

Abstract
Degeneration of locus coeruleus (LC) noradrenergic forebrain projection neurons is an early feature of Alzheimer’s disease (AD). The physiological consequences of this phenomenon are unclear, but observations correlating LC neuron loss with increased AD pathology in LC projection sites suggest that noradrenaline (NA) is neuroprotective. To investigate this hypothesis, we determined that NA protected both hNT human neuronal cultures and rat primary hippocampal neurons from amyloid-β (Aβ1–42 and Aβ25–35) toxicity. The noradrenergic co-transmitter galanin was also effective at preventing Aβ-induced cell death. NA inhibited Aβ25–35-mediated increases in intracellular reactive oxygen species, mitochondrial membrane depolarization, and caspase activation in hNT neurons. NA exerted its neuroprotective effects in these cells by stimulating canonical β1 and β2 adrenergic receptor signaling pathways involving the activation of cAMP response element binding protein (CREB) and the induction of endogenous nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). Treatment with functional blocking antibodies for either NGF or BDNF blocked NA’s protective actions against Aβ1–42 and Aβ25–35 toxicity in primary hippocampal and hNT neurons, respectively. Taken together, these data suggest that the neuroprotective effects of noradrenergic LC afferents result from stimulating neurotrophic NGF and BDNF autocrine or paracrine loops via β adrenoceptor activation of the CREB pathway.
Introduction
A major feature of Alzheimer’s disease (AD) is the selective degeneration of subcortical projection neurons mediating higher cognitive processes including noradrenergic locus coeruleus (LC) neurons (Adolfsson et al. 1979; Mann et al. 1983; Zarow et al. 2003; Grudzien et al. 2007). LC neurons provide the sole source of noradrenaline (NA) to the hippocampus and neocortex (Foote et al. 1983) and NA signaling plays an important role in various behaviors including selective attention, memory storage and retrieval, general arousal, vigilance and mood (Foote et al. 1983; Levine et al. 1990; Ressler and Nemeroff 1999; Berridge and Waterhouse 2003; Weinshenker 2008; Sara 2009). Degeneration of LC neurons and reductions in NA levels in LC target fields (Adolfsson et al. 1979; Mann et al. 1980; Palmer et al. 1987) are associated with the onset and duration of AD (Mann et al. 1984; Forstl et al. 1994; Zarow et al. 2003), suggesting a neuroprotective role for NA. In this regard, in vivo studies show that a loss of LC-derived NA impacts multiple aspects of AD-like neuropathology. For example, N-(2-chloroethyl)-N-ethyl-2 bromobenzylamine (DSP4)-induced LC damage increases amyloid-β (Aβ) deposition in the hippocampus and cortex and up-regulates markers of gliosis and inflammation in amyloid overexpressing mouse models of AD (Heneka et al. 2002; Heneka et al. 2006; Kalinin et al. 2007). Moreover, in vitro experiments show that NA protects cultured neurons from amyloid induced toxicity (Madrigal et al. 2007), excitotoxicity (Madrigal et al. 2009), metabolic stress (Madrigal et al. 2009), and oxidative stress (Troadec et al. 2001; Traver et al. 2005).
Despite the diverse repertoire for NA neuroprotection, the mechanisms underlying this action are not well understood. To address this problem, we first demonstrated that NA protects human hNT and rat primary hippocampal neurons against Aβ. We explored the neurotoxic sequela activated by Aβ exposure and tested whether they were sensitive to NA or specific noradrenergic receptor ligands. We also asked whether NA neuroprotection against Aβ involved the activation of neurotrophin-mediated pro-survival pathways. Our experiments suggest that NA can protect neurons from amyloid toxicity by inducing either nerve growth factor (NGF) or brain-derived neurotrophic factor (BDNF) expression through the activation of canonical β-adrenoceptor signaling cascades.
Materials and Methods
Neuronal cell culture
hNT neuronal cultures were derived from the human teratocarcinoma NT2 cell line (a gift from Virginia Lee, Univ. Penn) (Andrews et al. 1984; Lee and Andrews 1986). NT2 cells were maintained in OptiMem (Invitrogen, Carlsbad, CA) with 5% fetal bovine serum (FBS). For differentiation, cells were seeded at 25,000/cm2 into T75 flasks in 1:1 DMEM/F-12 media (Invitrogen)/10% FBS, treated twice a week with 10 μM all-trans retinoic acid (Sigma; St. Louis, MO) for 4 weeks and then seeded to new T75 flasks at 65,000/cm2 and treated with the mitotic inhibitors cytosine arabinoside (1 μM) and fluorodeoxyuridine (10 μM, Sigma) for 2 weeks. This resulted in a layer of phase-bright, post-mitotic neuronal cells loosely attached atop a monolayer of non-neuronal cells. Neuronal enrichment was achieved by gently trypsinizing the top neuronal layer and replating at 125,000/cm2 onto 2% Matrigel (BD Biosciences, San Jose, CA) and 10 μM poly-D-lysine (Sigma)-coated black-walled 96 well plates (spectrophotometric assays), 24-well plates (PCR), 60 mm dishes (immunoblotting), or 18 mm2 cover slips (fluorescence microscopy). hNT neurons were cultured for an additional 2 weeks in 1:1 DMEM/F-12 media/10% FBS. Rat E18 primary hippocampal neurons were purchased from Neuromics (Edina, MN) and cultured at ~35,000/cm2 on poly-D-lysine using manufacturer protocols
Aβ neurotoxicity experiments
Differentiated hNT or primary hippocampal neurons were rinsed, pretreated with 10 μM NA (Sigma, dissolved in water) for 5 minutes and then challenged with 10 μM Aβ25–35, Aβ1–42, or reverse peptides (Sigma)in serum-free OptiMem. These concentrations were derived from comprehensive dose response testing during pilot studies (not shown). Aβ25–35 was dissolved in DMSO and applied without pre-aggregation, which results in the rapid formation of oligomeric and protofibril intermediates in aqueous solutions (Giuffrida et al. 2007; Millucci et al. 2009). Aβ1–42 was dissolved in DMSO and pre-aggregated for 16 hours at 37oC. Western blotting revealed an accumulation of SDS-soluble immunoreactive material migrating at ~40–48 kDa reminiscent of oligomeric amyloid (Walsh et al. 1999; Chromy et al. 2003) (not shown). In a parallel experiment, hNT cultures were pre-treated with 1 μM galanin (dissolved in 0.1% trifluoroacetic acid) (Counts et al. 2002; Elliott-Hunt et al. 2007), a noradrenergic peptide co-transmitter, prior to Aβ1–42 exposure. Neuronal viability was determined by the Live/Dead cell viability assay (Invitrogen) (Lambert et al. 1998) following 4 days of Aβ challenge or by propidium iodide (PI) (Elliott-Hunt et al. 2007) following 48 hours challenge. For all experiments in this study, control (CTL) conditions included vehicle.
Membrane depolarization, oxidative stress, and apoptosis assays
Plasma membrane depolarization
Differentiated hNT neurons were preloaded for 30 minutes with the voltage-sensitive fluorescent probe Bis - (1,3 - diethylthiobarbituric acid) trimethine oxonol (DiBAC2; Molecular Probes/Invitrogen) prior to NA and Aβ treatments. DiBAC2 enters depolarized cells where it binds to intracellular/membrane proteins or membrane and exhibits enhanced fluorescence (Loew 1982). Glutamate (Glu, 1 mM, dissolved in water) was used as a positive control. DiBAC2 fluorescence was measured 5 minutes after Aβ or Glu application.
Oxidative stress
hNT neurons were pretreated with NA, challenged with Aβ25–35 for 1 hour and assayed with the reactive oxidation-sensitive fluorescent probe H2DCFDA (2',7'-dichlorofluorescin diacetate, Molecular Probes/Invitrogen), which is nonfluorescent until the acetate groups are removed by intracellular esterases and the probe is oxidized (Babo and Charbonneau 1994). Hydrogen peroxide (100 μM/10 μM FeSO4) was used as a positive control.
Mitochondrial membrane depolarization
hNT neurons were pretreated with NA and challenged with Aβ25–35 for 24 hours, incubated with JC-1 reagent (Stratagene, La Jolla, CA), and then analyzed by spectrophotometry and fluorescence microscopy. JC-1 (5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolcarbocyanine iodide) aggregates within the mitochondria with a red fluorescence at ~585 nm; mitochondrial membrane depolarization allows JC-1 into the cytosol, where it remains a monomer with a green fluorescence at ~515 nm (Cossarizza et al. 1993).
Apoptosis
hNT neurons were pretreated with NA, challenged with Aβ25–35 for 24 hours, and then assayed using the ApoFluor fluorescent pan-caspase-sensitive probe FAM-VAD-FMK (MP Biomedicals, Irvine, CA), which binds covalently to active caspases 1–9 (Smolewski et al. 2001). Staurosporine (1 μM) was used as a positive control.
Adrenoceptor ligand experiments
The following adrenoceptor agonists were tested for their ability to mimic NA’s protective effects against Aβ neurotoxicity: isoproterenol (ISO; β1 and β2, dissolved in water, 1–100 μM dose range), salmeterol (SAL; β2, DMSO, 100 nM - 10 μM), cirazoline (CRZ; α1, water, 100 nM - 10 μM), and UK 14, 304 (UK; α2, DMSO, 100 nM - 10 μM). hNT cultures were pretreated with ligand for 5 minutes, challenged with Aβ25–35 for 48 hours and assessed for cell death by PI. The following adrenoceptor antagonists were tested for their ability to block NA’s protective effects against Aβ neurotoxicity: CGP 20712 (CGP; β1, dissolved in water, 100 nM - 10 μM), ICI 118, 551 (ICI; β2, water, 100 nM - 10 μM), prazosin (PRZ; α1, DMSO, 100 nM - 10 μM), and yohimbine (YHB; α2, water, 100 nM - 10 μM). hNT cultures were pretreated with ligand for 10 minutes prior to NA or vehicle administration, challenged with Aβ25–35 for 48 hours, and then cell death was assessed by PI. All ligands were purchased from Tocris (Ellisville, MO). Dose ranges were based on ligand-specific dissociation constants and pilot testing.
cAMP pathway experiments
hNT neurons were pretreated with ISO (100 μM), ISO + PKI [1 μM, an inhibitor of protein kinase A (PKA), dissolved in water], or dibutyryl cAMP (1 μM, a cell-permeable analog of cAMP, dissolved in water) for 5 minutes prior to challenge with Aβ25–35. Cell death was assessed by PI after 48 hours. Reagents were purchased from Tocris.
Trk inhibitor experiments
hNT neurons were co-treated with NA and K252a (1 μM, dissolved in DMSO), genistein (GEN, 10 μM, DMSO), the inactive genistein analog, genistin [GEN(-),10 μM, DMSO], or vehicle for 5 minutes and then challenged with Aβ25–35 for 48 hours. Cell death was measured by PI. Inhibitors were purchased from Tocris.
Neurotrophin antibody experiments
hNT neurons were pretreated with NA for 5 minutes and then challenged with Aβ25–35 alone or Aβ25–35 in the presence of 1) polyclonal NGF antiserum with functional blocking capacity (AB1528, 1:100; Chemicon/Millipore, Temecula, CA), 2) a functionally inert monoclonal NGF antibody (MAB5260, 1:100, Chemicon), or polyclonal BDNF antiserum with functional blocking capacity (AB1779SP, 1:100, Chemicon). Fresh antibody was added to culture media after 24 hours. Cell death was assessed by PI 48 hours after the start of the experiment.
Quantitative real-time PCR (qPCR)
Differentiated hNT neurons were treated with NA for 30 min, 1 hr, or 2 hr and then processed for qPCR analysis using human-specific primer sets (SA Biosciences, Frederick, MD) for ngf, bdnf, and the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (gapdh). Samples were loaded in triplicate on 96 well plates and analyzed using SYBR green reporter dye on an Opticon 2 DNA Engine (Bio-Rad, Hercules, CA). Standard curves and cycle threshold (Ct) were measured using standards obtained from reference human brain total RNA (SA Biosciences). For graphical representation, Ct values were converted to signal intensity values using Easy Engine software (Bio-Rad).
Western blotting
Differentiated hNT cultures were treated with vehicle, NA (10 μM), ISO (100 μM), or dbcAMP (1 μM) for a 5 minutes to 2 hours time range and then harvested in ice cold homogenization buffer (20 mM Tris, 1 mM EGTA, 1 mM EDTA, 10% sucrose, pH 7.4) containing protease inhibitors. Sample proteins (10μg/sample) were separated by SDS-PAGE (10% acrylamide) and transferred to polyvinylidene fluoride membranes (Immobilon P, Millipore) electrophoretically. Membranes were blocked in TBS/0.1% Tween-20/5% milk for 30 minutes at room temperature (RT) and incubated overnight at 4°C with rabbit antiserum to phosphorylated cAMP response element binding protein (pCREB; 1:1000, Upstate/Millipore). Blots were rinsed and incubated for 1 hour at RT with horseradish peroxidase-conjugated goat anti-rabbit 2° IgG (1:8,000; Pierce, Rockford, IL). pCREB was visualized by enhanced chemiluminescence (Pierce/Thermo, Rockford, IL) on a Kodak Image Station 440CF (Perkin Elmer, Wellesley. MA). Blots were then stripped and reprobed with rabbit CREB antiserum (Upstate/Millipore) and processed as described above. Immunoreactive bands were quantified by densitometry using Kodak 1D image analysis software. pCREB signals were normalized to CREB signals for quantitative analysis.
Statistical analysis
All experiments were repeated 3 times independently. Data was analyzed by one-way ANOVA with Tukey’s post hoc testing (α = 0.05) except for western blot experiments, which were analyzed with Dunnett’s post hoc testing (α = 0.05). Student’s t test was used for qPCR comparisons (α = 0.05).
Results
Noradrenaline provides neuroprotection against amyloid
Differentiated human hNT neurons exhibit morphological and neurochemical properties similar to primary hippocampal and cortical neurons (Fig. 1A) (Lee and Andrews 1986; Pleasure et al. 1992; Tamagno et al. 2000). To test whether these cells were vulnerable to Aβ toxicity, cultures were treated with 10 μM Aβ25–35 (see Methods). Cellular degeneration resembling apoptosis (e.g., pyknotic soma) was seen within 24 hours under light microscopy and peaked at ~ 96 hours (Fig. 1B). An approximately 50% increase in cell death was measured by the LIVE/DEAD assay after 96 hours of exposure to either Aβ25–35 or Aβ1–42 (Fig. 1D). To determine whether NA protects neuronal cells against this As toxicity cultures were pretreated with 10 μM NA prior to Aβ administration. Qualitative (Fig. 1C) and quantitative (Fig. 1D) analysis revealed that NA provided neuroprotection against both Aβ25–35 and Aβ1–42. Propidium iodide (PI) staining was used after 48 hours to corroborate the protective effects of NA against Aβ25–35 and Aβ1–42 toxicity (Fig. 1E). NA neuroprotection against the toxic effect of amyloid treatment was also shown in experiments using rat E18 primary hippocampal cultures (Fig. 1F). Despite the structural differences in Aβ25–35 and Aβ1–42, the similar toxicity profile of these two amyloid species in our model is reminiscent of findings from biochemical and electrophysiological comparative amyloid studies (Chen et al. 2000; Frozza et al. 2009) and may reflect a similar aggregation state, as previously described (Pike et al. 1993; Pike et al. 1995). Finally, we demonstrated that galanin, a neuropeptide which colocalizes with noradrenergic LC neurons and has neuroprotective properties (Elliott-Hunt et al. 2004; Hokfelt 2005; Counts et al. 2009), also provided neuroprotection against amyloid in the hNT cultures (Fig. 1G) similar to a report using septal cholinergic neurons (Ding et al. 2006).
Figure 1.
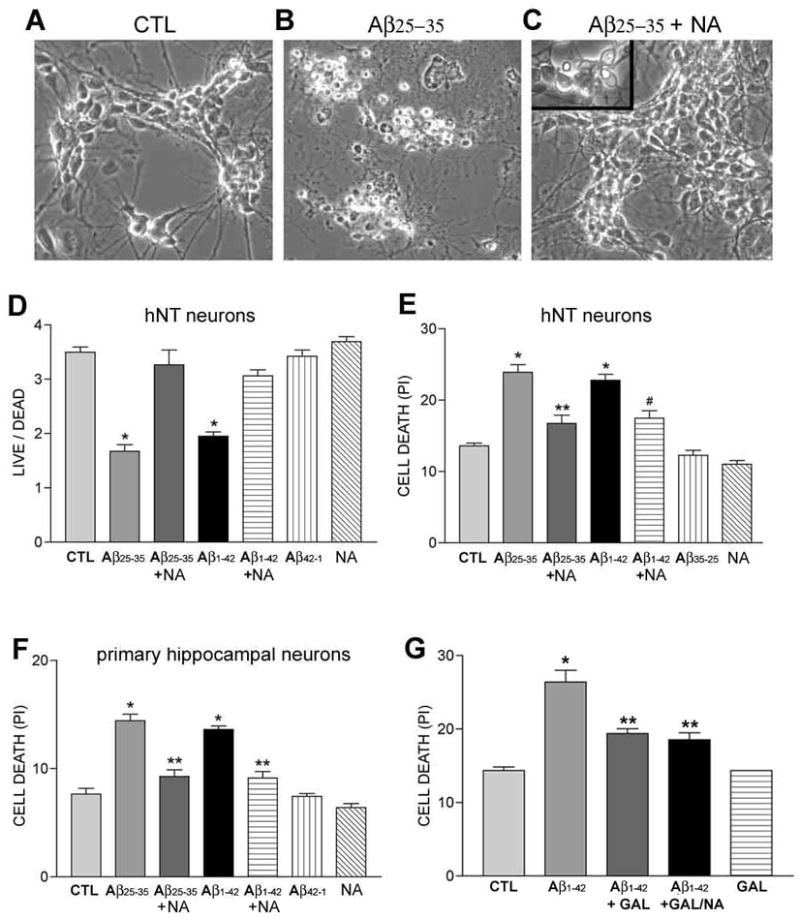
NA protects hNT and primary hippocampal neurons from Aβ toxicity.
Photomicrographs show human hNT neurons after (A) 4 days in control conditions (CTL), (B) 4 days exposure to 10 μM Aβ, or (C) 4 days exposure to 10 μM Aβ in the presence of 10 μM NA. Note the pyknotic cells (C, inset) suggesting Aβ toxicity to some neurons in the NA co-treated cultures. D) Bar graph shows NA protection of hNT neurons against both Aβ25–35 and Aβ1–42. Cultures were assayed by LIVE/DEAD. *, p < 0.001 vs. CTL E) Bar graph shows NA protection of hNT neurons after 2 days of Aβ25–35 or Aβ1–42 treatment via corroborative propidium iodide (PI) assays. *, p < 0.001 vs. CTL; **, p < 0.001 vs. Aβ25–35; #, p < 0.01 vs. Aβ1–42. F) Bar graph shows NA protection of rat primary hippocampal neurons after 2 days of Aβ25–35 or Aβ1–42 treatment via PI assay. *, p < 0.001 vs. CTL; **, p < 0.001 vs. Aβ; n = 8/treatment group in 3 independent experiments (i.e., n = 24 total/group) for each assay. Note that no effects were seen with reverse Aβ peptides in any of the assays. G) The NA peptide co-transmitter galanin (GAL) also conferred neuroprotection against 2 days of Aβ1–42 treatment. Co-treatment with GAL and NA did not significantly improve survival over GAL alone. Similar effects were observed with Aβ25–35 (not shown) *, p < 0.001 vs. CTL; **, p < 0.05 vs. Aβ Values in the Y-axis for LIVE/DEAD and PI experiments are arbitrary spectrophotometric units.
Noradrenaline does not prevent Aβ-induced membrane depolarization
Following the demonstration that NA was neuroprotective against Aβ in our culture systems, we explored the mechanism(s) by which NA prevented Aβ neurotoxicity. Depolarization of the plasma membrane is a well-established physiological property of Aβ (Good et al. 1996; Blanchard et al. 1997; Hartley et al. 1999) and may be an initial contributor to a neurotoxic cascade which includes oxidative stress, mitochondrial membrane depolarization, and apoptosis (Mattson 2006). We found that 5 minutes of Aβ25–35 treatment depolarized hNT cultures as measured by the voltage sensitive probe DiBAC2 and that pretreatment of the cultures with 10μM NA did not prevent Aβ-induced depolarization (Fig. 2).
Figure 2.
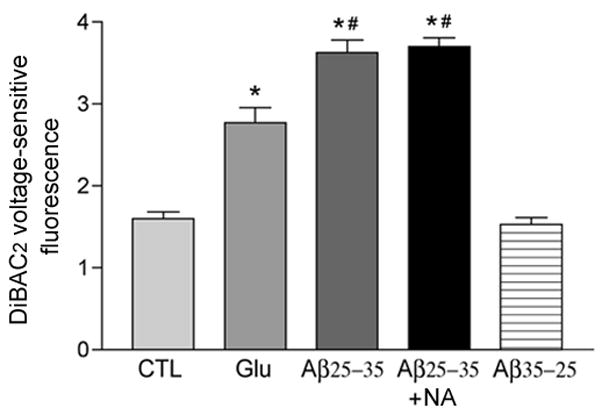
NA does not prevent Aβ-induced neuronal plasma membrane depolarization. A) Bar graph shows that 10 μM NA pre-treatment failed to prevent hNT plasma membrane depolarization induced after 1 hour of 10 μM Aβ25–35 exposure, as assayed using MTT. Glutamate (Glu) was used as a positive control. *, p < 0.001 vs. CTL; #, p < 0.01 vs. Glu. B) Bar graph shows that NA did not prevent Aβ-induced depolarization as measured by DiBAC2 after 5 minutes of Aβ treatment. No effect was observed with reverse Aβ35–25 peptide.*, p < 0.001 vs. CTL; #, p < 0.01 vs. Glu. Values in the Y-axis are arbitrary spectrophotometric units.
Noradrenaline inhibits Aβ-induced oxidative stress, mitochondrial depolarization, and caspase activation
Since NA did not prevent Aβ-mediated plasma membrane depolarization, we investigated its effects on more downstream events underlying Aβ neurotoxicity. To this end, 10 μM Aβ25–35 treatment of hNT neurons induced a significant 40% increase in reactive oxygen species (ROS) concentration after one-hour treatment (Fig. 3A). Pretreatment with 10 μM NA abolished the generation of ROS induced by Aβ (Fig. 3A). The effects of amyloid on mitochondrial membrane potential in hNT cultures were evaluated using the JC-1 mitochondrial reagent assay. Treatment of the cultures with 10 μM Aβ25–35 for 24 hours caused a significant 55% decrease in the ratio of intramitochondrial JC-1 to intracellular JC-1 (Fig. 3B), reflecting the depolarization of the mitochondrial membrane following amyloid exposure. Pretreatment with 10 μM NA restored the mitochondrial membrane potential to control levels (Fig. 3B). This protective effect of NA in mitochondria was corroborated by fluorescence microscopy. As shown in Fig. 3D, Aβ reduced whereas NA pretreatment maintained levels of intramitochondrial JC-1 aggregates, a proxy for intact mitochondria. Finally, we measured caspase activation in hNT cultures following 24 hours of 10 μM Aβ25–35 with or without NA. The 40% increase in caspase activation in the presence of Aβ alone was inhibited by NA pretreatment (Fig. 3C).
Figure 3.
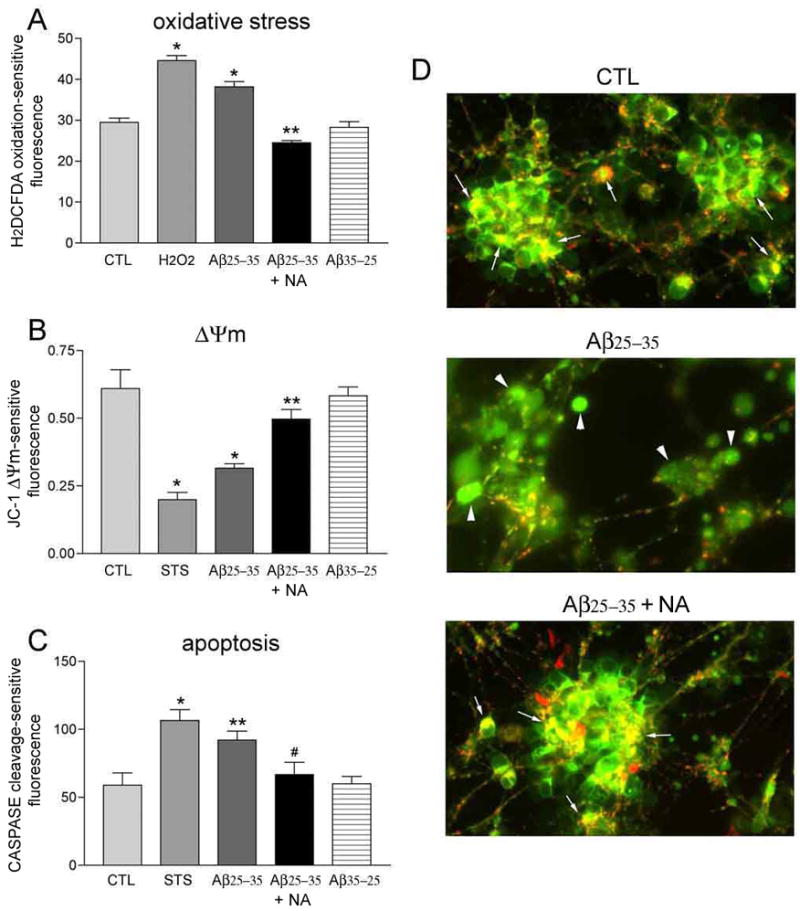
NA inhibits Aβ-induced oxidative stress, mitochondrial depolarization, and caspase activation. A) Bar graph shows that 1 hour of 10 μM Aβ25–35 results in increased reactive oxygen species (ROS) in hNT neurons. 10 μM NA restored ROS to CTL levels. Hydrogen peroxide (H2O2) was used as a positive control. No effect was seen with reverse Aβ. *, p < 0.001 vs. CTL; **, p < 0.001 vs. Aβ. B) NA prevented depolarization of the hNT mitochondrial membrane potential (Δψm) after 24 hours of treatment with Aβ. Δψm was measured using a JC-1-dependent ratio of intact (red/green) to depolarized (green) mitochondria. Staurosporine (STS) was the positive control. *, p < 0.001 vs. CTL; **, p < 0.05 vs. Aβ. C) 24 hours of Aβ exposure increases caspase activation, which was inhibited by NA. *, p < 0.001 vs. CTL; **, p < 0.05 vs. CTL; #, p < 0.05 vs. Aβ; n = 8/treatment group in 3 independent experiments for each assay. D) Fluorescent photomicrographs show overlays of intramitochondrial JC-1 aggregates (red) and intracellular JC-1 (green) in hNT cultures treated 24 hours in CTL (top), Aβ (middle), or Aβ with NA (bottom) conditions. Arrows in top and bottom panels show distinct red + green JC-1 colocalization indicating healthy mitochondria. Arrowheads in middle panel show diffuse green JC-1 staining of depolarized mitochondria. Values in the Y-axis for (A–C) are arbitrary spectrophotometric units.
Noradrenaline neuroprotection is mediated by β-adrenergic receptors
To test whether NA neuroprotection was mediated by adrenoceptor activation, hNT cultures were pretreated with α1, α2, β1, or β2-adrenoceptor specific agonists at a range of doses prior to the administration of Aβ (see Materials and Methods). As shown in Figure 4A, both the β-adrenoceptor agonist isoproterenol (ISO, 100 μM) and the β2-specific agonist salmeterol (SAL, 10 μM) mimicked the neuroprotective effects of 10 μM NA against 10 μM Aβ25–35, whereas the α1 agonist cirazoline (CRZ, 100 nM - 10 μM) and α2 agonist UK 14, 304 (UK; 100 nM -10 μM) were not protective. Likewise, pretreatment with the β1 antagonist CGP 20712 (CGP, 10 μM) or β2-antagonist ICI 118, 551 (ICI, 10 μM) completely prevented NA from reducing amyloid-mediated cell death (Fig. 4B). Neither the α1 antagonist prazosin (PRZ, 100 nM -10 μM) nor the α2 antagonist yohimbine (YHB, 100 nM - 10 μM) blocked NA neuroprotection (Fig. 4B).
Figure 4.

NA neuroprotection is mediated by B-adrenergic receptors. A) Bar graph shows cell death after 48 hours of 10 μM Aβ25–35 exposure following pretreatment of hNT neurons with 10 μM NA or adrenoceptor agonists isoproterenol (ISO, β), salmeterol (SAL, β2), cirazoline (CRZ, α1), or UK 14,304 (UK, α2) [all doses 10μM except ISO (100 μM)]. *, p < 0.001 vs. CTL; **, p < 0.001 vs. Aβ. B) Bar graph shows cell death after 48 hours of Aβ25–35 alone, pretreated with NA, or co-treated with NA following pretreatment with adrenoceptor antagonists CGP 20712 (CGP, β1), ICI 118, 551(ICI, β2), prazosin (PRZ, α1), or yohimbine (YHB, α2) (all doses 10 μM). *, p < 0.001 vs. CTL; **, p < 0.001 vs. Aβ; #, p < 0.05 vs., Aβ; n = 8/treatment group in 3 independent experiments for each assay.
To explore the role of β-adrenoceptor activation in preventing downstream events related to Aβ neurotoxicity, we pretreated hNT cultures with 10 μM CGP or ICI prior to NA and evaluated their effects on Aβ-induced oxidative stress, mitochondrial depolarization, and caspase activation, as described above. In the presence of either antagonist, 10 μM NA failed to prevent 10 μM Aβ25–35-induced mitochondrial depolarization (Fig. 5B) or caspase activation (Fig. 5C); however, the antagonists did not prevent NA from reducing Aβ-mediated oxidative stress (Fig. 5A).
Figure 5.
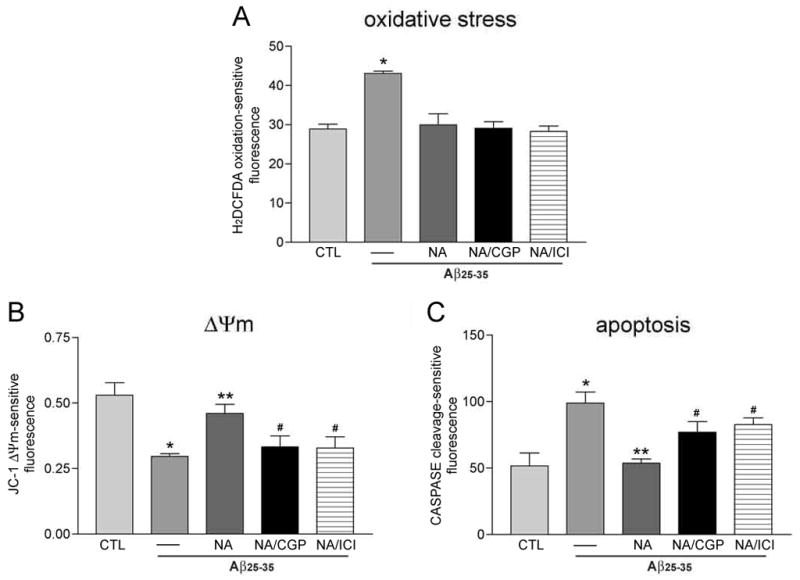
NA uses both receptor-dependent and independent signaling to inhibit cell death pathways. A. β-adrenoceptor antagonists CGP and ICI (10 μM each) failed to block 10 μM NA protection of hNT neurons against ROS induced by 1 hour of 10 μM Aβ25–35 exposure. *, p < 0.01 vs. CTL. On the other hand, CGP and ICI pretreatment did inhibit NA’s ability to block Aβ-mediated mitochondrial membrane depolarization (B) and caspase activation (C) after 24 hours. *, p < 0.01 vs. CTL; **, p < 0.01 vs. Aβ; #, p < 0.05 vs. NA; n = 4/treatment group in 3 independent experiments.
NA activates the cAMP 2nd messenger pathway
Since hNT neurons express β1 and β2 adrenoceptors (Fennell et al. 1998) and NA stimulation of these receptors mediated most aspects of neuroprotection in our assays, we tested whether the canonical β-adrenoceptor cAMP 2nd messenger pathway was activated in our paradigm. First, we demonstrated that 100 μM ISO-mediated protection of hNT cultures against amyloid was reversed by ~70% in the presence of the cAMP-dependent protein kinase A inhibitor PKI (1 μM, Fig. 6A). Furthermore, treatment of the hNT cultures with the membrane soluble cAMP mimetic dibutyryl cAMP (dbcAMP, 1 μM) mimicked ISO by preventing amyloid-induced cell death by ~85% (Fig. 6A).
Figure 6.
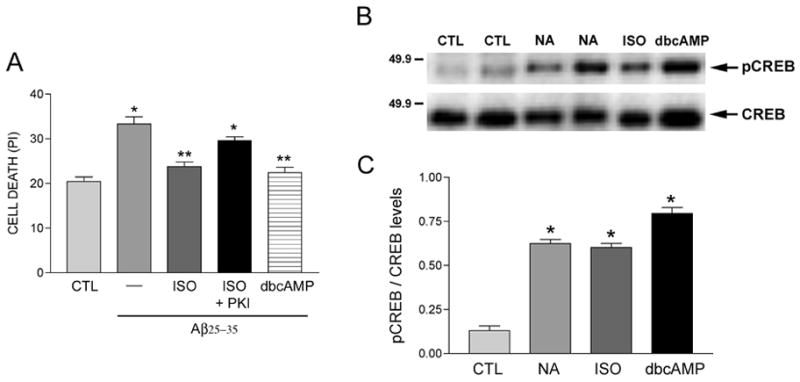
NA activates the cAMP 2nd messenger pathway. A) Bar graph shows that ISO (100 μM)-mediated neuroprotection against 10 μM Aβ in hNT neurons is blocked by PKI (1 μM), an inhibitor of PKA. Application of dbcAMP (1 μM), a permeable cAMP analog, mimicked ISO treatment. *, p < 0.001 vs. CTL; **, p < 0.001 vs. Aβ; n = 8/treatment group in 3 independent experiments. B) Western blot showing increased phosphorylated CREB (pCREB) immunoreactivity after 30 minutes in NA-treated hNT cultures. This effect was mimicked by ISO and dbcAMP. C) Quantitative analysis revealed an approximately 6–8 fold increase in pCREB immunoreactivity following NA, ISO, or dbcAMP treatments (n = 6). *, p < 0.001 compared to CTL.
We also immunoblotted detergent lysates of hNT cultures treated with 10 μM NA, 100 μM ISO, or 1 μM dbcAMP to measure levels of phosphorylated cAMP response element binding protein (pCREB), a major downstream target of cAMP involved in neuroprotective signaling (Mantamadiotis et al. 2002; Jancic et al. 2009). pCREB levels were elevated ~6–8 fold after 30 minutes of NA, ISO, or dbcAMP exposure (Fig. 6B, C), suggesting that activation of this pathway mediates NA neuroprotection.
NA confers neuroprotection against amyloid via neurotrophic pathways
When considering the possible CRE-containing gene targets of pCREB which might mediate NA neuroprotection in our model, we noted that 1) genes encoding the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) contain CREs (Tabuchi et al. 2002; McCauslin et al. 2006) and are up-regulated in response to CREB activation (Tabuchi et al. 2002; McCauslin et al. 2006; Chen and Russo-Neustadt 2009) and 2) hNT cultures express the Trk high affinity neurotrophin receptors for NGF (TrkA) and BDNF (TrkB) (Piontek et al. 1999). Therefore, we tested the hypothesis that NA induces NGF and/or BDNF expression to stimulate Trk-mediated neurotrophic signaling. To this end, quantitative real-time PCR (qPCR) was performed to measure ngf and bdnf mRNA levels in hNT cultures following one hour of 10 μM NA treatment. Bdnf mRNA levels were ~40% higher at this endpoint in NA-treated compared to untreated cultures (Fig. 7A), whereas ngf mRNA levels were increased ~25% following NA exposure (Fig. 7B). To test the involvement of Trk pathway stimulation in NA neuroprotection of hNT neurons, the Trk inhibitors K252a (1 μM) or genistein (10 μM) were co-incubated with 10 μM NA prior to the administration of 10 μM Aβ25–35-induced cell death. These inhibitors effectively blocked NA protection against amyloid (Fig. 8A). Moreover, functional blocking antibodies to NGF or BDNF prevented NA neuroprotection against amyloid, providing more direct evidence for the involvement of these neurotrophic pathways (Fig. 8B). Significantly, NGF and BDNF functional antibodies also reduced NA neuroprotection by ~50% when Aβ1–42 was applied to rat E18 primary hippocampal cultures (Fig. 8C)
Figure 7.
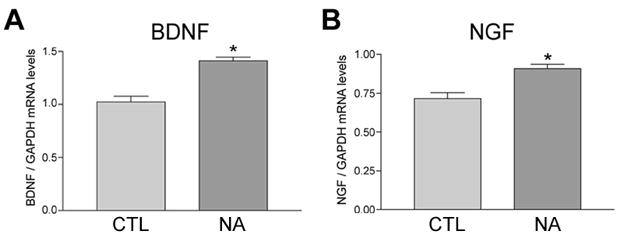
NA treatment increases BDNF and NGF mRNA levels in hNT neurons. A) Bar graph shows increased bdnf mRNA levels in hNT cultures following 1 hour of 10 μM NA, as measured by quantitative real-time PCR (qPCR) using human bdnf-specific primers. *, p < 0.05 vs. CTL. B) One hour of NA treatment also resulted in increased ngf mRNA levels in the hNT cultures. *, p < 0.05 vs. CTL; n = 3/treatment group in 3 independent experiments.
Figure 8.
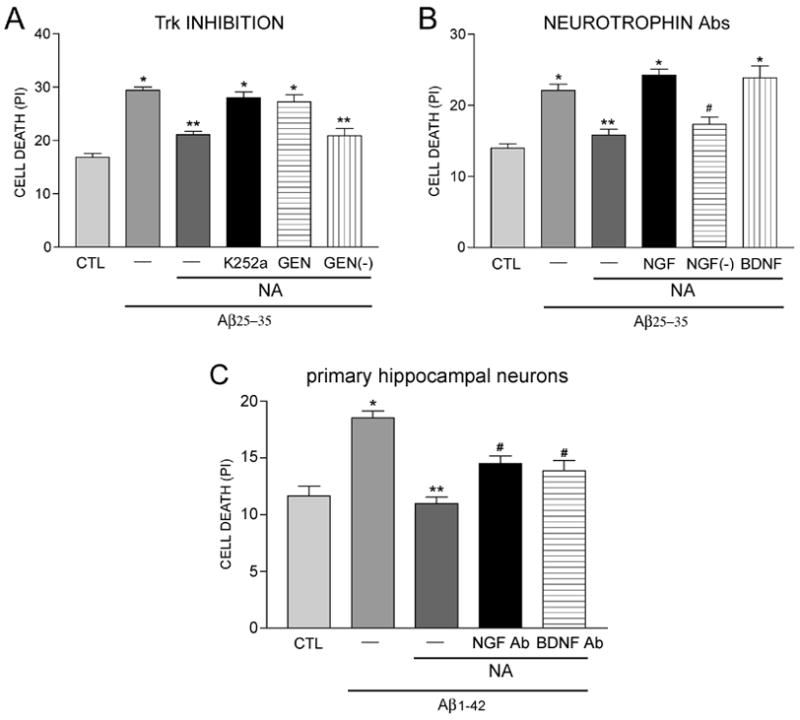
NA neuroprotection is dependent on BDNF and NGF. A) Trk high-affinity neurotrophin receptor pathways are involved in NA neuroprotection. Bar graph shows that 10 μM NA protection against 10 μM Aβ25–35 neurotoxicity was blocked by the Trk pathway inhibitors K252A (1 μM) and genistein (10 μM). Genistin [GEN(−), 10 μM)], an inactive genistein analog, served as a negative control. *, p < 0.001 vs. CTL; **, p < 0.001 vs. Aβ. B) Bar graph shows that functional blocking antibodies for NGF (1:100) and BDNF (1:100) reverse NA neuroprotection against Aβ in hNT neurons. Another NGF antibody without functional blocking capability [NGF(−), 1:100] served as a negative control. *, p < 0.001 vs. CTL; **, p < 0.01 vs. Aβ, #, p < 0.05 vs. Aβ; n = 8/treatment group in 3 independent experiments. C) Bar graph shows that functional NGF and BDNF blocking antibodies reverse 10 μM NA neuroprotection against 10 μM Aβ1–42 in rat E18 primary hippocampal cultures. *, p < 0.001 vs. CTL; **, p < 0.01 vs. Aβ, #, p < 0.05 vs. NA. n = 4/treatment group in 3 independent experiments.
Discussion
In the present study, we show that NA protects target neurons from amyloid toxicity by preventing Aβ-induced increases in oxidative stress, mitochondrial dysfunction and the activation of apoptotic pathways. Moreover, we found that NA exerts its neuroprotective properties primarily through β-adrenergic stimulation of cAMP production and pCREB signaling, which in turn activates both NGF and BDNF-mediated pro-survival pathways. Taken together, these findings support the hypothesis that NA confers neuroprotection against amyloid toxicity in target fields through the activation of a neurotrophic autocrine or paracrine loop. Hence, a potential consequence of LC neurodegeneration may be the reduction in NGF and/or BDNF signaling in LC projection sites in AD.
A recent investigation has shown that NA protects rat primary cortical neurons against amyloid and that one potential mechanism involved antioxidant protection through the stimulation of glutathione levels and activation of peroxisome proliferator activated receptor delta, which boosts antioxidant systems through the detoxification of superoxide radicals (Madrigal et al. 2007). Whether these effects were receptor-mediated is unclear. We found evidence that NA prevented amyloid-induced ROS in a receptor-independent manner in a well-established cell culture model for human hippocampal neurons (Lee and Andrews 1986; Pleasure et al. 1992; Tamagno et al. 2000). These observations complement a previous study demonstrating a receptor-independent role for NA in preventing neuronal cell death via low-level oxidative stress, perhaps via the catechol ring structure of NA since its diphenolic moiety pyrocatechol was also protective (Traver et al. 2005). On the other hand, our adrenoceptor pharmacological and protein signaling analysis indicated that NA’s role in stabilizing mitochondrial membrane permeability, preventing caspase activation, and promoting neuronal survival in Aβ-treated cultures was dependent on β-adrenoceptor activation of neurotrophins. This suggests a dual function for NA in neuroprotection via receptor-dependent and independent pathways.
Direct application of recombinant neurotrophins like BDNF blocks amyloid toxicity in neuronal cultures (Arancibia et al. 2008). However, the present investigation revealed that NA stimulates either an adrenoceptor-mediated autocrine or paracrine loop to induce endogenous neurotrophin production resulting in neuroprotection. Previous work demonstrated that NA up-regulates BDNF and phosphorylated pan-Trk levels in hippocampal cultures and that these responses can be reduced by Trk pathway inhibitors, suggesting a neuronal feed-forward BDNF production loop which involve adrenoceptor-Trk transactivation through G proteins (Chen et al. 2007). While we cannot rule out this latter signaling event in the present study, the prevention of NA’s neuroprotective effects by BDNF and NGF functional blocking antibodies places the induction and release of these neurotrophins upstream of Trk-mediated pro-survival functions. Intriguingly, noradrenergic innervation of glia and glial expression of β-adrenoceptors (Schubert et al. 1976; Hosli et al. 1982), combined with the role of astro- and microglia in neurotrophin production (Miklic et al. 2004; Riley et al. 2004), raises the possibility that NA activates a paracrine loop wherein β-adrenoceptor-expressing glial cells are stimulated to produce NGF and/or BDNF (Juric et al. 2008), which in turn are secreted and activate neuronal TrkA and TrkB receptors to provide protection against amyloid. This is in line with a recent finding that astroglial release of the chemokine MCP-1 can mediate NA neuroprotective properties (Madrigal et al. 2009). However, further studies are needed to test whether a paracrine neurotrophic loop is stimulated by NA.
A critical question arising from the present findings and previous reports of NA anti-inflammatory action (Feinstein et al. 2002; Heneka et al. 2002; Heneka et al. 2003) is whether NA both suppresses Aβ production and protects against Aβ toxicity. For instance, NA signaling reduces nitric oxide (NO) synthase expression in astrocytes (Feinstein 1998) in a cAMP-dependent manner (Galea and Feinstein 1999). This suggests the involvement of β-adrenoceptor and CREB in NA-mediated reductions of NO levels. Since NO activates Aβ production (Blasko and Grubeck-Loebenstein 2003), which in turn can stimulate a neurotoxic inflammatory cascade through the further induction of NO (Puzzo et al. 2006), NA may act to prevent a vicious cycle of NO and Aβ production. Furthermore, the observation that NA signaling induces BDNF production via cAMP and CREB to prevent Aβ toxicity suggests that activation of β-adrenoceptors by this neurotransmitter regulates the balance between amyloid and neurotrophin production to maintain neuronal survival. This would provide a unifying theory for NA’s putative role in regulating amyloidosis and neurotrophism and shed even greater insight on the mechanistic consequences for LC degeneration during the pathogenesis of AD.
The present findings are also informative in light of NA’s long-recognized role in higher cognitive processes such as memory and attention (Berridge and Waterhouse 2003; Weinshenker 2008; Sara 2009). Relevant to the memory deficits which are central to the clinical diagnosis of AD, NA is critical for certain types of hippocampal learning and memory tasks that are impaired in this disease. Electrophysiological studies show that NA stimulation of β-adrenoceptors profoundly influences long term potentiation - a model for memory consolidation - and is required for hippocampal dendritic protein expression (Gelinas and Nguyen 2005) and memory formation (Katsuki et al. 1997; Gelinas et al. 2008; Sara 2009), whereas rodents lacking NA signaling have specific defects in memory retrieval (Thomas and Palmiter 1997; Schimanski et al. 2007). Given the established role for CREB-BDNF (Korte et al. 1996; Patterson et al. 1996) and NGF (Woolf et al. 2001; Conner et al. 2009) in hippocampal synaptic plasticity, NA regulation of these neurotrophins through β-adrenoceptors may contribute to hippocampal learning and memory function in addition to essential neuronal survival signaling.
We also found that the LC noradrenergic co-transmitter GAL protected hNT cultures from Aβ toxicity. The mechanism for this is unclear, but GAL has been shown to protect hippocampal organotypic cultures from excitotoxicity (Elliott-Hunt et al. 2004; Elliott-Hunt et al. 2007) and primary septal neurons from amyloid (Ding et al. 2006) via the GAL receptor GALR2. GALR2 couples to Gq/11 proteins to activate phospholipase C and protein kinase C (Wang et al. 1998; Wittau et al. 2000), suggesting a different G protein-coupled pathway for cell survival relative to NA-stimulated Gs proteins. While the neuroprotective effects of NA and GAL co-administration were not additive in our model, these findings also support the hypothesis that LC deafferentation of projection zones in AD may impact several neurodegenerative sequela underlying the disease process.
In summary, the present findings provide data to a growing list of potential mechanisms by which noradrenergic signaling might exert neuroprotective properties (Troadec et al. 2001; Heneka et al. 2002; Traver et al. 2005; Chen et al. 2007; Madrigal et al. 2007; Madrigal et al. 2009), particularly against diffusible amyloid in brain areas associated with cognitive dysfunction during the progression of AD. With respect to AD therapy, current β-adrenoceptor agonists improve cognition but have a side effect profile that includes tachycardia and other arrythmias (Friedman et al. 1999), limiting their use as cognitive enhancing agents despite their integral involvement in memory formation and hippocampal cholinergic activity (Watabe et al. 2000) as well as regulating amyloid and neurotrophin production. However, our findings offer alternative targets which activate the same neuroprotective pathways elucidated here but circumscribe supraphysiological β-adrenoceptor activation, including strategies such as targeted BDNF delivery (Nagahara et al. 2009) or NA replacement (Weinshenker 2008). Considering the up-regulation of β-adrenoceptors observed in end-stage AD (Kalaria et al. 1989), this latter alternative might maintain endogenous receptor levels and activity early in the disease process without deleterious physiological side effects.
Acknowledgments
Supported by NIH AG26032 (SEC) and AG10688 (EJM).
Abbreviations
- Aβ
amyloid-β peptide
- BDNF
brain-derived neurotrophic factor
- CREB
cAMP response element binding protein
- GAL
galanin
- NA
noradrenaline
- NGF
nerve growth factor
References
- Adolfsson R, Gottfries CG, Roos BE, Winblad B. Changes in the brain catecholamines in patients with dementia of Alzheimer type. Br J Psychiatry. 1979;135:216–223. doi: 10.1192/bjp.135.3.216. [DOI] [PubMed] [Google Scholar]
- Andrews PW, Damjanov I, Simon D, Banting GS, Carlin C, Dracopoli NC, Fogh J. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab Invest. 1984;50:147–162. [PubMed] [Google Scholar]
- Arancibia S, Silhol M, Mouliere F, Meffre J, Hollinger I, Maurice T, Tapia-Arancibia L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis. 2008;31:316–326. doi: 10.1016/j.nbd.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Babo S, Charbonneau M. Measurement of Rat Mitochondrial Hydroperoxides Using in Situ Liver Perfusion of 2',7'-Dichlorodihydrofluorescein Diacetate. Toxic Meth. 1994;4:224. [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Blanchard BJ, Konopka G, Russell M, Ingram VM. Mechanism and prevention of neurotoxicity caused by beta-amyloid peptides: relation to Alzheimer's disease. Brain Res. 1997;776:40–50. doi: 10.1016/s0006-8993(97)01003-2. [DOI] [PubMed] [Google Scholar]
- Blasko I, Grubeck-Loebenstein B. Role of the immune system in the pathogenesis, prevention and treatment of Alzheimer's disease. Drugs Aging. 2003;20:101–113. doi: 10.2165/00002512-200320020-00002. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Russo-Neustadt AA. Running exercise-induced up-regulation of hippocampal brain-derived neurotrophic factor is CREB-dependent. Hippocampus. 2009 doi: 10.1002/hipo.20579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA. Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal. 2007;19:114–128. doi: 10.1016/j.cellsig.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res. 2000;60:65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Abeta(1–42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- Conner JM, Franks KM, Titterness AK, Russell K, Merrill DA, Christie BR, Sejnowski TJ, Tuszynski MH. NGF is essential for hippocampal plasticity and learning. J Neurosci. 2009;29:10883–10889. doi: 10.1523/JNEUROSCI.2594-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolcarbocyanine iodide (JC-1) Biochem Biophys Res Commun. 1993;197:40–45. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ginsberg SD, Mufson EJ. Galanin Fiber Hyperinnervation Preserves Neuroprotective Gene Expression in Cholinergic Basal Forebrain Neurons in Alzheimer's Disease. J Alzheimers Dis. 2009;18:885–896. doi: 10.3233/JAD-2009-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, McGuire SO, Sortwell CE, Crawley JN, Collier TJ, Mufson EJ. Galanin inhibits tyrosine hydroxylase expression in midbrain dopaminergic neurons. J Neurochem. 2002;83:442–451. doi: 10.1046/j.1471-4159.2002.01148.x. [DOI] [PubMed] [Google Scholar]
- Ding X, MacTavish D, Kar S, Jhamandas JH. Galanin attenuates beta-amyloid (Abeta) toxicity in rat cholinergic basal forebrain neurons. Neurobiol Dis. 2006;21:413–420. doi: 10.1016/j.nbd.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Elliott-Hunt CR, Pope RJ, Vanderplank P, Wynick D. Activation of the galanin receptor 2 (GalR2) protects the hippocampus from neuronal damage. J Neurochem. 2007;100:780–789. doi: 10.1111/j.1471-4159.2006.04239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott-Hunt CR, Marsh B, Bacon A, Pope R, Vanderplank P, Wynick D. Galanin acts as a neuroprotective factor to the hippocampus. Proc Natl Acad Sci U S A. 2004;101:5105–5110. doi: 10.1073/pnas.0304823101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein DL. Suppression of astroglial nitric oxide synthase expression by norepinephrine results from decreased NOS-2 promoter activity. J Neurochem. 1998;70:1484–1496. doi: 10.1046/j.1471-4159.1998.70041484.x. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Heneka MT, Gavrilyuk V, Dello Russo C, Weinberg G, Galea E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int. 2002;41:357–365. doi: 10.1016/s0197-0186(02)00049-9. [DOI] [PubMed] [Google Scholar]
- Fennell M, Khawaja XZ, Cockett MI, Wood A. Enhanced neuronal differentiation of NTera-2 cells expressing neuronally restricted beta2 adrenergic receptor. Brain Res. 1998;799:243–249. doi: 10.1016/s0006-8993(98)00289-3. [DOI] [PubMed] [Google Scholar]
- Foote SL, Bloom FE, Aston-Jones G. Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63:844–914. doi: 10.1152/physrev.1983.63.3.844. [DOI] [PubMed] [Google Scholar]
- Forstl H, Levy R, Burns A, Luthert P, Cairns N. Disproportionate loss of noradrenergic and cholinergic neurons as cause of depression in Alzheimer's disease--a hypothesis. Pharmacopsychiatry. 1994;27:11–15. doi: 10.1055/s-2007-1014267. [DOI] [PubMed] [Google Scholar]
- Friedman JI, Adler DN, Davis KL. The role of norepinephrine in the pathophysiology of cognitive disorders: potential applications to the treatment of cognitive dysfunction in schizophrenia and Alzheimer's disease. Biol Psychiatry. 1999;46:1243–1252. doi: 10.1016/s0006-3223(99)00232-2. [DOI] [PubMed] [Google Scholar]
- Frozza RL, Horn AP, Hoppe JB, Simao F, Gerhardt D, Comiran RA, Salbego CG. A comparative study of beta-amyloid peptides Abeta1–42 and Abeta25–35 toxicity in organotypic hippocampal slice cultures. Neurochem Res. 2009;34:295–303. doi: 10.1007/s11064-008-9776-8. [DOI] [PubMed] [Google Scholar]
- Galea E, Feinstein DL. Regulation of the expression of the inflammatory nitric oxide synthase (NOS2) by cyclic AMP. Faseb J. 1999;13:2125–2137. doi: 10.1096/fasebj.13.15.2125. [DOI] [PubMed] [Google Scholar]
- Gelinas JN, Nguyen PV. Beta-adrenergic receptor activation facilitates induction of a protein synthesis-dependent late phase of long-term potentiation. J Neurosci. 2005;25:3294–3303. doi: 10.1523/JNEUROSCI.4175-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JN, Tenorio G, Lemon N, Abel T, Nguyen PV. Beta-adrenergic receptor activation during distinct patterns of stimulation critically modulates the PKA-dependence of LTP in the mouse hippocampus. Learn Mem. 2008;15:281–289. doi: 10.1101/lm.829208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida ML, Grasso G, Ruvo M, Pedone C, Saporito A, Marasco D, Pignataro B, Cascio C, Copani A, Rizzarelli E. Abeta(25–35) and its C- and/or N-blocked derivatives: copper driven structural features and neurotoxicity. J Neurosci Res. 2007;85:623–633. doi: 10.1002/jnr.21135. [DOI] [PubMed] [Google Scholar]
- Good TA, Smith DO, Murphy RM. Beta-amyloid peptide blocks the fast- inactivating K+ current in rat hippocampal neurons. Biophys J. 1996;70:296–304. doi: 10.1016/S0006-3495(96)79570-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28:327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Gavrilyuk V, Landreth GE, O'Banion MK, Weinberg G, Feinstein DL. Noradrenergic depletion increases inflammatory responses in brain: effects on IkappaB and HSP70 expression. J Neurochem. 2003;85:387–398. doi: 10.1046/j.1471-4159.2003.01694.x. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci. 2002;22:2434–2442. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M. Locus Ceruleus Degeneration Promotes Alzheimer Pathogenesis in Amyloid Precursor Protein 23 Transgenic Mice. J Neurosci. 2006;26:1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokfelt T. Galanin and its receptors: Introduction to the Third International Symposium, San Diego, California, USA, 21–22 October 2004. Neuropeptides. 2005;39:125–142. doi: 10.1016/j.npep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Hosli L, Hosli E, Zehntner C, Lehmann R, Lutz TW. Evidence for the existence of alpha- and beta-adrenoceptors on cultured glial cells--an electrophysiological study. Neuroscience. 1982;7:2867–2872. doi: 10.1016/0306-4522(82)90109-9. [DOI] [PubMed] [Google Scholar]
- Jancic D, Lopez de Armentia M, Valor LM, Olivares R, Barco A. Inhibition of cAMP response element-binding protein reduces neuronal excitability and plasticity, and triggers neurodegeneration. Cereb Cortex. 2009;19:2535–2547. doi: 10.1093/cercor/bhp004. [DOI] [PubMed] [Google Scholar]
- Juric DM, Loncar D, Carman-Krzan M. Noradrenergic stimulation of BDNF synthesis in astrocytes: mediation via alpha1- and beta1/beta2-adrenergic receptors. Neurochem Int. 2008;52:297–306. doi: 10.1016/j.neuint.2007.06.035. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Andorn AC, Tabaton M, Whitehouse PJ, Harik SI, Unnerstall JR. Adrenergic receptors in aging and Alzheimer's disease: increased beta 2-receptors in prefrontal cortex and hippocampus. J Neurochem. 1989;53:1772–1781. doi: 10.1111/j.1471-4159.1989.tb09242.x. [DOI] [PubMed] [Google Scholar]
- Kalinin S, Gavrilyuk V, Polak PE, Vasser R, Zhao J, Heneka MT, Feinstein DL. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007;28:1206–1214. doi: 10.1016/j.neurobiolaging.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Izumi Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol. 1997;77:3013–3020. doi: 10.1152/jn.1997.77.6.3013. [DOI] [PubMed] [Google Scholar]
- Korte M, Staiger V, Griesbeck O, Thoenen H, Bonhoeffer T. The involvement of brain-derived neurotrophic factor in hippocampal long-term potentiation revealed by gene targeting experiments. J Physiol Paris. 1996;90:157–164. doi: 10.1016/s0928-4257(97)81415-5. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Andrews PW. Differentiation of NTERA-2 clonal human embryonal carcinoma cells into neurons involves the induction of all three neurofilament proteins. J Neurosci. 1986;6:514–521. doi: 10.1523/JNEUROSCI.06-02-00514.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Litto WJ, Jacobs BL. Activity of cat locus coeruleus noradrenergic neurons during the defense reaction. Brain Res. 1990;531:189–195. doi: 10.1016/0006-8993(90)90773-5. [DOI] [PubMed] [Google Scholar]
- Loew LM. Design and characterization of electrochromic membrane probes. J Biochem Biophys Methods. 1982;6:243–260. doi: 10.1016/0165-022x(82)90047-1. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Kalinin S, Richardson JC, Feinstein DL. Neuroprotective actions of noradrenaline: effects on glutathione synthesis and activation of peroxisome proliferator activated receptor delta. J Neurochem. 2007;103:2092–2101. doi: 10.1111/j.1471-4159.2007.04888.x. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Leza JC, Polak P, Kalinin S, Feinstein DL. Astrocyte-derived MCP-1 mediates neuroprotective effects of noradrenaline. J Neurosci. 2009;29:263–267. doi: 10.1523/JNEUROSCI.4926-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM, Yates PO, Hawkes J. The pathology of the human locus ceruleus. Clin Neuropathol. 1983;2:1–7. [PubMed] [Google Scholar]
- Mann DM, Yates PO, Marcyniuk B. A comparison of changes in the nucleus basalis and locus caeruleus in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1984;47:201–203. doi: 10.1136/jnnp.47.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM, Lincoln J, Yates PO, Stamp JE, Toper S. Changes in the monoamine containing neurones of the human CNS in senile dementia. Br J Psychiatry. 1980;136:533–541. doi: 10.1192/bjp.136.6.533. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid Redox Signal. 2006;8:1997–2006. doi: 10.1089/ars.2006.8.1997. [DOI] [PubMed] [Google Scholar]
- McCauslin CS, Heath V, Colangelo AM, Malik R, Lee S, Mallei A, Mocchetti I, Johnson PF. CAAT/enhancer-binding protein delta and cAMP-response element-binding protein mediate inducible expression of the nerve growth factor gene in the central nervous system. J Biol Chem. 2006;281:17681–17688. doi: 10.1074/jbc.M600207200. [DOI] [PubMed] [Google Scholar]
- Miklic S, Juric DM, Carman-Krzan M. Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci. 2004;22:119–130. doi: 10.1016/j.ijdevneu.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Millucci L, Ghezzi L, Bernardini G, Santucci A. Conformations and biological activities of Amyloid Beta Peptide 25–35. Curr Protein Pept SciI. 2009 doi: 10.2174/138920310790274626. in press. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AM, Wilcock GK, Esiri MM, Francis PT, Bowen DM. Monoaminergic innervation of the frontal and temporal lobes in Alzheimer's disease. Brain Res. 1987;401:231–238. doi: 10.1016/0006-8993(87)91408-9. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Piontek J, Chen CC, Kempf M, Brandt R. Neurotrophins differentially regulate the survival and morphological complexity of human CNS model neurons. J Neurochem. 1999;73:139–146. doi: 10.1046/j.1471-4159.1999.0730139.x. [DOI] [PubMed] [Google Scholar]
- Pleasure SJ, Page C, Lee VM. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci. 1992;12:1802–1815. doi: 10.1523/JNEUROSCI.12-05-01802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Palmeri A, Arancio O. Involvement of the nitric oxide pathway in synaptic dysfunction following amyloid elevation in Alzheimer's disease. Rev Neurosci. 2006;17:497–523. doi: 10.1515/revneuro.2006.17.5.497. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Nemeroff CB. Role of norepinephrine in the pathophysiology and treatment of mood disorders. Biol Psychiatry. 1999;46:1219–1233. doi: 10.1016/s0006-3223(99)00127-4. [DOI] [PubMed] [Google Scholar]
- Riley CP, Cope TC, Buck CR. CNS neurotrophins are biologically active and expressed by multiple cell types. J Mol Histol. 2004;35:771–783. doi: 10.1007/s10735-004-0778-9. [DOI] [PubMed] [Google Scholar]
- Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci. 2009;10:211–223. doi: 10.1038/nrn2573. [DOI] [PubMed] [Google Scholar]
- Schimanski LA, Ali DW, Baker GB, Nguyen PV. Impaired hippocampal LTP in inbred mouse strains can be rescued by beta-adrenergic receptor activation. Eur J Neurosci. 2007;25:1589–1598. doi: 10.1111/j.1460-9568.2007.05376.x. [DOI] [PubMed] [Google Scholar]
- Schubert D, Tarikas H, LaCorbiere M. Neurotransmitter regulation of adenosine 3',5'-monophosphate in clonal nerve, glia, and muscle cell lines. Science. 1976;192:471–473. doi: 10.1126/science.176728. [DOI] [PubMed] [Google Scholar]
- Smolewski P, Bedner E, Du L, Hsieh TC, Wu JM, Phelps DJ, Darzynkiewicz Z. Detection of caspases activation by fluorochrome-labeled inhibitors: Multiparameter analysis by laser scanning cytometry. Cytometry. 2001;44:73–82. doi: 10.1002/1097-0320(20010501)44:1<73::aid-cyto1084>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Tabuchi A, Sakaya H, Kisukeda T, Fushiki H, Tsuda M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J Biol Chem. 2002;277:35920–35931. doi: 10.1074/jbc.M204784200. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Aragno M, Parola M, Parola S, Brignardello E, Boccuzzi G, Danni O. NT2 neurons, a classical model for Alzheimer's disease, are highly susceptible to oxidative stress. Neuroreport. 2000;11:1865–1869. doi: 10.1097/00001756-200006260-00013. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Palmiter RD. Disruption of the dopamine beta-hydroxylase gene in mice suggests roles for norepinephrine in motor function, learning, and memory. Behav Neurosci. 1997;111:579–589. doi: 10.1037//0735-7044.111.3.579. [DOI] [PubMed] [Google Scholar]
- Traver S, Salthun-Lassalle B, Marien M, Hirsch EC, Colpaert F, Michel PP. The neurotransmitter noradrenaline rescues septal cholinergic neurons in culture from degeneration caused by low-level oxidative stress. Mol Pharmacol. 2005;67:1882–1891. doi: 10.1124/mol.104.007864. [DOI] [PubMed] [Google Scholar]
- Troadec JD, Marien M, Darios F, Hartmann A, Ruberg M, Colpaert F, Michel PP. Noradrenaline provides long-term protection to dopaminergic neurons by reducing oxidative stress. J Neurochem. 2001;79:200–210. doi: 10.1046/j.1471-4159.2001.00556.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- Wang S, Hashemi T, Fried S, Clemmons AL, Hawes BE. Differential intracellular signaling of the GalR1 and GalR2 galanin receptor subtypes. Biochemistry. 1998;37:6711–6717. doi: 10.1021/bi9728405. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Zaki PA, O'Dell TJ. Coactivation of beta-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci. 2000;20:5924–5931. doi: 10.1523/JNEUROSCI.20-16-05924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Curr Alzheimer Res. 2008;5:342–345. doi: 10.2174/156720508784533286. [DOI] [PubMed] [Google Scholar]
- Wittau N, Grosse R, Kalkbrenner F, Gohla A, Schultz G, Gudermann T. The galanin receptor type 2 initiates multiple signaling pathways in small cell lung cancer cells by coupling to G(q), G(i) and G(12) proteins. Oncogene. 2000;19:4199–4209. doi: 10.1038/sj.onc.1203777. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Milov AM, Schweitzer ES, Roghani A. Elevation of nerve growth factor and antisense knockdown of TrkA receptor during contextual memory consolidation. J Neurosci. 2001;21:1047–1055. doi: 10.1523/JNEUROSCI.21-03-01047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]