine LPL preparation across GPIHBP1- and vector-transduced RHMVECs. Once again, LPL was placed in the basolateral medium; after 1 h, heparin was added to the apical chamber and the LPL concentration in the apical medium was measured with an ELISA. LPL transport to the apical surface of cells was absent in vector-transduced cells but was easily detectable with GPIHBP1-transduced cells (Figure 5B).
When the LPL transport studies were carried out at 4°C, GPIHBP1-specific transport of LPL was blocked (Figure 5C). At 37°C, adding heparin or PIPLC to the basolateral chamber largely blocked the transport of LPL (Figure 5D).
Gin et al. (Gin et al., 2008) showed that the acidic domain of GPIHBP1 is required for LPL binding. We predicted that a mutant GPIHBP1 lacking the acidic domain would be unable to transport LPL but would retain the ability to transport antibody 11A12 (which binds to an epitope outside of the acidic domain) (Beigneux et al., 2009b). To test this prediction, we generated a mutant GPIHBP1 lentivirus in which all of the aspartates and glutamates in the acidic domain were replaced with alanine [D,E(24–48)A]. Endothelial cells transduced with this mutant GPIHBP1 transported antibody 11A12 normally (Figure 5E) but their ability to transport LPL was markedly impaired (Figure 5F).
GPIHBP1 Can Function as a Transporter In Vivo
We sought evidence that GPIHBP1 was present on both the basolateral and apical (luminal) surfaces of capillary endothelial cells in vivo and that it could function as a transporter. To assess the presence of GPIHBP1 on the apical and basolateral surfaces of capillary endothelial cells, we examined cross sections of capillaries in brown adipose tissue by confocal microscopy. The basolateral and apical surfaces of endothelial cells could be easily resolved in cross sections of capillaries containing a cell nucleus. In those sections, GPIHBP1 was present on both the apical and basolateral surfaces. The amount of GPIHBP1 on the basolateral and apical surfaces of endothelial cells was roughly equivalent (Figure 6A).
Figure 6. GPIHBP1 is present at both the basolateral and apical surfaces of endothelial cells in vivo and transports monoclonal antibody 11A12 across capillary endothelial cells.
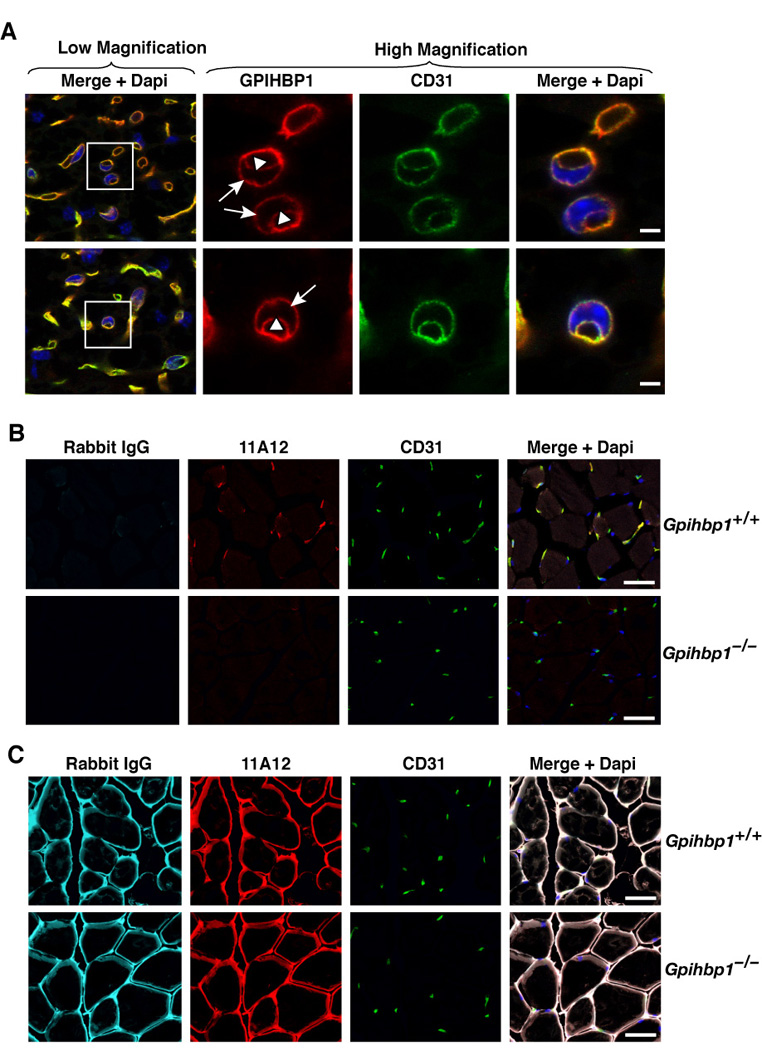
(A) Confocal microscopy showing the binding of GPIHBP1- and CD31-specific antibodies to brown adipose tissue of a wild-type mouse. Images were taken with a 100× objective without optical zoom (low magnification) or with 4× optical zoom (high magnification). To visualize both the apical and basolateral surfaces of capillaries, cross sections of capillaries that cut through an endothelial cell nucleus (blue) were identified (boxed areas) and viewed at high magnification. Both GPIHBP1 (red) and CD31 (green) were expressed at the apical (arrowheads) and basolateral (arrows) surfaces of endothelial cells. The scale bar represents a distance of 2.5 µm.
(B) Microscopy showing the transport of antibody 11A12 from the interstitial spaces to the lumens of capillaries of skeletal muscle. One quadriceps muscle of a wild-type and a Gpihbp1−/− mouse was injected with 10 µl of normal saline containing 15 µg each of the rat GPIHBP1-specific monoclonal antibody 11A12 and nonimmune rabbit IgG. After 30 min, the mice were injected intravenously with 200 µg each of fluorescent-labeled goat anti-rabbit IgG (pseudo-color cyan) and fluorescent-labeled goat anti-rat IgG (red). Three minutes later, the mice were extensively perfused with PBS and tissues fixed in situ with 3% PFA. Frozen sections were prepared and stained with antibodies against CD31 (green) to identify endothelial cells, and DAPI to identify nuclei. In wild-type mice, fluorescent-labeled anti-rat antibody was easily detectable within the capillaries of the injected muscle, whereas no staining was present in the skeletal muscle of Gpihbp1−/− mice. The scale bar represents a distance of 100 µm.
(C) Microscopy showing the presence of injected antibodies in the interstitial spaces of skeletal muscle. Frozen sections adjacent to the sections shown in Panel B were stained with fluorescent-labeled anti-rat antibody (red) and anti-rabbit antibody (pseudo-color cyan) to document the presence of antibody 11A12 and rabbit IgG within the interstitial spaces of the injected muscles. As expected, antibody 11A12 and rabbit IgG were present in both wild-type and Gpihbp1−/− mice. The scale bar represents a distance of 100 µm.
To determine if GPIHBP1 could function as a transporter in vivo, we performed an intramuscular injection of one quadriceps muscle in wild-type and Gpihbp1−/− mice with the GPIHBP1-specific monoclonal antibody 11A12, along with nonimmune rabbit IgG. After 30 min, the mice were injected intravenously with fluorescently-labeled secondary antibodies, with the goal of ascertaining whether either antibody had been taken up by endothelial cells and transported into the lumen of capillaries. Three minutes after the intravenous injection of the fluorescent antibodies, the mice were extensively perfused with PBS, and tissues fixed in situ with 3% PFA. Frozen sections were prepared and stained with an antibody against CD31 to identify endothelial cells. In wild-type mice, the fluorescent anti-rat antibody was detected exclusively in capillaries, colocalizing with CD31. No such staining was observed in Gpihbp1−/− mice (Figure 6B). The fluorescently labeled antibody against rabbit IgG was not detected in either wild-type or Gpihbp1−/− capillaries (Figure 6B). Also, in the wild-type mice, no staining of capillaries was observed in the contralateral (noninjected) quadriceps muscle. As expected, when adjacent sections of skeletal muscle were directly stained with fluorescently labeled anti-rat and anti-rabbit secondary antibodies, we observed abundant amounts of antibody 11A12 and the rabbit IgG in the interstitial spaces (surrounding the myocytes of wild-type and Gpihbp1−/− muscle) (Figure 6C).
Absence of LPL at the Luminal Surface of Capillaries in Gpihbp1−/− MIce
The fact that GPHBP1 functions as a transporter in vivo, combined with the fact that LPL is mislocalized in Gpihbp1−/− mice, made us suspect that LPL would be absent from the luminal surface of capillaries of Gpihbp1−/− mice. To address this issue in a direct fashion, wild-type and Gpihbp1−/− mice were injected intravenously with an anti-LPL antibody as well as FITC-labeled tomato lectin (which binds to the luminal surface of capillaries). Five minutes later, mice were euthanized and their tissues were extensively perfused with PBS and the heart was harvested. In the heart of wild-type mice, LPL was visible within capillaries, but LPL was nearly undetectable in the capillaries of Gpihbp1−/− mice (Figure 7A).
Figure 7. LPL is absent from capillary lumens in Gpihbp1−/− mice.
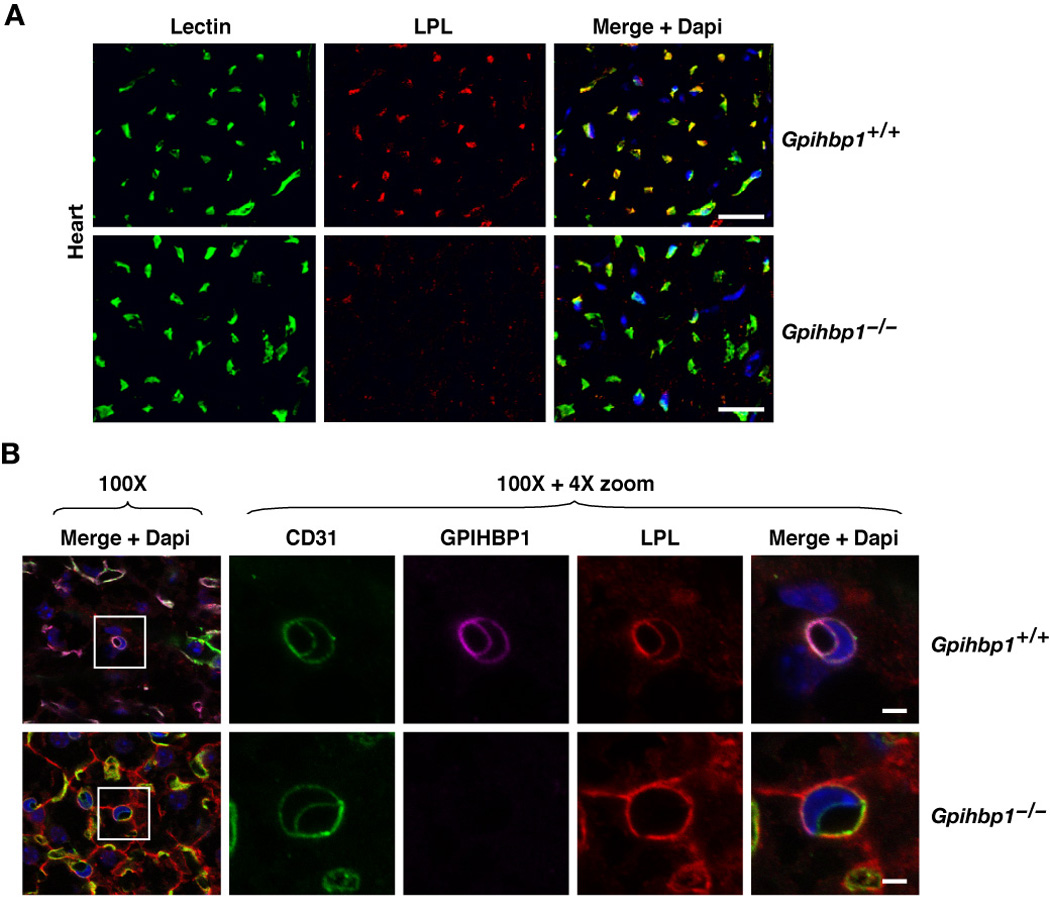
(A) Confocal images of heart showing the binding of FITC-labeled tomato lectin and a goat antibody against mouse LPL. Both the tomato lectin and the LPL-specific antibody were injected intravenously into a wild-type mouse and a Gpihbp1−/− mouse. Three min later, mice were perfused extensively with PBS and then perfusion-fixed with 3% PFA. The scale bars represent a distance of 50 µm.
(B) Confocal microscopy showing the binding of CD31-, GPIHBP1- and LPL-specific antibodies to brown adipose tissue of a wild-type mouse and a Gpihbp1−/− mouse. Images were taken with a 100× objective without optical zoom or with 4× optical zoom. To visualize both the apical and basolateral surfaces of capillaries, cross sections of capillaries that cut through an endothelial cell nucleus (blue) were identified (boxed areas) and viewed at high magnification. GPIHBP1 (purple), LPL (red) and CD31 (green) were all expressed at the apical and basolateral surfaces of endothelial cells in wild-type mice, but no LPL was present on the apical (luminal) surface of the capillary endothelium in Gpihbp1−/− mice. The scale bar represents a distance of 2.5 µm. (See also Figure S5.)
We also examined LPL localization in Gpihbp1−/− mice with confocal microscopy of cross sections of capillaries in brown adipose tissue of wild-type and Gpihbp1−/− mice. As expected, GPIHBP1 was present on both the luminal and abluminal surfaces of endothelial cells in wild-type mice (Figure 7B). Of note, LPL was present on both the luminal and abluminal surfaces of capillaries in wild-type mice, but was absent from the lumen in Gpihbp1−/− mice (Figure 7B, Figure S5). These findings are consistent with the ability of GPIHBP1 to serve as a transporter from the basolateral to the apical surfaces of endothelial cells (Figures 4–6) and strongly support the idea that GPIHBP1 is the critical molecule for shuttling LPL from the interstitial spaces into the capillary lumen.
DISCUSSION
That LPL is synthesized by adipocytes and myocytes but hydrolyzes triglycerides of lipoproteins within capillaries has been known for decades (Cryer et al., 1976; Havel and Kane, 2001). Precisely how LPL finds its way into capillaries, however, has been a mystery. In the current study, we provide evidence that GPIHBP1 is essential for LPL entry into capillaries. First, we found that LPL is mislocalized away from capillaries in the absence of GPIHBP1. GPIHBP1 is expressed exclusively in capillary endothelial cells, and in tissue sections of wild-type mice, most of the LPL colocalizes with GPIHBP1. In Gpihbp1−/− mice, most of the LPL is found on the outside of parenchymal cells, colocalizing with collagen IV (in the case of brown adipose tissue) or β-dystroglycan or laminin (in the case of skeletal muscle and heart tissue). Second, we found that GPIHBP1 is present on both the basolateral and apical surfaces of endothelial cells, both in cultured cells and in vivo within the capillaries of brown adipose tissue. Third, we show that the expression of GPIHBP1 in endothelial cells confers the ability to transport LPL and a GPIHBP1-specific monoclonal antibody from the basolateral to the apical surface of cells. This transport could be blocked by adding PIPLC to the basolateral chamber (an intervention that releases GPIHBP1 from the surface of cells). Also, the transport of LPL, but not the transport of antibody 11A12, was blocked by mutating the long stretch of negatively charged amino acids within GPIHBP1’s acidic domain. Mutating the acidic domain eliminates the ability of GPIHBP1 to bind LPL but has no effect on the epitope for antibody 11A12 (Beigneux et al., 2009b). Fourth, we show that GPIHBP1 transports a GPIHBP1-specific monoclonal antibody across endothelial cells in vivo. Finally, we show that LPL is absent from the capillary lumen in Gpihbp1−/− mice. All of these findings point to a direct role for GPIHBP1 in transporting LPL from the interstitial spaces to the lumen of capillaries.
Our current studies explain enigmatic findings from earlier studies. First, the mechanism for the striking phenotype of Gpihbp1−/− mice, severe hypertriglyceridemia (Beigneux et al., 2007), was puzzling, but it now makes perfect sense. With little or no LPL inside the capillaries of Gpihbp1−/− mice, lipolytic processing of triglyceride-rich lipoproteins cannot occur. Second, our findings help to explain the abnormal kinetics of LPL release in Gpihbp1−/− mice after an injection of heparin. Weinstein et al. (Weinstein et al., 2008) found that the release of LPL into the plasma of Gpihbp1−/− mice is delayed after a bolus of heparin, while LPL release is rapid in wild-type mice. In the current studies, we showed that LPL in Gpihbp1−/− mice is located in close proximity to parenchymal cells, whereas it is largely associated with capillaries in wild-type mice. When LPL is located in capillaries, it makes sense that an intravenous bolus of heparin would release LPL quickly. In the case of Gpihbp1−/− mice, it presumably takes longer for the heparin to reach the interstitial spaces and release that pool of LPL. Third, our findings provide an explanation for the normal LPL mass and activity measurements in Gpihbp1−/− mice. By immunohistochemistry, we found substantial amounts of LPL in the tissues of Gpihbp1−/− mice, but it was strikingly mislocalized.
Very recently, mutations in GPIHBP1 have been identified in humans with severe hypertriglyceridemia (Beigneux et al., 2009a; Franssen et al., 2010; Olivecrona et al., 2009; Wang and Hegele, 2007). All are missense mutations involving highly conserved amino acid residues within GPIHBP1’s Ly6 domain. Functional studies on the mutant proteins have revealed that the mutations result in a loss of GPIHBP1’s capacity to bind LPL.
The exact mechanism by which GPIHBP1 transports LPL across endothelial cells is not clear. Given the known association of GPI-anchored proteins with caveolae (Brown and Rose, 1992; Nosjean et al., 1997; Rajendran and Simons, 2005; Varma and Mayor, 1998) and the abundance of caveolae in endothelial cells (Anderson, 1993), one possibility is that LPL is transported by caveolar-dependent transcytosis. This possibility needs to be investigated thoroughly.
Our immunochemical studies of GPIHBP1 were made possible by a new monoclonal antibody against mouse GPIHBP1, 11A12 (Beigneux et al., 2009b). Antibody 11A12 binds avidly to GPIHBP1 in conventional immunohistochemistry studies and when injected intravenously into mice; these experiments revealed that GPIHBP1 is restricted to capillary endothelial cells and is absent in larger vessels. Our studies of LPL localization were aided by two antibodies against LPL, one generated against native bovine LPL and the other against a recombinant mouse LPL fragment; both antibodies were specific for LPL as judged by western blots and immunohistochemical studies with Lpl knockout mice. Both antibodies worked well in immunohistochemical studies, detecting LPL in parenchymal cells and in capillary endothelial cells (but not larger vessels), colocalizing with GPIHBP1. The availability of these antibodies provides for new approaches to investigate mislocalization of LPL away from capillaries as a potential explanation for hypertriglyceridemia.
The ability to visualize GPIHBP1 and LPL on the luminal surface of capillaries and the ability to assess transport from the interstitial space to the capillary could open the door to a better understanding of hypertriglyceridemia. For many years, scientists interested in hypertriglyceridemia have focused on measuring LPL levels 15 min after an injection of heparin, LPL levels in tissue extracts, and LPL mRNA levels. Unfortunately, there is little reason to believe that any of these measurements provide an accurate representation of the amount of LPL in capillaries. Indeed, in the case of Gpihbp1−/− mice, none of those measurements provided insights into the mechanism of hyperlipidemia. The phenotype of Gpihbp1−/− mice only made sense with the realization that GPIHBP1 functions as a transporter, and that no LPL is present in the lumens of capillaries of those mice. We speculate that assessing intracapillary LPL levels could prove to be worthwhile in other forms of hypertriglyceridemia—aside from that caused by specific structural abnormalities in GPIHBP1.
In humans, mechanisms for severe cases of hypercholesterolemia have come into focus (Goldstein et al., 1985; Goldstein et al., 2001; Horton et al., 2009), but the same cannot be said for many cases of severe hypertriglyceridemia. Many patients with severe hypertriglyceridemia have no mutations in LPL, GPIHBP1, APOCII, or APOAV and no obvious explanation for their disease (Beigneux et al., 2009a; Wang and Hegele, 2007). For some patients with hypertriglyceridemia, there are no obvious abnormalities in tissue stores of LPL or levels of LPL in the postheparin plasma—a situation reminiscent of Gpihbp1-deficient mice. Thus, it seems possible that defective transport of LPL into the lumen of capillaries could underlie at least some cases of hypertriglyceridemia in humans. A structurally intact GPIHBP1 is a cornerstone of this transport, but we suspect that other proteins participate in this process. Elucidating those factors might represent a productive area for future research.
EXPERIMENTAL PROCEDURES
Mice
Gpihbp1−/− and L0-MCK mice [Lpl-deficient mice expressing human LPL from a muscle specific promoter (MCK)] have been described previously (Beigneux et al., 2007; Weinstock et al., 1997). Mice were fed a chow diet and housed in a barrier facility with a 12-h light-dark cycle. All studies were approved by UCLA’s Animal Research Committee.
Antibodies, Immunohistochemistry, and Antibody Injection Experiments
A rat monoclonal antibody against mouse GPIHBP1 (11A12) has been described previously (Beigneux et al., 2009b). In some experiments, antibody 11A12 was coupled to fluorescent dye Alexa555 (Invitrogen, Carlsbad, CA) or infrared dye IR800 (Li-Cor, Lincoln, NE). We also used a chicken antibody against bovine LPL (Olivecrona and Bengtsson, 1983) and a goat antibody against a recombinant mouse LPL fragment produced in Escherichia coli (Weinstein et al., 2008). The specificity of the LPL-specific antibodies was assessed by western blot and by immunocytochemistry (see Supplemental Methods).
To detect GPIHBP1 in mouse tissues, 8–10 µm-thick frozen sections were prepared and incubated with primary antibodies [3 µg/ml for monoclonal antibody 11A12, 1:400 for the hamster anti-CD31 antibody (Millipore, Billerica, Mass), 1:200 for the rabbit anti-β-dystroglycan antibody (Santa Cruz Biotechnology, Santa Cruz, CA), 1:1000 for the rabbit anti-collagen type IV antibody (Cosmo Bio USA, Carlsbad, CA), 1:160 for the rabbit anti-laminin antibody (Millipore), 7 µg/ml for the chicken anti–bovine LPL antibody, and 10 µg/ml for the goat anti–mouse LPL antibody]. Secondary antibodies (Alexa555- or Alexa649-labeled anti-rat IgG, Alexa488- or Alexa649-labeled anti-hamster IgG, Alexa488- or Alexa649-labeled anti-rabbit IgG, Alexa546-labeled anti-chicken IgG, and Alexa549- or Alexa488-labeled anti-goat IgG) were used at a dilution of 1:200 and were incubated with slides at room temperature for 1 h. Images were obtained with an Axiovert 200 MOT microscope equipped with an Apotome (both from Zeiss, Germany) or by confocal fluorescence microscopy with a Leica SP2 1P-FCS microscope (Heidelberg, Germany) as described in the Supplemental Methods.
In some experiments, mice were injected intravenously with 50 µg of GPIHBP1- or 200 µg LPL-specific antibodies in a volume of 0.1 ml. After 3 min, mice were perfused with 10 ml PBS (2–3 ml/min) through the left ventricle followed by 10 ml of 3% paraformaldehyde (PFA) in PBS. The tissues were embedded in OCT and processed for immunohistochemistry.
For in vivo antibody transport experiments, one quadriceps muscle of wild-type and Gpihbp1−/− mice was injected intramuscularly with 10 µl of normal saline containing 15 µg each of the rat anti-GPIHBP1 antibody 11A12 and nonimmune rabbit IgG. After 30 min, the mice were injected intravenously with 200 µg each of Alexa488-labeled goat anti-rabbit IgG and Alexa568-labeled goat anti-rat IgG. Three minutes later, the mice were perfused with PBS and tissues fixed in situ with 3% PFA. The tissues were embedded in OCT and processed for immunohistochemistry.
Production of GPIHBP1-expressing Endothelial Cell Lines
Rat heart microvessel endothelial cells (RHMVECs) and bovine aortic endothelial cells were obtained from VEC Technologies and grown in MCDB-131 complete medium with antibiotics (VEC Technologies, Rensselaer, NY). The coding sequences for an S-protein–tagged GPIHBP1 (Beigneux et al., 2007) were cloned into the lentiviral vector pRRL-cPPT-MCS-IRES as described in the Supplemental Methods. A lentivirus for a S-protein–tagged mouse GPIHBP1 mutant [D,E(24–48)A] in which all of the aspartates and glutamates between residues 24 and 48 were replaced with alanine (EDGDADPEPENYNYDDDDDEEEEEE to AAGAAAPAPANYNYAAAAAAAAAAA) was created by site-directed mutagenesis with Phusion mutagenesis (Finnzymes, Espoo, Finland).
To assess levels of GPIHBP1 expression, total RNA was prepared from mouse tissues with TRI Reagent (Sigma, St. Louis, MO) and from transduced cells with the RNeasy kit (QIAGEN, Valencia, CA). RNA was treated with DNase I (Ambion, Austin, TX) and reversed transcribed into cDNA with a mixture of random primers, oligo(dT), and Superscript III (Invitrogen). Primers 5′-AGCAGGGACAGAGCACCTCT-3′ and 5′-AGACGAGCGTGATGCAGAAG-3′ were used to amplify the mouse Gpihbp1 cDNA, and primers 5′-CAGCGAGATCCTGAGGGTCA-3′ and 5′-TCCCCTCTTTCACAGAGCACC-3′ were used to amplify mouse and rat Cd31 cDNAs. PCR reactions were performed in triplicate on a 7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA). Gene-expression levels were calculated with the comparative cycle threshold (CT) method.
Production of LPL for LPL Transport Assays
Human LPL was concentrated from the medium of a Chinese hamster ovary (CHO) cell line stably expressing V5-tagged human LPL (a gift from Dr. Mark Doolittle, UCLA) (Beigneux et al., 2009b). The production of bovine LPL is described in the Supplemental Methods.
Endothelial Cell–based Assays of LPL Transport
Endothelial cells were grown on Millicell filters (polyethylene terephthalate, 1.1-cm2 filtration area, 1-µm pore size; Millipore) that had been coated with fibronectin (BD Biosciences, San Jose, CA). Cells were grown until they formed tight monolayers as judged by electrical resistance (tested with a Millicell Electrical Resistance System; Millipore) or by observing water tightness over 24 h. For each assay, monolayers were monitored for leakage by observing the transport of either an irrelevant antibody (IRDye680-labeled donkey anti-goat IgG; Li-Cor) or BSA (1 µg/ml; Amresco, Solon, OH).
For antibody transport assays, IRDye800-labeled antibody 11A12 and an IRDye680-labeled donkey anti-goat IgG antibody (Li-Cor) were added to the top (apical) or bottom (basolateral) chamber in PBS at a concentration of 0.25 µg/ml. After incubating the cells for the specified times at 37°C, 1 U/ml of PIPLC was added to either the apical or basolateral chamber to release GPIHBP1. Antibody in apical and basolateral media was detected by dot blotting or western blots (Supplemental Methods).
For LPL transport assays, human or bovine LPL (hLPL or bLPL, respectively) was added to the basolateral chamber and PBS was added to the apical chamber. Cells were incubated at 37°C for 1 h and treated with either heparin (100 U/ml for 15 min at room temp; APP Pharmaceuticals, Schaumburg, IL) or PIPLC (2.5 U/ml for 30 min at 37°C) to release LPL from the cell surface. Apical and basolateral samples were then collected and relative amounts of LPL were assessed by dot blotting, western blots, or an ELISA (Supplemental Methods).
HIGHLIGHTS
GPIHBP1 is required for proper targeting of LPL to the lumen of capillaries
GPIHBP1 is located at both the basolateral and apical surfaces of endothelial cells
GPIHBP1 transports a GPIHBP1-specific monoclonal antibody across endothelial cells
GPIHBP1 also transports LPL, providing a mechanism for LPL entry into capillaries
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Richard Anderson for helpful advice; the UCLA Vector Core for help with lentivirus production and transduction; and the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA (supported with funding from NIH-NCRR shared resources grant CJX1-44385-WS-29646 and NSF Major Research Instrumentation grant CHE-0722519) for the use of the confocal microscope. This work was supported by the American Heart Association, Western States Affiliate and National Office, and National Institutes of Health grants R01 HL094732, P01 HL090553, RO1 HL086683 and R01 HL087228.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest to declare.
REFERENCES
- Anderson RG. Plasmalemmal caveolae and GPI-anchored membrane proteins. Curr Opin Cell Biol. 1993;5:647–652. doi: 10.1016/0955-0674(93)90135-d. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A, Young SG. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. doi: 10.1016/j.cmet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Franssen R, Bensadoun A, Gin P, Melford K, Peter J, Walzem RL, Weinstein MM, Davies BS, Kuivenhoven JA, Kastelein JJ, Fong LG, Dallinga-Thie GM, Young SG. Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler Thromb Vasc Biol. 2009a;29:956–962. doi: 10.1161/ATVBAHA.109.186577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Gin P, Davies BS, Weinstein MM, Bensadoun A, Fong LG, Young SG. Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J Biol Chem. 2009b;284:30240–30247. doi: 10.1074/jbc.M109.046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 2789–2816. [Google Scholar]
- Cryer A, Riley SE, Williams ER, Robinson DS. Effect of nutritional status on rat adipose tissue, muscle and post-heparin plasma clearing factor lipase activities: their relationship to triglyceride fatty acid uptake by fat-cells and to plasma insulin concentrations. Clin Sci Mol Med. 1976;50:213–221. doi: 10.1042/cs0500213. [DOI] [PubMed] [Google Scholar]
- Davies BS, Waki H, Beigneux AP, Farber E, Weinstein MM, Wilpitz DC, Tai LJ, Evans RM, Fong LG, Tontonoz P, Young SG. The expression of GPIHBP1, an endothelial cell binding site for lipoprotein lipase and chylomicrons, is induced by peroxisome proliferator-activated receptor-gamma. Mol Endocrinol. 2008;22:2496–2504. doi: 10.1210/me.2008-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen R, Young SG, Peelman F, Hertecant J, Sierts JA, Schimmel AW, Bensadoun A, Kastelein JJ, Fong L, Dallinga-Thie GM, Beigneux AP. Chylomicronemia with Low Postheparin Lipoprotein Lipase Levels in the Setting of GPIHBP1 Defects. Circ Cardiovasc Genet. 2010 doi: 10.1161/CIRCGENETICS.109.908905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frykman PK, Brown MS, Yamamoto T, Goldstein JL, Herz J. Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1995;92:8453–8457. doi: 10.1073/pnas.92.18.8453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gin P, Yin L, Davies BS, Weinstein MM, Ryan RO, Bensadoun A, Fong LG, Young SG, Beigneux AP. The acidic domain of GPIHBP1 is important for the binding of lipoprotein lipase and chylomicrons. J Biol Chem. 2008;283:29554–29562. doi: 10.1074/jbc.M802579200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS, Anderson RGW, Russell DW, Schneider WJ. Receptor-mediated endocytosis: Concepts emerging from the LDL receptor system. Annu. Rev. Cell Biol. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. In The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 2863–2913. [Google Scholar]
- Havel RJ, Kane JP. Introduction: Structure and metabolism of plasma lipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. In The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 2705–2716. [Google Scholar]
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50 Suppl:S172–S177. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosjean O, Briolay A, Roux B. Mammalian GPI proteins: sorting, membrane residence and functions. Biochim Biophys Acta. 1997;1331:153–186. doi: 10.1016/s0304-4157(97)00005-1. [DOI] [PubMed] [Google Scholar]
- Obunike JC, Lutz EP, Li Z, Paka L, Katopodis T, Strickland DK, Kozarsky KF, Pillarisetti S, Goldberg IJ. Transcytosis of lipoprotein lipase across cultured endothelial cells requires both heparan sulfate proteoglycans and the very low density lipoprotein receptor. J Biol Chem. 2001;276:8934–8941. doi: 10.1074/jbc.M008813200. [DOI] [PubMed] [Google Scholar]
- Olivecrona G, Ehrenborg E, Semb H, Makoveichuk E, Lindberg A, Hayden MR, Gin P, Davies BS, Weinstein MM, Fong LG, Beigneux AP, Young SG, Olivecrona T, Hernell O. Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J Lipid Res. 2009 doi: 10.1194/jlr.M002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivecrona T, Bengtsson G. Immunochemical properties of lipoprotein lipase. Development of an immunoassay applicable to several mammalian species. Biochim Biophys Acta. 1983;752:38–45. doi: 10.1016/0005-2760(83)90230-8. [DOI] [PubMed] [Google Scholar]
- Olivecrona T, Olivecrona G. The Ins and Outs of Adipose Tissue. In: Ehnholm C, editor. In Cellular Lipid Metabolism. Berlin Heidelberg: Springer; 2009. pp. 315–369. [Google Scholar]
- Rajendran L, Simons K. Lipid rafts and membrane dynamics. J Cell Sci. 2005;118:1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- Varma R, Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–E288. doi: 10.1152/ajpendo.90920.2008. [DOI] [PubMed] [Google Scholar]
- Wang J, Hegele RA. Homozygous missense mutation (G56R) in glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPI-HBP1) in two siblings with fasting chylomicronemia (MIM 144650) Lipids Health Dis. 2007;6:23. doi: 10.1186/1476-511X-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein MM, Yin L, Beigneux AP, Davies BS, Gin P, Estrada K, Melford K, Bishop JR, Esko JD, Dallinga-Thie GM, Fong LG, Bensadoun A, Young SG. Abnormal Patterns of Lipoprotein Lipase Release into the Plasma in GPIHBP1-deficient Mice. J Biol Chem. 2008;283:34511–34518. doi: 10.1074/jbc.M806067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock PH, Levak-Frank S, Hudgins LC, Radner H, Friedman JM, Zechner R, Breslow JL. Lipoprotein lipase controls fatty acid entry into adipose tissue, but fat mass is preserved by endogenous synthesis in mice deficient in adipose tissue lipoprotein lipase. Proc Natl Acad Sci U S A. 1997;94:10261–10266. doi: 10.1073/pnas.94.19.10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.