

Abstract
The role of the cerebral cortex in the cognitive deficits in preterm survivors is poorly understood. Periventricular leukomalacia (PVL), the key feature of encephalopathy of prematurity, is characterized by periventricular necrotic foci and diffuse gliosis in the surrounding cerebral white matter. Here, we tested the hypothesis that reductions in the density of layer I neurons and/or pyramidal neurons in layers III and/or V are associated with PVL, indicating cortical pathology potentially associated with cognitive deficits in long‐term survivors. In controls (23 gestational weeks to 18 postnatal months) (n = 15), a lack of significant differences in pyramidal density among incipient Brodmann areas suggested that cytoarchitectonic differences across functional areas are not fully mature in the fetal and infant periods. There was a marked reduction (38%) in the density of layer V neurons in all areas sampled in the PVL cases (n = 17) compared to controls (n = 12) adjusted for postconceptional age at or greater than 30 weeks, when the six‐layer cortex is visually distinct (P < 0.024). This may reflect a dying‐back loss of somata complicating transection of layer V axons projecting through the necrosis in the underlying white matter. This study underscores the potential role of secondary cortical injury in the encephalopathy of prematurity.
Keywords: Brodmann area, Cajal–Retzius cell, excitotoxicity, microtubule‐associated protein‐2, prematurity
INTRODUCTION
Despite critical advances in the survival of premature infants, long‐term cognitive deficits are increasingly recognized as a major adverse outcome with enormous personal and societal burden (64). Of the 63 000 very low‐birth weight infants (≤1500 g) born in the United States each year, 25%–50% develop cognitive, behavioral, attentional and associational deficits 47, 64. Moreover, of the approximately 350 000 late preterm infants (34–36 gestational weeks) born yearly, 2.1% (7000 infants) develop developmental disabilities, cerebral palsy and/or seizures, a twofold increase over such neurological disability in term infants (53). Abnormalities in working memory, planning and attention have all been reported in premature infants who survive into childhood 1, 3, 6, 8, 9, 15, 18, 27, 41, 58, 59, 62, 63, 66, 67. Nevertheless, the cellular basis of these cognitive abnormalities is poorly understood, particularly in relationship to the cerebral cortex, the key neuroanatomic substrate of cognitive processing. The rubric that best defines the known brain abnormalities in premature infants is the “encephalopathy of prematurity,” an entity comprised of white and gray matter lesions in various combinations that have been defined by neuropathological and neuroimaging studies 32, 64. Its most notable component is the white matter lesion, periventricular leukomalacia (PVL), which occurs in association with neuronal/axonal deficits involving the cerebral cortex, thalamus, basal ganglia, hippocampus, cerebellum and/or brain stem 5, 14, 21, 32, 40, 54. The cause of PVL is likely cerebral ischemia/reperfusion with excitoxicity and free radical toxicity compounded in certain instances by infection/inflammation and cytokine toxicity (32). In a recent survey of the neuropathology of prematurity in 41 infants dying in the perinatal period, we found that approximately one‐third of PVL cases had obvious focal neuronal loss and/or gliosis in the cerebral cortex, primarily in the frontal lobe (54). In addition, premature infants studied in childhood, adolescence and adulthood demonstrate persistent cortical volume deficits that correlate with a wide spectrum of cognitive deficits 9, 13, 25, 26, 52, 67. In the following study, our main objective was to determine the role of pyramidal neurons in perinatal cortical pathology associated with PVL, the defining lesion of the encephalopathy of prematurity.
The microcircuitry of the cerebral cortex underlying cognitive processing is dependent upon precise interrelationship between variable numbers of excitatory pyramidal neurons and inhibitory non‐pyramidal (granular) neurons in cortical modules depending upon specific connectivity. As a first step in the neuropathological analysis of the cerebral cortex in the encephalopathy of prematurity, we focused upon pyramidal neurons because they account for 75%–80% of the cortical neurons 11, 29, and they play a critical role in cellular injury caused by hypoxia–ischemia via excitoxic mechanisms. We also analyzed neurons in layer I, that is, putative Cajal–Retzius cells, because these neurons are important in the development and function of pyramidal neurons 12, 43. We assessed three main cytoarchitectonic regions: (i) the granular (koniocortex) cortex of sensory areas characterized by small densely packed neurons in the middle laminae; (ii) agranular cortex of the motor and premotor cortical areas characterized by large pyramidal cells in layer V, and relative absence of granular cells; and (iii) homotypical cortex of association regions with varying populations of granule cells. Here, we tested the hypothesis that the density of layer I neurons, layer III pyramidal neurons and/or layer V pyramidal neurons is decreased in the cerebral cortex overlying PVL compared to controls adjusted for postconceptional age. For cell quantitation in archival tissue sections, we employed the two‐dimensional method of Benes et al used by them in the analysis of the cerebral cortex in schizophrenia 4, 62. To help identify the pyramidal neurons of interest, we applied immunostaining with the antibody to the microtubule‐associated protein‐2 (MAP2), which demonstrates the somatodendritic compartment of virtually all neurons but especially pyramidal neurons 22, 38, 50. We report here a laminar‐specific reduction in pyramidal neurons of layer V, without severe or widespread neuronal damage in the cortex itself, suggesting de‐afferentiation of these pyramidal neurons by the underlying white matter necrosis and secondary cell body loss via dying‐back mechanisms.
MATERIALS AND METHODS
Clinical database
Cerebral cortical samples were accrued from PVL cases and controls between 1993 and 2007 in the Department of Pathology, Children's Hospital Boston, MA. We studied cases in two groups: PVL (n = 20) and control (non‐PVL) (n = 15) based upon neuropathological findings. Of this cohort, 17 PVL cases and 12 control cases were ≥30 postconceptional weeks, that is, the time frame when the six‐layer cortex is visually distinct (see below). Cases were classified as PVL based upon the histopathological criteria of periventricular focal necrosis in the deep white matter with surrounding diffuse reactive gliosis and microglial activation 5, 20, 31, 32. The stage of periventricular necrosis was categorized by histopathological criteria: (i) acute with coagulative necrosis, cytoplasmic hyper‐eosinophilia and nuclear pyknosis of all tissue elements, and hyper‐eosinophilic axonal spheroids; (ii) organizing with infiltrating macrophages and reactive astrocytes in the necrotic foci; and (iii) chronic, with focal cavitation or scar formation with axonal mineralization 31, 32. Reactive astrocytes were present in the surrounding white matter at all histopathological stages of the focal necrosis. Control cases did not have PVL or other significant brain pathology upon standard histological examination. Cases with disorders of known cortical maldevelopment, for example, Down syndrome, were excluded from analysis. All cases were studied with approval of the Human Protection Committee and Institutional Review Board at Children's Hospital Boston. The autopsy reports were reviewed for major clinical findings, systemic autopsy diagnoses and neuropathological findings. The age of each case was expressed in postconceptional (gestational plus postnatal) weeks.
Immunocytochemistry
To assess cerebral cortical pathology in all study cases and controls, we applied antibodies to MAP2 and fractin. We applied immunohistochemistry with the neuron‐specific MAP2 antibody for quantitation of selected neuronal density in layers I, III and V. For MAP2 analysis, antigen retrieval involved 10 minutes of microwave at 195°F in citrate buffer, pH 6.0. After retrieval, standard methods were applied in deparaffinized tissue sections (4 µm) (20). The antibody MAP2 (MAP2, dilution: 1:2000, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, sc‐20172) is a rabbit polyclonal antibody raised against amino acids 1–300 mapping at the N‐terminus of MAP2 of human origin. The antibody fractin (BD Pharmingen, San Jose, CA) was used at 1:500 dilution. Negative immunohistochemical controls omitted the primary antibodies. We first qualitatively assessed immunostained sections with standard light microscopy. In a pilot study of the cerebral cortex in five control cases, we found that the MAP2 antibody labeled layer I neurons and pyramidal neurons in layers III and V in all incipient Brodmann areas sampled.
Quantitation of neuronal density in cortical layers I, III and V
We used the procedure of Benes et al for two‐dimensional counting in the cerebral cortex 4, 62, which we modified for the developing human brain in archival microscopic tissue sections from our pathology department. The following morphological criteria were applied for a neuron to be counted: (i) positive MAP2 cytoplasmic immunostaining; (ii) distinctly visible nucleus; and (iii) specific neuronal morphology of layer I neurons and pyramidal neurons (see below). The Neurolucida software program (MicroBrightField, Williston, VT) was used to quantify neuronal density in the selected layers. At low magnification (×40), we outlined the boundary of the entire section and then the boundary of the entire cortex in the section (1, 2). Next, we defined three rectangular “boxes” of the cortex that were each 600 µm wide, as per the procedure of Benes et al 4, 62, each involving a specific Brodmann area (Figure 2). Within this defined cortical region, the boundaries of the six cortical laminae were outlined (Figure 2). Once the outlines were set in place, the selected neurons in layers I, III and VI were counted at high magnification (×200). Prior to the study, we performed a pilot analysis in three control cases to determine the optimal width of the cortical rectangular boxes for assessment of neuronal density in each of the selected laminae. We counted neurons in the three cortical layers in boxes that were 100–800 µm in width in 100 µm increments, and neuronal density in each lamina was calculated. We found that widths greater than 500 µm, that is, at 600, 700 and 800 µm, provided minimal variation in neuronal density (<4%) (data not shown). These pilot data indicated that boxes greater than 500 µm in width provided the most accurate density measurements in the developing brain, and we selected 600 µm for use in the study. Neuronal density was calculated as the number of specific neuronal subtype divided by the area of the laminar box of interest (mm2). The Brodmann area was determined by comparison of the region counted to standard available cytoarchitectonic maps (Figure 1). In the PVL cases, we sampled the cortex overlying foci of periventricular necrosis and diffuse white matter gliosis, and compared it to the cortex in control cases without PVL and from similar Brodmann areas. Because of the obvious white matter lesions in the study group, the analysis could not be performed blinded; it was nevertheless performed without detailed clinicopathological information, including age.
Figure 1.
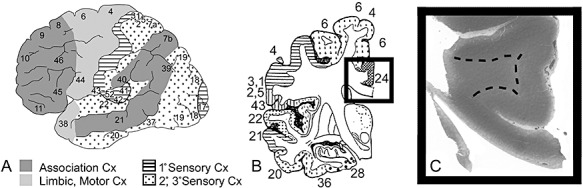
The Brodmann area was determined by comparison of the region counted to standard available maps. A. The cortex is subdivided for analysis into regions based upon cytoarchitectonic patterns: koniocortex (sensory related) with stripes (primary sensory) and dots (secondary and tertiary sensory); agranular (motor and limbic related), light gray; and homotypical (association), dark gray. The specific Brodmann area is defined from standard maps (courtesy of the Harvard Brain Bank) (B) in the tissue section (C). Examination of the area 24 (cingulate gyrus) is exemplified in a representative control at 72 postconceptional weeks (2 postnatal months).
Figure 2.
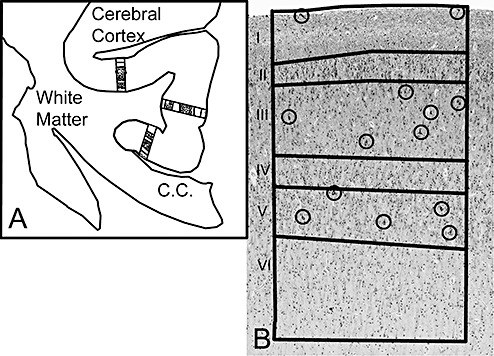
The Neurolucida software program (MicroBrightField) was used to quantify the density of layer I neurons and pyramidal neurons in layers III and V with MAP2 immunostaining in a two‐dimensional sampling column from the pial surface to the gray‐white border. A. We first selected the Brodmann area for study, as demonstrated in area 24 (cingulate gyrus). At low magnification (×40), we outlined the boundary of the entire section and then the boundary of the entire cortex in the section (Figure 1). Next, we defined three rectangular “boxes” of the cortex that were each 600 µm wide in the Brodmann area. B. Within this defined cortical region, the boundaries of the six cortical laminae were outlined, as illustrated in a representative control cases at 40 postconceptional weeks (term). Once the outlines were set in place, the selected neurons in layers I, III and VI were counted at high magnification (×200) in the MAP2‐immunostained section. The circles identify the neurons counted in each lamina of interest.
Percent of MAP2‐immunostained neurons in layer V
Antibodies against MAP2 are considered sensitive probes of ischemic injury, with reductions in the intensity of MAP2 immunostaining associated with hypoxic–ischemic conditions in human autopsied brain 36, 38, 42, 50. In this study, we counted the pyramidal neurons immunostaining with MAP2, irrespective of the degree of intensity. We then combined this information with data involving the percent pyramidal neurons in layer V that are immunostained for MAP2 relative to the total number of pyramidal neurons in order to determine if the hypothesized reduction in the density of MAP2 neurons was caused by a loss of cell bodies and/or immunostaining. We counted 100 pyramidal neurons in layer V in the cerebral cortex overlying PVL compared to the same anatomic region in controls adjusted for postconceptional age. We then determined the percent of total pyramidal neurons that were immunopositive for MAP2, and compared the measures between PVL cases and controls.
Analysis of fractin‐immunopositive neurons in the cerebral cortex
We utilized an antibody to fractin as an apoptotic marker to test the hypothesis that there is increased injury in layer I neurons and/or pyramidal neurons in layer III and/or layer V overlying PVL with or without an associated reduction in neuronal density (18). We surveyed for fractin‐positive neurons in the entire cerebral cortex overlying PVL compared to similar regions in the control cases adjusted for postconceptional age. We qualitatively determined the incidence (presence or absence) of positive fractin immunostaining.
Cortical and laminar thickness
The cortical and laminar thickness was determined in microscopic sections stained with hematoxylin‐and‐eosin/Luxol‐fast‐blue (H & E/LFB), adjacent to the sections immunostained and assessed with MAP2. The H & E/LFB‐stained sections were used because the demarcations between the cortex and underlying white matter were more distinct than in those immunostained with MAP2. A measuring tool in the Neurolucida program called “quick measure line” was used to make the thickness measurement.
Neuronal size
The neurons were measured with the use of the computer program Neurolucida and the “quick measure line” tool. The size of the neuron was determined by measuring the largest diameter of the cell at midpoint. The mean of the largest diameter of five neurons for each subtype sampled was used for statistical analysis.
Statistical analysis
Demographic characteristics were compared between PVL cases and controls using t‐tests for continuous variables and Fisher's exact tests for categorical variables. The normative development in the (non‐PVL) controls was examined by regression analysis of age, brain weight, percent MAP2‐immunopositive cells, neuronal size, neuronal density and laminar thickness. For neuronal size and density, repeated measures regression was used as there were measurements from three regions per case. The effect of functional cortical region, Brodmann area and cerebral lobe upon neuronal density in controls was tested using repeated measures analysis of covariance (ANCOVA) controlling for age. In the comparison of PVL cases to controls, ANCOVA controlling for age was used to test for differences between study groups in brain weight, percent MAP2‐immunopositive cells, neuronal size, neuronal density and laminar thickness. For neuronal size and density, repeated measures ANCOVA was used as there were measurements from three regions per case. Differences in the incidence of histopathological features and fractin immunostaining between PVL cases and controls were tested by Fisher's exact test. Analysis was conducted using SAS v9.2 (Cary, NC). In all analyses, a P value <0.05 was considered statistically significant.
RESULTS
Baseline development of the human cerebral cortex
The analysis of the cerebral cortical data in the control group provided baseline information about normative human development. We analyzed the cerebral cortex in 15 control cases ranging in postconceptional age from 23 weeks (midgestation) to 112 weeks (18 postnatal months) with a median of 34 weeks in which MAP2‐immunopositive neurons were identified (Supporting Information Table S1). These control cases did not have PVL or other significant neuropathological features. The primary causes of death were: prematurity with respiratory distress syndrome, n = 4; congenital heart disease, n = 2; primary pulmonary hypertension, n = 1; hydrops fetalis caused by placental chorioangiomas, n = 1; hydrops fetalis caused by parvo virus, n = 1; sacral teratoma, n = 1; cystic lymphatic malformation of the neck, n = 1; Werdnig–Hoffmann disease, n = 1; foreign body aspiration, n = 1; Blackfan–Diamond syndrome, n = 1; and bronchiolitis, n = 1. The following Brodmann regions (with available histological sections) were assigned the following subtypes of cortex: (i) koniocortex (sensory) (n = 7): primary sensory—17 (visual), 22, 41 (auditory), 3 (somatosensory); and secondary/tertiary sensory—18 and 19 (visual), 20 (auditory); (ii) agranular cortex (motor cortex) (n = 2)—4; and (iii) homotypical cortex: frontal association (n = 22)—8, 9, 23, 24, 31, 32, 46, 47; parietal association (n = 5)—7, 21, 40; and limbic association (n = 2)—28, 29. Not all Brodmann areas were available in all cases.
The brain weight increased significantly with increasing gestational age, as expected [slope = 37.1 ± 2.4 (standard error) g/week, P = 0.011]. The pyramidal cells were first detected with MAP2 immunostaining in layer V at 23 gestational weeks (3, 4); of note, they were not appreciated at this age in the H & E/LFB‐stained sections. We did not identify any MAP2‐immunopositive neurons at 20 weeks in a fetus dying of pulmonary immaturity (Figure 4). At 23 weeks, layers I and II were readily distinguished from one another, as was layer II from the deeper layers (layers III–VI), but distinctions between the deeper layers were not obvious (Figure 5). At 27 weeks, pyramidal neurons were present in layers III and V. By 30 weeks, the six laminae were fully differentiated from one another (Figure 5). By the second half of gestation, the distinct morphology of the pyramidal neurons in layers III and V, as well as layer I neurons, was fully developed. In layer I, MAP2‐immunostained neurons had abundant perikarya and were horizontal in orientation, consistent with the known morphology of Cajal–Retzius neurons (Figure 3) (41). Pyramidal neurons were identified by the classic triangular‐shaped cytoplasm (Figure 3). Immunostaining to MAP2 was identified predominately in pyramidal neurons of all sizes compared to negligible staining in non‐pyramidal (granular) neurons (data not shown). The percent pyramidal neurons in layer V that immunostained for MAP2 relative to the total number of pyramidal neurons was 86.7% ± 5.9% in the controls, and did not change significantly with increasing postconceptional age [slope = 0.011, P = 0.70 (regression). The size of pyramidal neurons increased significantly in only layer V (slope = 0.044, P = 0.0003) with increasing postconceptional age (Figure 3), and not in layer I neurons (slope = 0.013, P = 0.80) or pyramidal neurons in layer III (slope = 0.046, P = 0.23).
Figure 3.

Selected neurons in the frontal association (homotypical) cortex are illustrated in representative controls at 43 postconceptional weeks (A), 53 weeks (B and D) and 23 weeks (C). These neurons are: A. MAP2‐immunostained horizontal neurons in layer I which are putative Cajal–Retzius cells; B. MAP2‐immunostained pyramidal neurons in layer III; C. MAP2‐immunostained pyramidal neurons in layer V at 23 weeks; and D. MAP2‐immunostained pyramidal neurons in layer V at 53 weeks. The pyramidal neurons in layer III (B) tend to be smaller than those in layer V (D) at the same age. The pyramidal neurons in layer V increase in size and process complexity from 23 weeks (C) to 53 weeks (D). Scale bar = 50 µm.
Figure 4.
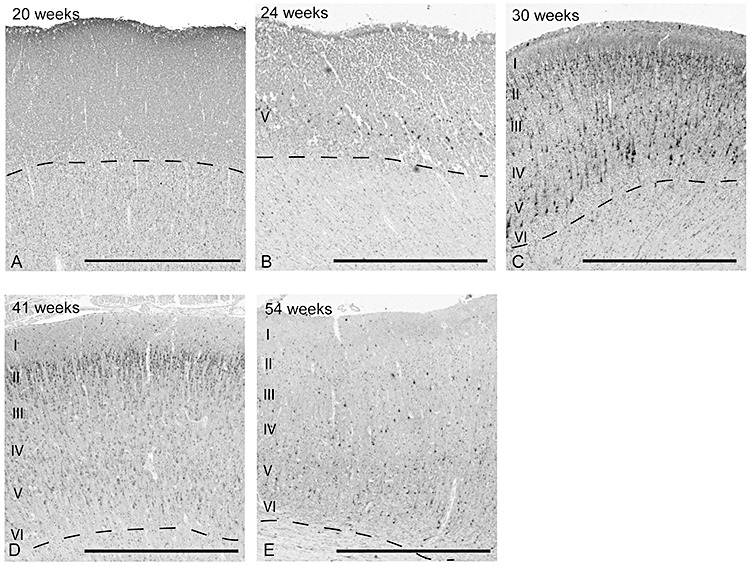
There is a developmental change in the appearance of MAP2‐immunopositive neurons in the frontal association cortex from 20 postconceptional weeks to 54 weeks in the control group. At 20 weeks, MAP2‐immunopositive neurons are not identified, but are present in the incipient layer V at 23 weeks. At 30 weeks, MAP2‐immunopositive neurons are prominent in layers II–V, and in layers II–VI thereafter. Scale bar = 1 mm.
Figure 5.

There is a striking change in the laminar pattern between 23 (A) and 43 (C) postconceptional weeks, with differentiation of layers III–VI appearing around 30 weeks (B), as illustrated in representative controls in the frontal association cortex. Over this time course, layers III–VI are increasingly detected, first appearing around 30 weeks. At 23 weeks, layers I and II are discernible; the latter characterized by dense cellular packing. The change in the laminar pattern is caused by an increase in the neuropil and differentiation of neuronal subtypes. The dotted line represents the border between layer VI and underlying white matter. Scale bar = 1 mm.
Although differences in pyramidal neuronal density in layers III and/or V were suggested among the various regions and Brodmann areas analyzed, there were no significant differences upon adjustment for gestational age (Supporting Information Table S2). Of note, a sufficient number of sections of the primary sensory cortex in the control group were not available for analysis (Supporting Information Table S2). Given that we did not visually identify six laminae until 30 postconceptional weeks, we excluded controls less than 30 weeks in a comparative analysis of the koniocortex vs. the homotypical cortex, but still did not identify a statistically significant difference in the density of layer I neurons and/or of pyramidal neurons in layer III and/or layer V between these areas (data not shown). The lack of statistical significance is likely due, at least in part, to the wide density variations among individual cases in each anatomic category and the small sample size for several individual Brodmann areas. Consequently, we felt justified in making no adjustments for these anatomical regions in the analyses examining total cortical neuronal density in all regions sampled in PVL cases and controls. In other analyses, we combined the Brodmann areas into koniocortex and homotypical cortex in order to increase the sample size for the major types of cytoarchitectonic patterns (Supporting Information Table S2). Developmental changes in density were sought based upon increasing gestational age. With increasing gestational age, there was no significant change in the density of layer I neurons or pyramidal neurons in layers III and V in koniocortex or homotypical cortex (see above). Yet, when all areas were combined, there was a significant mild decrease in the density of pyramidal neurons in layer V (slope = −0.68, P = 0.048) (Figure 6). The cerebral cortex increased in overall thickness with increasing gestational age (P < 0.001). The individual laminae all increased in thickness as well, with the exception of layer IV (Supporting Information Table S3).
Figure 6.
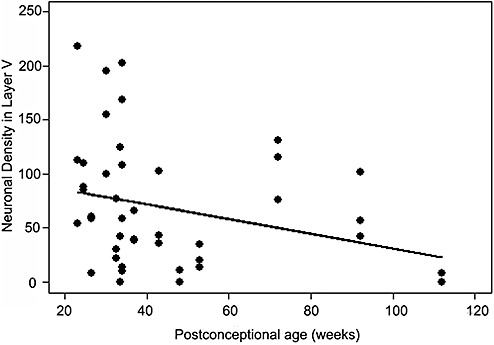
There is a normative significant decrease in the density of pyramidal neurons in layer V with increasing postconceptional age for all layers cortical regions combined (control cases) (P = 0.048).
The cerebral cortex in PVL
Demographics
We analyzed the cerebral cortex in 20 PVL cases and 15 controls (Supporting Information Table S1). Both groups were comprised of premature infants born on average near‐term with PVL cases born at 33.9 ± 4.3 weeks, and controls at 33.1 ± 6.2 weeks (P = 0.67) (Supporting Information Table S1). In this cohort, 75% (15/20) of the PVL cases were born prematurely (<37 gestational weeks) compared to 67% (10/15) of the controls (P = 0.71). Fifty percent (10/20) of the PVL cases in this series were very early‐to‐mid preterm (<32 weeks) compared to 67% of the controls (10/15) (P = 0.49) (Supporting Information Table S1). The PVL cases lived on average a shorter postnatal period (5.9 ± 14.0 weeks) compared to the controls that lived 13.2 ± 23.6 weeks, but the difference was not significant (P = 0.30) (Supporting Information Table S1). There was no significant difference between the postconceptional age between the PVL cases (39.8 ± 14.0) and controls (46.3 ± 26.1) (P = 0.39) (Supporting Information Table S1). There was also no difference between the two groups in the mean Apgar scores at 5 minutes (6.8 for both groups) or incidence of congenital heart disease, necrotizing enterocolitis, sepsis (defined by documented positive blood culture or bacterial infection in any organ), seizures or mechanical ventilation (Supporting Information Table S1). Of note, 100% of the PVL cases and 87% of the controls received mechanical ventilation (P = 0.19) (Supporting Information Table S1). The primary causes of death in the PVL group were: prematurity with respiratory distress syndrome, n = 7; congenital heart disease, n = 6; congenital diaphragmatic hernia, n = 2; mesangial sclerosis of the kidney, n = 1; inherited urea cycle defect, n = 1; translocation of chromosomes 12 and 14, n = 1; skeletal dysplasia, n = 1; tegmental necrosis and hypoplasia of the medulla oblongata, n = 1. There were no stillbirth cases in the PVL or control groups. There was no difference in the postmortem interval among the two groups (P = 0.11) (Supporting Information Table S1), and there was no obvious visual effect of postmortem interval upon immunostaining for the two antibodies analyzed.
Neuropathology of PVL
PVL in our affected cases was characterized by the diagnostic features of focal periventricular necrosis associated with diffuse gliosis and microglial activation in the white matter and relative sparing of the cerebral cortex utilizing standard neuropathological criteria 31, 32 (Figure 7). In the PVL cases, the stage and degree of severity of the white matter damage varied, reflecting the typical spectrum of this pathology in our autopsy service. At the time of brain cutting, 35% (7/20) of the PVL cases had gross periventricular necrotic foci that were either cystic cavities (4/7) or chalky‐white foci (3/7). Thus, grossly cystic lesions accounted for only 20% (4/20) of the autopsy cases; these cysts ranged in size from 0.75 to 2.2 cm. The chalky‐white foci of necrosis, that is, the classic so‐called “white spots” of PVL 31, 32, accounted for 15% (3/20) of the PVL cases; these lesions were typically 2–3 mm in diameter. In the 65% of cases without gross lesions, the microscopic necrotic foci varied in size up to approximately 2 mm. There was a significant difference between the PVL and control cases in the incidence of germinal matrix hemorrhages with 30% (6/20) in the PVL cases compared to 0% (0/15) in the controls (P = 0.03); this finding was not unexpected given that the control brains were selected based upon the absence of significant neuropathological findings in the forebrain. The brain weight adjusted by postconceptional age tended to be smaller in the PVL cases (349.5 ± 25.4 g) compared to the controls (400.7 ± 25.4), but the difference was not statistically significant (P = 0.17).
Figure 7.
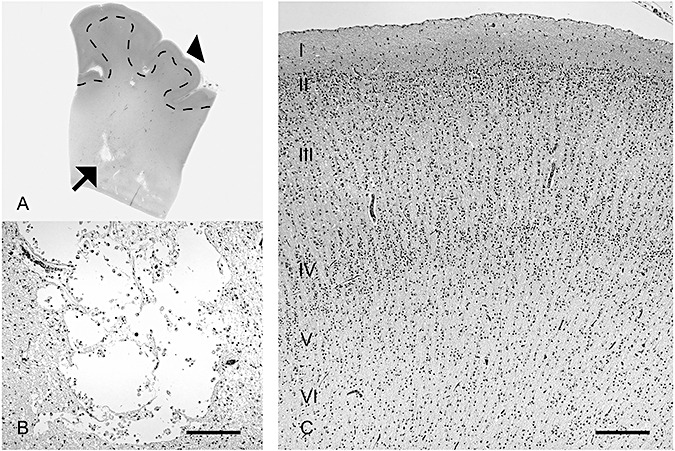
The cerebral cortex overlying periventricular leukomalacia (PVL) is relatively intact histologically, as illustrated in the PVL case of an infant dying at 39 postconceptional weeks. A. A low‐power histological section provides orientation, with the microcysts of focally necrotic lesions in the deep (periventricular) white matter (arrow) illustrated in high magnification (B) and with the overlying cerebral cortex (arrowhead) (C). Scale bar = 200 µm.
Histopathological features in the cerebral cortex
In general, the cerebral cortex overlying PVL was histologically intact (Figure 7). Nevertheless, the PVL group demonstrated mild degrees of histopathology, including reactive astrogliosis characterized by hypertrophic astrocytes upon immunostaining for glial fibrillary acidic protein in 38% of the cases, and reactive “naked” glial change with enlarged nuclei and no cytoplasm in 39% of the cases (Figure 8); however, there was no significant difference in these findings between the PVL and control groups (Supporting Information Table S4). There was also no difference in the presence of acute (agonal) neuronal necrosis or microglial activation (Figure 8) (Supporting Information Table S4). There was no significant difference in the presence of fractin‐immunopositive neurons in any layer of the cortex between the PVL cases (3/12, 25%) and controls (2/10, 20%) (P = 1.00). There was no difference in the incidence of the percent of MAP2‐stained pyramidal cells in layer V between the PVL cases (88.0 ± 5.0%) and controls (86.7 ± 5.9%) (P = 0.88). There was also no obvious cortical anomaly in the PVL cases compared to controls, including in radial (vertical) organization.
Figure 8.

Between 22% and 39% of the periventricular leukomalacia cases demonstrated mild and/or non‐specific histopathological changes in the cerebral cortex that were not significantly different in incidence or features from controls. These changes included: (A) astrogliosis characterized by naked glial nuclei (arrows) (H & E); (B) acutely necrotic, hyper‐eosinophilic neurons with pyknotic nuclei (arrows) (H & E); (C) reactive astrocytes diffusely present throughout all layers (arrows) (glial fibrillary acid protein immunocytochemistry); and (D) diffusely activated microglia (arrows) (CD68 immunocytochemistry). Scale bar = 100 µm.
Quantitative changes in neuronal density and size
The cerebral cortex in the specified (sensory and homotypical) types of cerebral cortex was analyzed in the same tissue sections with focal periventricular necrosis in the PVL cases, that is, the cerebral cortex overlying focal white matter damaged was assessed. The sample size of the cortical subtypes in the PVL cases was: frontal association, n = 10; parietal association, n = 17; limbic association, n = 0; agranular, n = 2; and sensory, n = 21. The analysis of the density of layer I neurons and pyramidal neurons in layers III and V revealed a significant reduction (67%) in density of pyramidal neurons in layer V in the koniocortex in all PVL cases (n = 20) (32.21 ± 6.12 neurons/mm2) compared to all controls (97.88 ± 11.11 neurons/mm2) (n = 15) adjusted for postconceptional age (P < 0.001). There was also a marginal reduction (30.0%) of layer V neurons in all areas combined in PVL cases (49.35 ± 7.21 neurons/mm2) compared to controls (70.49 ± 8.36 neurons/mm2) adjusted for postconceptional age (P = 0.060). In a second analysis, the PVL cases and controls that were <30 postconceptional weeks were excluded as a distinct six‐layer cortex was not appreciated visually until 30 weeks and older. This analysis of the density of layer I neurons and pyramidal neurons in layers III and V revealed a significant reduction (83%) in density of pyramidal neurons in layer V in the koniocortex in PVL cases (24.49 ± 5.69 neurons/mm2) (n = 17) compared to the controls (144.92 ± 15.44 neurons/mm2) (n = 12) adjusted for postconceptional age (P < 0.001) (Supporting Information Table S5). There was also a significant reduction (38%) of layer V neurons in all areas combined in PVL cases (40.59 ± 6.91 neurons/mm2) compared to controls (65.69 ± 8.27 neurons/mm2) adjusted for postconceptional age (P = 0.024) (Supporting Information Table S5). There were no changes in layer I neurons or layer III pyramidal neurons in either the koniocortex or homotypical cortex (Supporting Information Table S5). Of note, Brodmann Area 4 (primary motor) was not specifically analyzed because of the small sample size of the controls (n = 2 PVL case; n = 2 controls). There was no difference in cell diameter between PVL cases and controls, adjusted for age.
Quantitative changes in cortical and laminar thickness
The overall thickness of the cerebral cortex (layers I–VI) was not significantly different between the PVL cases (1685.0 ± 84.0 mm) and controls (1667.9 ± 89.28) (P = 0.88) adjusted for regions and postconceptional age in H & E‐stained sections, nor were there differences in thickness in the individual laminae (Supporting Information Table S6).
DISCUSSION
There is increasing appreciation of the neuropathological complexity of the encephalopathy of prematurity, yet the role of the cerebral cortex in this entity is incompletely understood. Historically, the conventional teaching has been that the cerebral cortex in premature infants, including overlying PVL, is spared from damage (2), and that the cerebral white matter of the preterm brain is developmentally susceptible to injury, whereas the cortex is resistant 28, 29. Over a decade ago, this view was challenged by the seminal Golgi study of the cerebral cortex in long‐term survivors of severe white matter necrosis by Marin‐Padilla in which secondary neuronal abnormalities, including in layer V, were first defined (45). Subsequently, our group demonstrated in a semiquantitative neuropathological survey of premature brains that non‐specific gliosis is present in the cerebral cortex in approximately one‐third of cases with PVL (51), and that immunomarkers of oxidative stress are present in some, but not all cortical neurons in PVL (17). The latter finding is indicative of a potential primary injury to the cortex complicating hypoxia–ischemia (17). We now report a highly significant reduction in the density of pyramidal neurons in layer V in all areas combined and in sensory cortex in particular in PVL that may not be a primary injury, but rather secondary to de‐afferentiation of the projecting axons through the underlying white matter necrosis. The detection of this abnormality, like the cortical abnormalities described by Marin‐Padilla, was dependent upon special neuropathological techniques (immunocytochemistry and computer‐based quantitation as opposed to the Golgi technique employed by Marin‐Padilla) (45). In the following discussion, we highlight this cortical finding in layer V neurons in the context of our observations about normative cortical development during the period of high PVL risk, that is, the end of the second trimester, third trimester and perinatal period. We begin with a consideration of the potential limitations of the study.
Potential limitations of the study
Issues related to the database
We emphasize that the controls in this study are autopsied infants who died in extremis, including treatment with mechanical ventilation, and thus, they may not be truly representative of “normal” living infants. Nevertheless, normal preterm brains are extraordinarily rare, given that “normal” preterm infants do not die, and preterm infants rarely, if ever, die without intensive care and terminal hypoxia–ischemia and other metabolic derangements. Thus, it was not unanticipated that the control group in our study demonstrated minor cortical changes, for example, astrogliosis. Given the caveats in interpretation of normality, we nevertheless found a specific difference between PVL and control brains in the density of layer V neurons that suggests a disease‐related phenomenon that is hypothesis generating for testing in animal models. An additional caveat in the interpretation of our data is the small sample size of PVL cases and controls for the different Brodmann areas that required combining koniocortex and homotypical cortex from different Brodmann areas for certain analyses. Thus, the finding of a reduced density in pyramidal neurons that is laminar specific in PVL in this study requires confirmation in an independent data set and/or in animal models.
Two‐dimensional cell counting approach
A potential limitation is our use of a two‐dimensional as opposed to three‐dimensional approach to cell counting in the cerebral cortex. It is argued that three‐dimensional counting provides “unbiased” counts of neurons, whereas two‐dimensional approaches are “assumption based” and therefore potentially yield inaccurate results 4, 62. Yet, all approaches are essentially assumption based and involve inherent biases, and thus the selection of two‐ vs. three‐dimensional approaches for a particular study is based upon relative strengths and weaknesses 4, 62. In this study, the need to obtain a large data set to uncover potentially quantitative abnormalities in PVL mandated the use of the archives in our pathology department in which multiple cases and controls have been accrued over the last decade, that is, the period of modern intensive care. The use of archival tissue in turn required the application of a quantitative method in formalin‐fixed, paraffin‐embedded tissue as fresh brain samples were not historically harvested and prepared for stereological (three‐dimensional) analysis. Because of our dependence on archival tissue in which stereology is not an option, coupled with the validity of two‐dimensional methods in general 4, 62, we believe the use of a two‐dimensional approach is reasonable and valid.
Relationship of MAP2 to hypoxia–ischemia
A third potential limitation of the study was the selection of MAP2 as the antibody to recognize pyramidal neurons. The cytoskeleton phosphoprotein MAP2 regulates the microtubule assembly, providing scaffolding for organelle distribution and localization of signal transduction in dendrites, particularly near spines (10). It is well recognized that all cortical neurons do not stain with MAP2, as experimental studies suggest that immunonegative neurons may reflect MAP2 levels below the level of detection, or, alternatively, molecular forms of MAP2 that are poorly recognized by particular antibodies (22). Hypoxic–ischemic injury is also known to reduce the intensity of MAP2 immunostaining in cortical neurons in human disorders 33, 46 and animal models (40). In this study, we considered the possibility that a decrease in the density of MAP2‐immunopositive pyramidal neurons in layer V was caused by a loss of immunostaining intensity as a complication of hypoxia–ischemia in PVL, and was not necessarily caused by neuronal loss. We found, however, no significant percent difference in the incidence of MAP2‐immunostained neurons between the PVL and control cases, indicating to us that any potential changes in MAP2 pyramidal density were caused by loss of neurons and not immunostaining.
Baseline development of the human cerebral cortex in early life
In our baseline analysis of the cortical development at midgestation, we found that the neocortex transforms from an undifferentiated cortical plate to a highly specialized structure around 30 gestational weeks. At this age, the cortical plate becomes comprised of six layers in which each layer is characterized by a specific composite of pyramidal and non‐pyramidal neurons. Thus, the peak time frame of the encephalopathy of prematurity, 28–34 weeks, coincides with a rapid period of cortical development that may enhance its vulnerability to injury. Indeed, our findings of dramatic changes in lamination, laminar thickness and pyramidal cell differentiation and density in the controls are consistent with the neuroimaging finding of a fourfold increase in cortical volume over this same time period 24, 30, 31, 64. Pyramidal neurons are known to originate from radial progenitors (radial glial cells) in the subventricular zone (SVZ); these early precursors produce neurons particularly in the deeper cortical layers that reach the cortex by radial migration before the second half of gestation 7, 39, 57, 64, 68. The early differentiation of pyramidal neurons in layer V is consistent with these early migrators. Over the second half of gestation and into infancy, we found a striking increase in the overall thickness of the cortex, as well as in layers I–III, V and VI. The increase in thickness in layers I–III in particular over the last half of gestation likely reflects their expansion by late migrating (GABAergic) interneurons, given that the SVZ continues to actively generate mainly GABAergic neurons beyond midgestation 7, 39, 57, 64, 68. There is also expansion of the neuropil during the time frame of PVL because of dendritic arborization, spine density formation and arrival of preterminal afferents. The lack of change in the thickness of layer IV over late gestation may indicate the development of neuropil prior to the second half of gestation because of the early arrival of thalamic afferents, but this possibility requires further study.
The question arises: At what age are Brodmann areas distinguishable in the developing human cortex? Rare human analysis suggests that the neuronal subtypes and their density change with age across the different regions of the cerebral cortex during development, but such analysis is limited by a focus upon infancy and not late gestation, as in our study (37). Given that the definition of a particular Brodmann area is dependent upon the recognition of pyramidal and non‐pyramidal cells in the six discrete laminae, we found that it is not possible to determine such areas before approximately 30 gestational weeks, that is, the age when the six‐layer cortex was first discernible to us. Yet, gyration is not complete in the human brain until term, at which time all primary, secondary and tertiary gyri have formed (12). Thus, prior to term and from 30 weeks onward (the period at which pyramidal and non‐pyramidal neurons are readily distinguished), only early differentiating Brodmann areas can be ascertained with certainty as the distinct gyri and sulci for definitive anatomic localization are not present.
Overall, we did not find a significant difference in the density of layer I neurons and/or pyramidal neurons in layers III and V in the fetal and infant cortex in incipient/differentiating or defined (term and thereafter) based upon known functionality of the cortex (eg, sensory vs. association), Brodmann area or cerebral lobe in the time frame analyzed, that is, midgestation through infancy. This lack of clear‐cut cytoarchitectonic differences may reflect structural immaturity of the cortex at this age period, extensive biological variation among cases (as reflected in the large standard deviations of our results) and/or the small sample size available to us. Considerable variability has also been found in the architectonics and/or folding of the human adult cortex that underscores the variability found by us here in the developing human cortex 16, 55, 56. Our findings of laminar development in different areas of the neocortex are generally consistent with those of others 33, 34, 35, 48, 60. Of note, Kostovic and Judas reported that the developmental appearance or disappearance of layer IV, the major recipient of thalamocortical afferents, is an important indicator of the architectonic maturation of the cortical plate: before 34 weeks, the prospective granular layer IV is well developed in all neocortical areas; after 34 weeks, however, the distinctiveness and granularity of the layer IV gradually diminish in the premotor cortex and disappear completely in the primary motor cortex (34). Our sample of the premotor and primary motor cortices was too small to confirm this finding, but their observation of a relative uniformity of cortical structure prior to 30 weeks is in agreement with the findings reported here.
The cerebral cortex in PVL
The main abnormality found in the cerebral cortex overlying PVL in the perinatal period is 83% in the density of pyramidal neurons in layer V of the sensory cortex, and a 38% decrease in the sensory and associative cortices combined compared to controls adjusted for postconceptional age. These abnormalities are associated with reactive astroglial changes in approximately 40% of PVL cases, a finding that is not significantly different from that in the control group. The non‐specific gliosis in the PVL group indicates cortical injury, but it does not distinguish primary from secondary insults as gliosis can occur in both. Indeed, the neuronal injury may be primary as it is associated with immunomarkers of intrinsic neuronal damage (increased fractin immunostaining), albeit in only 25% of the PVL cases and not specific to PVL (as these changes occur to approximately the same degree in controls). In addition, markers of oxidative stress have been reported by us in cortical neurons in a previous study in PVL (16). Given that human pyramidal neurons are known to be glutamatergic, and express NDMA and non‐NMDA receptors 49, 61, 65, the decrease in the density of layer V pyramidal neurons in a cell‐, region‐ and laminar‐specific pattern suggests that these particular neurons are exquisitively sensitive to hypoxia–ischemia. Layer III pyramidal neurons, however, are likewise glutamatergic, but we did not find a reduction in their density in the cortex overlying PVL as we did for layer V pyramidal neurons. Thus, the possibility of secondary loss of pyramidal neurons in layer V, which are long‐projecting neurons 44, 46, warrants consideration.
In the Golgi analysis of the cerebral cortex in survivors of prematurity with white matter damage, Marin‐Padilla found that the developing cortex was deprived of afferent terminals because of their severance from the cortex by the extensive white matter necrosis. Moreover, some of the cortical neurons failed to reach their subcortical targets because their axons were also destroyed by the white matter lesion (45). In addition, the axotomized pyramidal cells transformed from long‐projecting into local‐circuit neurons (45). Based upon these Golgi observations, Marin‐Padilla proposed that the neurological sequelae of perinatal white matter lesions are a direct consequence of post‐injury, gray matter transformations. In our study, the only laminar‐specific change was in the pyramidal neurons in layer V which, unlike layer III pyramidal neurons, have long projections to subcortical regions, and therefore transverse the deep cerebral white matter, the site of focal necrosis and diffuse gliosis in PVL. These projecting neurons likely experience axotomy as Marin‐Padilla described and undergo cell death caused by a dying‐back mechanism. Germaine to this possibility is our report of widespread axonal injury in the deep and subcortical white matter in PVL beyond the periventricular necrotic foci (19). Of note, we did not find a major depopulation of pyramidal neurons indicative of a developmental delay in migration from the SVZ. Given that the pyramidal neurons migrate and differentiate early before midgestation (see above), the lack of a major developmental disorder of these neurons is expected, as the peak age of PVL is in late gestation and early infancy.
What is the functional significance of reduced density of pyramidal neurons in layer V, given that these neurons play a key role in the canonical glutamate circuit (51)? In this circuit in the rodent barrel cortex, for example, sensory (glutamatergic) information from the thalamus relating to deflections of an individual whisker is processed primarily in the layer IV barrel, which in turn innervates and depolarizes layer II/III in a strictly columnar fashion 23, 51. The glutamatergic output of layer II/III pyramidal neurons then forms a prominent input to layer V pyramidal neurons, which in turn depolarize neurons widely distributed across the barrel cortex (51). Layer V pyramidal neurons also project to the basal ganglia, brain stem and spinal cord depending upon the connectivity of the particular cortical column. We speculate that if there is a preferential reduction in the density of layer V pyramidal neurons, loss of the major outflow of the glutamate circuit impairs the function subserved by the particular cortical area.
CONCLUSIONS
The present study defines subtle neuropathological changes in pyramidal neurons in the cerebral cortex of the encephalopathy of prematurity and a potential role for secondary injury in its pathogenesis. This study underscores the idea that brain injury in the preterm infant is a “complex amalgam” of primary and secondary disturbances (59) with the destructive consequences of white matter necrosis upon the development of cortical neurons and their axons. It is uncertain if a loss of layer V pyramidal neurons alone could account for the substantial degree of cortical volume reductions reported in premature infants at term equivalent or beyond 9, 13, 25, 26, 52, 67. Indeed, damage to non‐pyramidal (GABAergic) (11) and/or neuropil elements (dendrites, axonal terminals) in the cerebral cortex may occur in combination with the layer V neuronal deficit. In this regard, we did not find a reduction in the upper cortical laminae in the PVL cases in this study that would suggest a deficit of GABAergic neurons or neuropil. Nevertheless, further research is needed to determine the role of granular neurons, neuropil elements and cortical architectonics (eg, minicolumns) in the cortical pathology of PVL.
Supporting information
Table S1. Demographics of the study population.
Table S2. The effect of functional cortical region, Brodmann area and cerebral lobe upon neuronal density in layers I, III and V of the controls (n = 15).*
Table S3. Thickness of the cortical laminae [individual and combined (I–VI)] laminae with increasing gestational age in controls (n = 15) in hematoxylin‐and‐eosin‐stained sections.
Table S4. Histopathological features in the cerebral cortex in the periventricular leukomalacia cases compared to controls.
Table S5. The density of layer I neurons and pyramidal neurons in layers III and V in periventricular leukomalacia cases compared with controls (postconceptional age ≥30 weeks) adjusted for postconceptional age.
Table S6. The thickness of individual laminae and combined I–VI laminae in periventricular leukomalacia cases compared to controls adjusted for postconceptional age.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (PO1‐NS38475 and P30‐HD18655). We are grateful to Dr Francine Benes, McLean Hospital, Belmont, MA, for help with the quantitative procedures in the developing human cerebral cortex in this study. We thank Mr Richard A. Belliveau for assistance in study preparation, and Ms Lena L. Liu for help with histological preparations.
REFERENCES
- 1. Anderson PJ, Doyle LW (2004) Executive functioning in school‐aged children who were born very preterm or with extremely low birth weight in the 1990s. Pediatrics 114:50–57. [DOI] [PubMed] [Google Scholar]
- 2. Banker BQ, Larroche JC (1962) Periventricular leukomalacia of infancy. A form of neonatal anoxic encephalopathy. Arch Neurol 7:386–410. [DOI] [PubMed] [Google Scholar]
- 3. Bayless S, Stevenson J (2007) Executive functions in school‐age children born very prematurely. Early Hum Dev 83:247–254. [DOI] [PubMed] [Google Scholar]
- 4. Benes FM, Lange N (2001) Two‐dimensional versus three‐dimensional cell counting: a practical perspective. Trends Neurosci 24:11–17. [DOI] [PubMed] [Google Scholar]
- 5. Billiards SS, Haynes RL, Folkerth RD, Borenstein NS, Trachtenberg FL, Rowitch DH et al (2008) Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol 18:153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bohm B, Smedler AC, Forssberg H (2004) Impulse control, working memory and other executive functions in preterm children when starting school. Acta Paediatr 93:1363–1371. [DOI] [PubMed] [Google Scholar]
- 7. Brazel CY, Romanko MJ, Rothstein RP, Levison SW (2003) Roles of the mammalian subventricular zone in brain development. Prog Neurobiol 69:49–69. [DOI] [PubMed] [Google Scholar]
- 8. Breslau N, Chilcoat HD, Johnson EO, Andreski P, Lucia VC (2000) Neurologic soft signs and low birthweight: their association and neuropsychiatric implications. Biol Psychiatry 47:71–79. [DOI] [PubMed] [Google Scholar]
- 9. Brown NC, Inder TE, Bear MJ, Hunt RW, Anderson PJ, Doyle LW (2009) Neurobehavior at term and white and gray matter abnormalities in very preterm infants. J Pediatr 155:32–38. [DOI] [PubMed] [Google Scholar]
- 10. Buddle M, Eberhardt E, Ciminello LH, Levin T, Wing R, DiPasquale K, Raley‐Susman KM (2003) Microtubule‐associated protein 2 (MAP2) associates with the NMDA receptor and is spatially redistributed within rat hippocampal neurons after oxygen–glucose deprivation. Brain Res 978:38–50. [DOI] [PubMed] [Google Scholar]
- 11. Buxhoeveden DP, Casanova MF (2002) The minicolumn hypothesis in neuroscience. Brain 125:935–951. [DOI] [PubMed] [Google Scholar]
- 12. Chi JG, Dooling EG, Gilles FH (1977) Gyral development of the human brain. Ann Neurol 1:86–93. [DOI] [PubMed] [Google Scholar]
- 13. Del Rio JA, Soriano E, Ferrer I (1992) Development of GABA‐immunoreactivity in the neocortex of the mouse. J Comp Neurol 326:501–526. [DOI] [PubMed] [Google Scholar]
- 14. Dyet LE, Kennea N, Counsell SJ, Maalouf EF, Ajayi‐Obe M, Duggan PJ et al (2006) Natural history of brain lesions in extremely preterm infants studied with serial magnetic resonance imaging from birth and neurodevelopmental assessment. Pediatrics 118:536–548. [DOI] [PubMed] [Google Scholar]
- 15. Espy KA, Stalets MM, McDiarmid MM, Senn TE, Cwik MF, Hamby A (2002) Executive functions in preschool children born preterm: application of cognitive neuroscience paradigms. Child Neuropsychol 8:83–92. [DOI] [PubMed] [Google Scholar]
- 16. Fischl B, Rajendran N, Busa E, Augustinack J, Hinds O, Yeo BT et al (2008) Cortical folding patterns and predicting cytoarchitecture. Cereb Cortex 18:1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Folkerth RD, Trachtenberg FL, Haynes RL (2008) Oxidative injury in the cerebral cortex and subplate neurons in periventricular leukomalacia. J Neuropathol Exp Neurol 67:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harvey JM, O'Callaghan MJ, Mohay H (1999) Executive function of children with extremely low birthweight: a case control study. Dev Med Child Neurol 41:292–297. [DOI] [PubMed] [Google Scholar]
- 19. Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC (2008) Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res 63:656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA et al (2003) Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol 62:441–450. [DOI] [PubMed] [Google Scholar]
- 21. Haynes RL, Folkerth RD, Trachtenberg FL, Volpe JJ, Kinney HC (2009) Nitrosative stress and inducible nitric oxide synthase expression in periventricular leukomalacia. Acta Neuropathol 118:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hendry SH, Bhandari MA (1992) Neuronal organization and plasticity in adult monkey visual cortex: immunoreactivity for microtubule‐associated protein 2. Vis Neurosci 9:445–459. [DOI] [PubMed] [Google Scholar]
- 23. Higashi S, Molnar Z, Kurotani T, Toyama K (2002) Prenatal development of neural excitation in rat thalamocortical projections studied by optical recording. Neuroscience 115:1231–1246. [DOI] [PubMed] [Google Scholar]
- 24. Huppi PS, Warfield S, Kikinis R, Barnes PD, Zientara GP, Jolesz FA et al (1998) Quantitative magnetic resonance imaging of brain development in premature and mature newborns. Ann Neurol 43:224–235. [DOI] [PubMed] [Google Scholar]
- 25. Inder TE, Huppi PS, Warfield S, Kikinis R, Zientara GP, Barnes PD et al (1999) Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Ann Neurol 46:755–760. [DOI] [PubMed] [Google Scholar]
- 26. Inder TE, Warfield SK, Wang H, Huppi PS, Volpe JJ (2005) Abnormal cerebral structure is present at term in premature infants. Pediatrics 115:286–294. [DOI] [PubMed] [Google Scholar]
- 27. Isaacs EB, Lucas A, Chong WK, Wood SJ, Johnson CL, Marshall C et al (2000) Hippocampal volume and everyday memory in children of very low birth weight. Pediatr Res 47:713–720. [DOI] [PubMed] [Google Scholar]
- 28. Jensen FE (2006) Developmental factors regulating susceptibility to perinatal brain injury and seizures. Curr Opin Pediatr 18:628–633. [DOI] [PubMed] [Google Scholar]
- 29. Jones EG (2000) Microcolumns in the cerebral cortex. Proc Natl Acad Sci U S A 97:5019–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kapellou O, Counsell SJ, Kennea N, Dyet L, Saeed N, Stark J et al (2006) Abnormal cortical development after premature birth shown by altered allometric scaling of brain growth. Plos Med 3:e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinney HC, Armstrong DD (2002) Perinatal neuropathology. In: Greenfield's Neuropathology, 7th edn. Graham DI, Lantos PL (eds), pp. 519–606. Arnold: London. [Google Scholar]
- 32. Kinney HC, Volpe JJ (2009) Perinatal panencephalopathy in premature infants: is it due to hypoxia–ischemia? In: Brain Hypoxia and Ischemia with Special Emphasis on Development. Haddad GG, Yu SP (eds), pp. 153–185. Humana Press: New York. [Google Scholar]
- 33. Kostović I (1990) Structural and histochemical reorganization of the human prefrontal cortex during perinatal and postnatal life. Prog Brain Res 85:223–240. [DOI] [PubMed] [Google Scholar]
- 34. Kostovic I, Judas M (2002) Correlation between the sequential ingrowth of afferents and transient patterns of cortical lamination in preterm infants. Anat Rec 267:1–6. [DOI] [PubMed] [Google Scholar]
- 35. Kostovic I, Rakic P (1990) Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J Comp Neurol 297:441–470. [DOI] [PubMed] [Google Scholar]
- 36. Kuhn J, Meissner C, Oehmichen M (2005) Microtubule‐associated protein 2 (MAP2)—a promising approach to diagnosis of forensic types of hypoxia–ischemia. Acta Neuropathol 110:579–586. [DOI] [PubMed] [Google Scholar]
- 37. Landing BH, Shankle WR, Hara J, Brannock J, Fallon JH (2002) The development of structure and function in the postnatal human cerebral cortex from birth to 72 months: changes in thickness of layers II and III co‐relate to the onset of new age‐specific behaviors. Pediatr Pathol Mol Med 21:321–342. [DOI] [PubMed] [Google Scholar]
- 38. Leifer D, Kowall NW (1993) Immunohistochemical patterns of selective cellular vulnerability in human cerebral ischemia. J Neurol Sci 119:217–228. [DOI] [PubMed] [Google Scholar]
- 39. Letinic K, Zoncu R, Rakic P (2002) Origin of GABAergic neurons in the human neocortex. Nature 417:645–649. [DOI] [PubMed] [Google Scholar]
- 40. Ligam P, Haynes RL, Folkerth RD, Liu L, Yang M, Volpe JJ, Kinney HC (2008) Thalamic damage in periventricular leukomalacia: novel pathologic observations relevant to cognitive deficits in survivors of prematurity. Pediatr Res 65:524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luciana M, Lindeke L, Georgieff M, Mills M, Nelson CA (1999) Neurobehavioral evidence for working‐memory deficits in school‐aged children with histories of prematurity. Dev Med Child Neurol 41:521–533. [DOI] [PubMed] [Google Scholar]
- 42. Malinak C, Silverstein FS (1996) Hypoxic–ischemic injury acutely disrupts microtubule‐associated protein 2 immunostaining in neonatal rat brain. Biol Neonate 69:257–267. [DOI] [PubMed] [Google Scholar]
- 43. Marin‐Padilla M (1990) Three‐dimensional structural organization of layer I of the human cerebral cortex: a Golgi study. J Comp Neurol 299:89–105. [DOI] [PubMed] [Google Scholar]
- 44. Marin‐Padilla M (1992) Ontogenesis of the pyramidal cell of the mammalian neocortex and developmental cytoarchitectonics: a unifying theory. J Comp Neurol 321:223–240. [DOI] [PubMed] [Google Scholar]
- 45. Marin‐Padilla M (1997) Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol 56:219–235. [DOI] [PubMed] [Google Scholar]
- 46. Marin‐Padilla M (1999) Developmental neuropathology and impact of perinatal brain damage. III: gray matter lesions of the neocortex. J Neuropathol Exp Neurol 58:407–429. [DOI] [PubMed] [Google Scholar]
- 47. Marlow N, Rose AS, Rands CE, Draper ES (2005) Neuropsychological and educational problems at school age associated with neonatal encephalopathy. Arch Dis Child Fetal Neonatal Ed 90:F380–F387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mrzljak L, Uylings BM, Kostovic I, Van Eden CG (1988) Prenatal development of neurons in the human prefrontal cortex: I. A qualitative Golgi study. J Comp Neurol 271:355–386. [DOI] [PubMed] [Google Scholar]
- 49. O'Connor JA, Hemby SE (2007) Elevated GRIA1 mRNA expression in layer II/III and V pyramidal cells of the DLPFC in schizophrenia. Schizophr Res 97:277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oehmichen M, Woetzel F, Meissner C (2009) Hypoxic–ischemic changes in SIDS brains as demonstrated by a reduction in MAP2‐reactive neurons. Acta Neuropathol 117:267–274. [DOI] [PubMed] [Google Scholar]
- 51. Petersen CC (2007) The functional organization of the barrel cortex. Neuron 56:339–355. [DOI] [PubMed] [Google Scholar]
- 52. Peterson BS, Anderson AW, Ehrenkranz R, Staib LH, Tageldin M, Colson E et al (2003) Regional brain volumes and their later neurodevelopmental correlates in term and preterm infants. Pediatrics 111:939–948. [DOI] [PubMed] [Google Scholar]
- 53. Petrini JR, Dias T, McCormick MC, Massolo ML, Green NS, Escobar GJ (2009) Increased risk of adverse neurological development for late preterm infants. J Pediatr 154:169–176. [DOI] [PubMed] [Google Scholar]
- 54. Pierson CR, Folkerth RD, Billiards SS, Trachtenberg FL, Drinkwater ME, Volpe JJ, Kinney HC (2007) Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol 114:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rajkowska G, Goldman‐Rakic PS (1995) Cytoarchitectonic definition of prefrontal areas in the normal human cortex: I. Remapping of areas 9 and 46 using quantitative criteria. Cereb Cortex 5:307–322. [DOI] [PubMed] [Google Scholar]
- 56. Rajkowska G, Goldman‐Rakic PS (1995) Cytoarchitectonic definition of prefrontal areas in the normal human cortex: II. Variability in locations of areas 9 and 46 and relationship to the Talairach Coordinate System. Cereb Cortex 5:323–337. [DOI] [PubMed] [Google Scholar]
- 57. Rakic P (2003) Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex 13:541–549. [DOI] [PubMed] [Google Scholar]
- 58. Rose SA, Feldman JF (1996) Memory and processing speed in preterm children at eleven years: a comparison with full‐terms. Child Dev 67:2005–2021. [PubMed] [Google Scholar]
- 59. Stjernqvist K, Svenningsen NW (1999) Ten‐year follow‐up of children born before 29 gestational weeks: health, cognitive development, behaviour and school achievement. Acta Paediatr 88:557–562. [DOI] [PubMed] [Google Scholar]
- 60. Takashima S, Chan F, Becker LE, Armstrong DL (1980) Morphology of the developing visual cortex of the human infant: a quantitative and qualitative Golgi study. J Neuropathol Exp Neurol 39:487–501. [DOI] [PubMed] [Google Scholar]
- 61. Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, Jensen FE (2006) Developmental regulation of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole‐propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J Comp Neurol 497:61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Todtenkopf MS, Vincent SL, Benes FM (2005) A cross‐study meta‐analysis and three‐dimensional comparison of cell counting in the anterior cingulate cortex of schizophrenic and bipolar brain. Schizophr Res 73:79–89. [DOI] [PubMed] [Google Scholar]
- 63. Vicari S, Caravale B, Carlesimo GA, Casadei AM, Allemand F (2004) Spatial working memory deficits in children at ages 3–4 who were low birth weight, preterm infants. Neuropsychology 18:673–678. [DOI] [PubMed] [Google Scholar]
- 64. Volpe JJ (2009) Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 8:110–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Woo TU, Shrestha K, Amstrong C, Minns MM, Walsh JP, Benes FM (2007) Differential alterations of kainate receptor subunits in inhibitory interneurons in the anterior cingulate cortex in schizophrenia and bipolar disorder. Schizophr Res 96:46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Woodward LJ, Edgin JO, Thompson D, Inder TE (2005) Object working memory deficits predicted by early brain injury and development in the preterm infant. Brain 128:2578–2587. [DOI] [PubMed] [Google Scholar]
- 67. Woodward LJ, Moor S, Hood KM, Champion PR, Foster‐Cohen S, Inder TE, Austin NC (2009) Very preterm children show impairments across multiple neurodevelopmental domains by age 4 years. Arch Dis Child Fetal Neonatal Ed 94:F339–344. [DOI] [PubMed] [Google Scholar]
- 68. Zecevic N, Chen Y, Filipovic R (2005) Contributions of cortical subventricular zone to the development of the human cerebral cortex. J Comp Neurol 491:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Demographics of the study population.
Table S2. The effect of functional cortical region, Brodmann area and cerebral lobe upon neuronal density in layers I, III and V of the controls (n = 15).*
Table S3. Thickness of the cortical laminae [individual and combined (I–VI)] laminae with increasing gestational age in controls (n = 15) in hematoxylin‐and‐eosin‐stained sections.
Table S4. Histopathological features in the cerebral cortex in the periventricular leukomalacia cases compared to controls.
Table S5. The density of layer I neurons and pyramidal neurons in layers III and V in periventricular leukomalacia cases compared with controls (postconceptional age ≥30 weeks) adjusted for postconceptional age.
Table S6. The thickness of individual laminae and combined I–VI laminae in periventricular leukomalacia cases compared to controls adjusted for postconceptional age.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item