

Abstract
Cellular senescence is a persistently growth-arrested phenotype in normal and transformed cells induced by non-cytotoxic stress. Cytostasis as a method of cancer treatment has recently generated significant interest. Research into the induction of cellular senescence as cancer therapy has been hindered by a lack of compounds that efficiently induce this response. We describe a semiautomated high-throughput method to identify library compounds that induce senescence using prostate cancer cells cultured in 96 well plates. Primary hits are identified by low cell numbers after 3 days in culture, measured by Hoechst 33342 fluorescence. A secondary visual assessment of senescence-associated β-galactosidase staining and cellular morphology in the same wells distinguishes senescence from quiescence, apoptosis and other false-positives. This method was used to screen a 4160 compound library of known bioactive compounds and natural products at a 10μM dose. Candidate compounds were further selected based on persistent growth arrest after drug removal and increased expression of previously described senescence marker genes. Four lead compounds not previously associated with senescence were identified for further investigation. This is the first successful assay to identify novel agents from compound libraries based on senescence-induction in cancer cells.
Keywords: cellular senescence, high-throughput screen, prostate, prostate cancer
Introduction
There is increasing interest in the development of oncostatic compounds that prevent the growth and progression of cancers without cytotoxicity. This strategy may increase patient survival while minimizing treatment side-effects and chemoresistance in prostate and other cancers. The induction of cellular senescence is one mechanism by which this effect may be achieved(1). Cellular senescence is a general program of persistent growth arrest in response to sub-lethal stresses in both normal non-transformed and immortalized transformed cells. Senescent cells cease dividing, become insensitive to mitogenic and certain apoptotic stimuli, and develop a phenotype similar to replicatively exhausted cells, exhibiting a characteristic enlarged and flattened morphology and increased senescence-associated β-galactosidase (SA-β-gal) staining activity (Fig 1A)(1, 2). While ongoing studies seek to identify universal markers and regulators of senescence, SA-β-gal staining remains a standard and accepted marker used to identify senescent cells.
Fig 1.

Screen for the identification of senescence in cancer cells. A. Senescent morphology and SA-β-gal activity. Phase contrast microscopy of DU145 cells cultured with DMSO (control) or 250nM AZQ, identified by this study, for 3 days, fixed and stained for SA-β-gal activity overnight, as previously described(2). Original image magnification: 400x. B. Multi-step screening strategy. Prostate cancer cell lines were exposed to a library of compounds for 3 days, fixed and stained overnight for SA-β-gal activity, followed by staining with Hoechst 33342. Compounds of interest were initially identified by decreased Hoechst 33342 fluorescence indicative of low cell numbers. These wells were then visually assessed for the extent of SA-β-gal activity and senescent morphology.
Agents that generate oxidative stress, DNA damage and/or stress-related signaling induce cellular senescence. These include both endogenous processes including telomere loss, accumulated oxidative damage, dysregulated oncogene activity, and exogenous factors such as chemicals, viral oncogenes, UV light, and ionic radiation. In aging organisms, cellular senescence represents an in vivo tumor suppressor mechanism that limits the proliferation of damaged cells(1). This frequently involves the activity of tumor suppressors p53 and pRb, and increased protein expression of cyclin-dependent kinase (CDK) inhibitors p21waf1/cip1, p16ink4a and p27kip1 (1). Cells exhibiting SA-β-gal staining and other senescence characteristics have been observed in benign lesions including lung adenomas(3), melanocytic naevi(4), and prostatic intraepithelial neoplasia(5). A similar senescent state can be chemically induced in prostate and other cancer cell lines in vitro, independent of p53, Rb and other tumor suppressor pathways(6, 7). In humans, SA-β-gal staining has been observed in lung tumors(8) and breast tumors after treatment with genotoxic drugs(9). Evidence in some studies suggest that the induction of senescence as a cancer treatment may benefit patients, including decreased incidence and severity of toxic side-effects, stimulation of immune responses and prolonged survival (1, 10, 11). However, the investigation of drug-induced senescence in tumor models has been hampered by the lack of identified compounds that effectively induce this response.
Toward this end, we have developed a rapid semi-automated high-throughput method to screen libraries for novel compounds that induce senescence in prostate cancer cells. Cells are stained concurrently with DNA-binding Hoechst 33342 and for SA-β-gal activity, and compounds are selected on the basis of both growth inhibition associated with senescence and the phenotypic changes that result from its induction. Candidate compounds can then be further validated for induction of persistent growth arrest and expression of senescence marker genes. Using this assay, we screened a library of 4160 known bioactive compounds and natural products at a 10μM dose, identifying 4 lead compounds not previously associated with senescence induction and demonstrating the utility of these methods.
Materials and Methods
Compound Library
Compounds used in this study were stored, maintained and handled by the Keck-University of Wisconsin Carbone Comprehensive Cancer Center (Keck-UWCCC) Small Molecule Screening Facility (hts.wisc.edu/Index.htm). The compound library used for screening consists of 3 commercially available collections totaling 4,160 compounds. This includes: 2000 diverse FDA approved drugs and natural products (Microscource Discovery Systems, Inc; Gaylordsville, CT); the 1280 compound LOPAC1280 library of diverse characterized compounds (Sigma; St Louis, MO); and 880 characterized compounds (Prestwick Chemicals; Illkirch, FR). Compounds were dissolved in DMSO and stored in 384 well plates at −80°C. Included on each 384 well plate are 64 DMSO negative controls. Further details can be obtained at http://hts.wisc.edu/Libraries.htm#kba. Compound structures were obtained from PubChem (http://pubchem.ncbi.nlm.nih.gov/).
Duplexed Cell Growth-Inhibition/SA-β-gal Assay
Biomek FX robotic high-throughput fluid handling instruments (Beckman-Coulter; Fullerton, CA) were operated by the Keck-UWCCC Small Molecule Screening Facility (hts.wisc.edu/Index.htm). DU145 cells were cultured as previously described(7), suspended at a density of 1×104 cells/100μl culture medium and 100μl/well added to 96 well plates(Corning #3906). Library compounds were administered to cells at a final concentration of 10μM and incubated for 3 days. Cells were then washed in warm PBS, fixed and stained for SA-β-gal activity overnight, as previously described(2) using 100μl/well. Cells were again washed in PBS and incubated at room temperature in PBS + 10μg/ml Hoechst 33342 (Invitrogen; Carlsbad, CA) for 10 min. Hoechst 33342 fluorescence (ex/em: 355nm/460nm) was measured using a Victor V-3 high-throughput stacking plate reader (Perkin-Elmer; Waltham, MA). In control experiments, cells were cultured in medium containing 25nM Doxorubicin (Sigma) to induce senescence (7), and Hoechst 33342 fluorescence was measured to calculate Z′ compared to proliferating cells, as previously described(12).
In the pilot screen, wells in which fluorescence was more than 1 standard deviation (SD) below the average of 384 data points (one drug plate) were then visually inspected by three independent observers to assess the intensity of SA-β-gal staining and the presence or absence of senescence morphology. Selected compounds were subsequently assessed for induction of persistent growth arrest by exposing cells to drug in 96 well plates for 3 days, and then assessing growth by Hoechst fluorescence 3 days after drug removal. Doxorubicin (25nM)-induced senescence arrest was used as a control and wells with Hoechst fluorescence less than the average of the Doxorubicin controls were visually inspected to confirm growth inhibition and development of a senescence-like morphology.
Prostate Senescence Marker Gene Expression
DU145 or LNCaP cells were cultured as previously described(7). These cells were split to duplicate wells in 96 well plates at density of 1×104 cells/100μl culture medium and 100μl/well and incubated overnight. Cells were then treated with 10μM concentration of each selected compound, and incubated for 3 days. RNA was isolated from cells, reverse transcribed, and Glb1, BRAK, CSPG2, and 18S gene expression were measured by quantitative real-time PCR (qPCR) using an iCycler thermocyler and MyiQ software (BioRad; Hercules, CA) as previously described(7). Expression of genes in each sample was standardized to 18S measurements, and relative expression of treated samples was normalized to that of untreated cells.
Results and Discussion
Method Development
Our larger interests lie in the characterization and development of compounds that induce senescence in prostate cancer models. However, this requires identification of compounds with this activity. To date, efforts to identify agents capable of inducing senescence have largely focused on testing individual compounds to determine the extent to which senescence is induced(6). Here, we describe a high-throughput method to identify compounds in chemical libraries that induce characteristics of cellular senescence in prostate cancer cells.
Development of these methods presented numerous challenges. While the importance of many molecular pathways regulating senescence-induction has been described, these mechanisms may vary among cell types and are frequently inactivated in cancers(1). Consequently, no standard universal markers of senescence beyond SA-β-gal staining/Glb1 expression have been identified for use in cancer cells(2, 13). As androgen-independent DU145 cells develop phenotypic senescence and SA-β-gal staining in response to chemical treatment independent of p53 and Rb, these were used as a model of advanced prostate cancer(7). However, while these cells were induced to senescence and increased SA-β-gal staining by exposure to 25nM Doxorubicin in control experiments, this did not significantly change the OD600 of whole or solubilized senescent cells when measured using a plate reader, and thus by itself is not amenable to high throughput screening (data not shown). Other attempts to identify senescence induction based on the expression of reporters regulated by the promoter of CSPG2, previously shown to be specifically upregulated in senescent prostate cancer cells(7), were not significantly different in wells of proliferating or senescent cells.
Lacking a reliable individual marker of senescence, we ultimately adopted a multi-step strategy based on identifying general phenotypic characteristics that define senescent cells, namely the induction of persistent growth arrest, SA-β-gal staining and morphological characteristics of senescent cells (Fig 1B). Our method is based on the pairing of two compatible staining techniques that allow detection of growth inhibition and assessment of SA-β-gal activity in the same well. Cells are plated into 96 well plates, drugs are added, and after 3 days incubation they are fixed and stained for SA-β-gal overnight followed immediately by staining with the fluorescent DNA-binding Hoechst 33342 (Fig 1B). Nuclear Hoechst 33342 staining can be measured in each well using a high-throughput plate reader to quickly identify wells with decreased fluorescence, indicative of low cell number. This dual staining did not interfere with either technique (data not shown). This relatively small set of wells could then be assessed visually to determine the extent of SA-β-gal staining, further selecting compounds for additional investigation.
Control experiments were performed to demonstrate the ability of Hoechst 33342 fluorescence to discriminate proliferating cells from senescent and apoptotic cells using increasing doses of Doxorubicin. Exposure of DU145 cells to 25nM Doxorubicin had been previously shown to induce senescent cell morphology and SA-β-gal activity (7). Our repeated experiments demonstrate that doses of Doxorubicin 25nM and higher reduce fluorescence significantly (p<0.003) when compared to untreated or 5nM Doxorubicin after 3 days exposure (Fig 2A). We then compared the fluorescence of untreated proliferating cells to cells induced to senescence (Fig 2B) and repeated experiments (n=4) generated an average Z′-factor of 0.53, ranging from 0.5 to 0.6(12), indicating a suitably high signal-to-noise ratio to identify growth inhibition using this technique. Increased concentrations of Doxorubicin (100nM and 250nM) induced apoptosis and cytotoxicity with low fluorescence similar to blank wells. Although the fluorescence of senescent cells (25nM) is statistically different from higher cytotoxic doses (p<0.05), the Z′-factor copmparing these data was less than 0.5 indicating the need for additional analyses to distinguish senescence from cell death. Therefore, we used visual observation of SA-β-gal and cellular morphology to identify senescence in those specific wells that were growth-inhibited (2). Wells found to contain robust staining and senescent morphology (Fig 1A) are selected for further assessment.
Fig 2.

Development of Hoechst 33342 fluorescence to identify senescence in treated cancer cells. A. DU145 cells were cultured in a 96-well plate treated with increasing doses of Doxorubicin (n=14). Cells were cultured three days, fixed, stained for SA-β-gal activity and Hoechst 33342. Blank wells were included as negative controls. B. Calculation of Z′ in senescent versus proliferating DU145 cells. Cells were cultured with 25nM Doxorubicin (DOX) or untreated (UN) for 3 days before being stained and Hoechst 33342 fluorescence measured by plate reader. The average Z′ of all four experiments was 0.53 (p<0.003). Error bars represent standard error.
Pilot Screen
We used this method to screen a library of 4160 known structurally diverse characterized bioactive compounds and natural products for senescence-inducing activity. After incubating cells with 10μM of each compound or DMSO for 3 days, cells were fixed, stained and Hoechst fluorescence measured. Wells with a decrease in fluorescence greater than 1 standard deviation of the average data resulted in 625 initial hits from the library (Fig 3A). Subsequent visual scoring of these wells for robust SA-β-gal expression and senescence morphology identified 226 compounds as cytotoxic at 10μM and 51 compounds as potentially inducing senescence (1.2% of the library).
Fig 3.

Screen of known bioactive compound library and confirmatory assays for senescence induction. A. Hoechst 33342 fluorescence measured in cells treated with library compounds or DMSO as a vehicle. Fluorescence data was analyzed in groups of 384 data points, and the average and standard deviation was calculated for each group. Wells with fluorescence decreased more than 1 SD below the average of all data (open circles) were selected to be visually assessed for SA-β-gal activity and senescence morphology. B. Persistence of proliferative arrest after drug removal. Wells with Hoechst 33342 fluorescence lower than the average of Doxorubicin-treated cells were visually inspected, identifying 9 compounds that maintained senescence (closed squares). All data are the average of duplicates and includes 12 untreated controls. C. Expression of senescence marker genes Glb1, BRAK and CSPG2 in DU145 cells treated with 9 candidate compounds versus untreated. In triplicate wells, cells were left untreated (UN) or exposed to 10μM of candidate compounds, 25nM Doxorubicin (DOX) as a positive control (+) and 10μM idoxyuridine (IDO), which induces quiescence, as a negative control (−), and analyzed by qPCR. Results were standardized to 18S expression and then normalized to expression in untreated cells. Candidate compounds: Methotrexate (MET); Chlorhexadine (CHL); Crassin Acetate (CRA); Cytarabine (CYT); IC261; AZQ; Bithionol (BIT); Dichlorophene (DIC); Pyrithione (PYR). These results are representative of two independent experiments. An asterisk(*) designates compounds inducing significant induction (p<0.05) of all 3 markers.
Confirmatory Assays
We tested whether these compounds induce a proliferation arrest in cells that persists after drug removal consistent with senescence. Cells were plated in duplicate wells, exposed to the 51 candidate compounds for 3 days then allowed to recover in drug-free media for an additional 3 days. After fixing and staining we found cells treated with 24 of the 51 compounds maintained decreased Hoechst 33342 fluorescence less than the average of cells cultured in 25nM Doxorubicin, used as a control for senescence induction (Fig 3B). Visual assessment of these wells confirmed development of robust SA-β-gal staining and morphology induced by 9 compounds.
Glb1, BRAK, and CSPG2 have been utilized as markers for induced cellular senescence in DU145 and other cancer cell lines(7, 13). We assessed expression of these genes in cells exposed to the 9 selected compounds. DU145 cells were treated for 3 days with 10μM of each compound and gene expression analyzed by qPCR. Controls included untreated cells, and cells exposed to Idoxyuridine (a compound that induced a quiescent growth arrest). Cells induced to senescence with 25nM Doxorubicin were included as a positive control. Of the candidate compounds, Methotrexate, Cytarabine, Chlorhexidine and IC 261 did not induce significant expression of all three markers (Fig 3C). This experiment was reproduced using the androgen-dependent cell line LNCaP at the 10μM screening dose (data not shown). These genes were similarly induced in this cell line by the remaining compounds except for Crassin Acetate. Finally, we confirmed that senescence-induction by these remaining compounds does not induce apoptosis, based on the absence of both Annexin V/Propidium Iodide staining and cleavage of poly(ADP)-ribosyl polymerase (data not shown).
Conclusions
In summary, we have developed these methods to screen compound libraries and identify those that induce senescence. Without ideal means to develop a completely automated screen, we developed a pragmatic semi-automated approach utilizing a high-throughput first step followed by visual assessment of individual wells of interest, achieved by dual-staining with Hoechst 33342 and for SA-β-gal activity. Consolidation of these two assays greatly reduces the time and resources required make this initial identification and although not completely automated, these methods are nonetheless rapid; a subsequent screen of 16,000 was completed within a month (data not shown). The diversity of compounds identified in the later stages of selection suggests the screen is not biased towards one particular pathway or mechanism. As understanding of induced cellular senescence in cancer cells continues to develop, reliable markers of senescence may be identified that both reliably identifies senescence and is more amenable to high throughput screening methods.
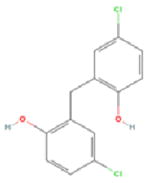
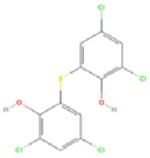
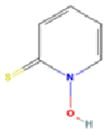
Our pilot screen of a known bioactive library at a single 10μM dose identified 4 compounds not previously associated with senescence-induction (Table 1). Several of these chemicals have demonstrated anti-proliferative activity in cancer cell lines, but have had limited in vivo testing. Mechanistically, AZQ is a DNA alkylating compound with limited cytotoxic activity in solid tumor models (14), and the Zn2+ ionophore Pyrithione induces oxidative stress(15). Both of these cellular stresses are associated with senescence induction. The mechanisms by which Bithionol and Dichlorophene induce senescence remain unknown. From these, we have further characterized the senescence-inducing activity of AZQ in prostate cancer cell lines in vitro and prostate tumor xenograft models in vivo, the results of which are forthcoming (Ewald et al., in preparation). Additionally, our screen identified over 226 compounds in the library that are cytotoxic at 10μM doses. As senescence-induction has been described in response to low doses of cytotoxic compounds, these could be screened at lower doses to further identify senescence-inducing activity. In all, this development allows the advancement of investigations into the nature and regulation of induced cellular senescence in cancer cells and its utility as a means to treat and manage cancers.
Table 1.
Senescence-inducing compounds identified by high throughput screening. (+) and (−) are indicative of presence and lack of anti-proliferative activity, respectively, as reported previously in PubChem and in the literature.
| Compound Name | AZQ | Bithionol | Dichlorophene | Pyrithione | |
|---|---|---|---|---|---|
| Structure | 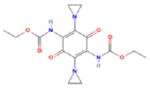 |
 |
 |
 |
|
| PubChem ID | 42616 | 2406 | 3037 | 1570 | |
| PubChem Anti-Cancer Activity | In Vitro | + | + | + | + |
| In Vivo | − | − | − | Not Reported | |
| Mechanism of Action | DNA Alkylation | Unknown | Unknown | Zn2+ Ionophore | |
Acknowledgments
This work was supported by the National Institutes of Health (R01CA97131), the University of Wisconsin George M. O’Brien Urology Research Center (1P50DK065303), the John Livesey endowment and the Department of Defense Prostate Cancer Research Program (DAMD17-02-1-0163). J.A.E. is supported through an NIH Ruth L. Kirchstein National Research Service Award (T32 CA009681-14).
Due to constraints on the number of references in this manuscript, we apologize to authors of many other works that are pertinent to this investigation but could not be cited.
Literature Cited
- 1.Schmitt CA. Cellular senescence and cancer treatment. Biochim Biophys Acta. 2007;1775(1):5–20. doi: 10.1016/j.bbcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 4.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 5.Majumder PK, Grisanzio C, O’Connell F, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14(2):146–155. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang BD, Broude EV, Dokmanovic M, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59(15):3761–3767. [PubMed] [Google Scholar]
- 7.Schwarze SR, Fu VX, Desotelle JA, Kenowski ML, Jarrard DF. The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia. 2005;7(9):816–823. doi: 10.1593/neo.05250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65(7):2795–2803. doi: 10.1158/0008-5472.CAN-04-1270. [DOI] [PubMed] [Google Scholar]
- 9.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62(6):1876–1883. [PubMed] [Google Scholar]
- 10.Schmitt CA, Fridman JS, Yang M, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 11.Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 13.Lee BY, Han JA, Im JS, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 14.Bender JF, Grillo-Lopez AJ, Posada JG., Jr Diaziquone (AZQ) Invest New Drugs. 1983;1(1):71–84. doi: 10.1007/BF00180194. [DOI] [PubMed] [Google Scholar]
- 15.Seo SR, Chong SA, Lee SI, et al. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J Neurochem. 2001;78(3):600–610. doi: 10.1046/j.1471-4159.2001.00438.x. [DOI] [PubMed] [Google Scholar]