

Abstract
The dsRNA-activated protein kinase, PKR, plays a pivotal role in the cellular antiviral response. PKR contains an N-terminal dsRNA binding domain (dsRBD) and a C-terminal kinase domain. An autoinhibition model has been proposed in which latent PKR exists in a closed conformation where the substrate binding cleft of the kinase is blocked by the dsRBD. Binding to dsRNA activates the enzyme by inducing an open conformation and enhancing dimerization. We have tested this model by characterizing the affinity and kinetics of nucleotide substrate binding to PKR. The fluorescent nucleotide mant-AMPPNP binds to unactivated PKR with Kd ~ 30 μM and the affinity is not strongly affected by autophosphorylation or binding to dsRNA. Biphasic binding kinetics are observed where the fast phase depends on nucleotide concentration but the slow phase is ligand-independent. The kinetic data fit to a two-step model of ligand binding followed by a slow conformation change. The kinetics are also not strongly affected by phosphorylation state or dsRNA binding. Thus, the equilibrium and kinetic data indicate that substrate accessibility of the kinase is not modulated by PKR activation state as predicted by the autoinhibition model. In atomic force microscopy images, monomers of the latent protein are resolved with three separate regions linked by flexible, bridge-like structures. Resolution of the individual domains in the images supports a model in which unactivated PKR exists in an open conformation where the kinase domain is accessible and capable of binding substrate.
PKR is a soluble protein kinase which is activated by dsRNA (1). PKR is constitutively expressed in most mammalian cell types but it is induced by type 1 interferons and plays a central role in the innate immunity defense against viral infection (2). In addition, PKR has been implicated in a variety of cellular signal transduction pathways (3). PKR is a member of the family of protein kinases which phosphorylate eIF2α, resulting in a blockage of translational initiation (4). Production of dsRNA during viral infection leads to PKR activation and inhibition of protein synthesis and apoptosis (5). PKR contains an N-terminal double-stranded RNA binding domain (dsRBD) and a C-terminal kinase domain. The dsRBD consists of two tandem copies of a widely distributed double stranded RNA binding motif (6). In an NMR structure of a PKR dsRBD construct, each of the dsRNA binding motifs adopts the canonical αβββα fold with a ~20 amino acid intervening region that is unstructured in the absence of dsRNA (7). The PKR kinase domain structure is typical of eukaryotic protein kinases with a substrate binding site lying at the interface of N- and C-terminal lobes (8).
An autoinhibition model for PKR has been proposed whereby dsRNA binding activates the enzyme by inducing a conformational change that relieves the latent enzyme of the inhibition that is mediated by interaction of the dsRBD with the kinase domain (9–12). Photoaffinity labeling of PKR with ATP derivatives requires either the presence of dsRNA (13, 14) or prior autophosphorylation (14), suggesting that enzymatic activation is associated with increased accessibility of the nucleotide binding site. Deletion of a portion of the N-terminal region of PKR encompassing the dsRBD can produce a constitutively active kinase capable of autophosphorylation and phosphorylation of eIF2α in the absence of dsRNA (12, 15–18). In the context of a dsRBD construct, NMR resonances associated with the second dsRNA binding motif are broadened or disappear upon addition of exogenous kinase domain, suggesting that it directly contacts the catalytic domain (10). Intrinsic tryptophan fluorescence (19) and electrophoretic mobility shift assays (20) support the existence of a dsRNA-induced conformational change.
In addition to increasing the accessibility of the active site, dsRNA binding is believed to enhance dimerization of PKR, which in turn facilitates intermolecular autophosphorylation (21). PKR is expressed in a latent form, but upon binding dsRNA it undergoes autophosphorylation at multiple serine and threonine residues (22–26) as well as tyrosines (27), producing an activated enzyme form. Following autophosphorylation, PKR is constitutively active, and is capable of phosphorylating exogenous substrates such as eIF2α in the absence of dsRNA (14, 21, 28). PKR exists in a weak monomer-dimer equilibrium with Kd = 450 μM and dimer stability is enhanced by ~500-fold upon autophosphorylation (29). Autophosphorylation can be induced without dsRNA by incubation of the enzyme at high concentrations, suggesting a model where dsRNA functions to bring PKR monomers into close proximity in a manner that is analogous to the dimerization of free PKR (29).
The activity of protein kinases is often tightly regulated. Structural studies reveal that the conformation of the catalytic core is conserved among protein kinases in the active state; however, the kinase domain adopts a diverse range of inactive conformations (30–32). Many kinases, including PKR, are activated by phosphorylation within the activation loop which may improve access of nucleotide or peptide substrates or realign critical catalytic residues. Kinetic and equilibrium binding measurements of diverse protein kinases indicate that activation loop phosphorylation consistently enhances the phosphoryl transfer rate by 2–4 orders of magnitude but the effects on nucleotide and peptide substrate binding affinities are highly variable (33). In the structure of a complex of the PKR kinase domain with eIF2α, threonine 446 in the activation loop is phosphorylated and interacts with helix αC to correctly position catalytic residues. Phosphorylation at Thr 446 also allosterically regulates eIF2α binding (8, 34).
A quantitative molecular description of PKR activation requires correlation of functional assays of PKR activation with direct measurement of dsRNA binding and enzymatic dimerization. However, detailed kinetic analysis of autophosphorylation reactions is generally difficult, and in the case of PKR the analysis is complicated by the presence of ~15 autophosphorylation sites. Thus, we have investigated PKR activation by monitoring the affinity and kinetics of nucleotide binding to PKR in latent and activated states. Somewhat surprisingly, we find that the affinity and kinetics are largely unaffected by autophosphorylation or binding to dsRNA. These data argue against models where dsRNA binding activates PKR by opening up the substrate binding cleft.
Materials and Methods
Reagents and Materials
Wild type unphosphorylated PKR and the catalytically-inactive K296R mutant were expressed in E. coli and purified as previously described (29). Protein concentrations were measured by absorbance using ε280= 4.33 × 104 M−1cm−1. At enzyme concentrations greater than 0.5 μM, PKR undergoes dsRNA – independent autophosphorylation upon addition of ATP (29). PKR was phosphorylated by incubation in phosphorylation buffer (20 mM HEPES, 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, pH 7.5) with 5 mM ATP at a protein concentration of 10–15 μM. The dimeric phosphoenzyme was purified by gel filtration on Superdex-200. We have previously demonstrated that this procedure results in essentially complete phosphorylation of PKR (29).
All solutions were prepared using nanopure (resistivity at least 18.2 MΩ cm) water. Mant-ATP and mant-AMPNP were purchased from Molecular Probes, mant-dATP was from Jena Bioscience (Jena, Germany) and unlabeled nucleotides (ATP, ADP, AMPPNP) were obtained from Sigma. Nucleotide concentrations were measured by absorption using ε255 = 2.33 × 104 M−1cm−1 for mant-nucleotides and ε259=1.54 × 104 M−1cm−1 for unlabeled nucleotides. The complementary 40 nt RNA sequences shown below were purchased from Dharmacon, deprotected using the manufacturer’s instructions and stored in TE buffer at −80°C.
TS40: 5′-GGA GAA CUU CAU GCC CUU CGG AUA AGG ACU CGU AUG UAC C-3′
BS40: 3′-CCU CUU GAA GUA CGG GAA GCC UAU UCC UGA GCA UAC AUG G-5′
The RNA sequences were annealed in AU 200-Mg2+ buffer (20mM HEPES, 200mM NaCl, 5 mM MgCl2, 0.1mM EDTA, 0.1mM TCEP, pH 7.5) by heating to 70°C and cooling slowly to room temperature. RNA concentrates were determined using ε260= 6.62 × 105 M−1cm−1 PKR was mixed with dsRNA at a molar ratio of 2.5 PKR: dsRNA.
Equilibrium Fluorescence Measurements
Fluorescence intensity and anisotropy data were collected with a FluoroMax-3 fluorimeter equipped with Glan-Thompson polarizers (Jobin Yvon Inc., New Jersey). All measurements were performed at 20°C with samples in AU 200-Mg2+ buffer using a 1 × 0.2 cm microcuvette. Titrations were recorded with sequential additions of either enzyme or ligand with 4 minutes between additions. Buffer blanks were subtracted from the measured intensities and intensities were corrected for dilution. Typically, equilibrium binding of mant-nucleotides is measured by fluorescence energy transfer from tryptophans to mant (35, 36). However, energy transfer in PKR is relatively inefficient and binding was characterized by monitoring the fluorescence intensity or emission anisotropy increase with direct excitation of the mant-nucleotide at 360 nm. The anisotropy is defined by
| (1) |
where I|| and I⊥ are the intensities with the emission polarizers in the parallel or perpendicular orientation with the excitation polarizer in the vertical (parallel) orientation. The G factor corrects for polarization bias of the gratings and is given by the ratio of the intensities (I||/I⊥) with the excitation polarizer in the perpendicular orientation. G was determined initially for each experiment and fixed in subsequent measurements. Buffer backgrounds were subtracted for each polarizer orientation.
Dissociation constants were measured using a fixed concentration of mant-nucleotide titrated with PKR (forward titration) or a fixed enzyme concentration with addition of mant-nucleotide (reverse titration). Both types of titrations were fit using a 1:1 binding model with a single equilibrium dissociation constant, Kd. Under conditions where the enzyme and ligand concentrations are comparable, the concentration of the enzyme-ligand complex ([EL]) is given by
| (2) |
where [E]0 and [L]0 are the total concentration of enzyme and ligand, respectively. Binding isotherms were analyzed using nonlinear least-squares fitting with Igor Pro (Wavemetrics, Lake Oswego, OR). For models where a 1:1 stoichiometry was assumed, [E]0 was fixed at the true enzyme concentration. Alternatively, [E]0 was treated as an adjustable parameter and the stoichiometry (N) was calculated as the ratio of [E]0 to the actual concentration of PKR. Fluorescence intensity data were analyzed using the following equation:
| (3) |
where I is the fluorescence intensity (corrected for dilution), I0 is the fluorescence intensity in the absence of PKR and ΔI is the limiting fluorescence increase due to PKR binding. The intensity data were fit by treating I0, ΔI and Kd as adjustable parameters. For analysis of anisotropy titrations, the measured anisotropy is the average anisotropies of the free ligand and the ligand bound to enzyme, weighted by their concentrations and their relative fluorescence intensities:
| (4) |
where rL is the anisotropy of free ligand, rEL is the anisotropy of ligand in the EL complex and IEL/IL is the fluorescence intensity ratio. IEL/IL was separately measured for mant nucleotide binding to each form of PKR studied. The anisotropy data were fit by treating rL, rEL, and Kd as adjustable parameters.
Competitive anisotropy titrations were used to determine the affinities of unlabeled nucleotides using a fixed concentration of PKR and mant-nucleotide and variable concentration of unlabeled nucleotide. Because of the relatively weak binding of nucleotides to PKR, it is not feasible to completely saturate the enzyme with mant-nucleotide and the analysis of the competition titration requires solution of a cubic equation to obtain [EL]. The competitive anisotropy titrations were analyzed using the solution described in Appendix 2 of Talavera and De la Cruz (36), constraining the total concentrations of labeled and unlabeled nucleotides, Kd (dissociation constant of the mant-nucleotide) and IEL/IL, while allowing rL, rEL, KU (dissociation constant of the unlabeled nucleotide) and [E]o to float.
Stopped-Flow kinetics measurements
The kinetics of mant-nucleotide binding and dissociation were measured using an Applied Photophysics fluorescence stopped flow instrument model SX.18MV. Mant-ATP was employed because autophosphorylation is not a complication on the short timescale of these measurements. All measurements were performed in AU 200-Mg2+ buffer at 20°C with excitation at 295 nm. Emission was measured using 400 nm long-pass glass filter (Edmund Industrial Optics, New Jersey). For measurement of association kinetics, 2 μM of enzyme was mixed at a 1:1 ratio with variable concentrations of mant-nucleotide. Data were collected using 1000 points over 10 s. Between 10 and 24 traces were averaged at each nucleotide concentration and the data were fit to single and multiple exponential decay functions using KaleidaGraph (Synergy Software). The dissociation kinetics were measured by displacement of bound mant-nucleotide with a large excess of ATP. Enzyme (2 μM) was pre-equilibrated with 80 μM mant-ATP and then mixed at a 1:1 ratio with 4 mM ATP. Data were collected using 1000 points over 10 s and the resulting kinetic traces were fit to single and multiple exponential decay functions. Kinetic simulations (37) and global data analysis (38) were performed using KinTekSim.
Atomic force microscopy
Images were obtained with a Nanoscope III DI atomic force microscope (AFM) (Veeco, Santa Barbara, CA) with 512 × 512 pixel resolution and a scan rate of 3 Hz, using an E type scanner (15×15 μm2 maximum scan size). AFM tips employed were Nanosensors NCL cantilevers with spring constants between 40 and 55 N/m and nominal resonance frequencies of 182–199 kHz (Molecular Imaging, Phoenix, AZ, USA). All images were plane fitted and 3rd order flattened using DI imaging software before analysis. Experiments were carried out in 20 mM HEPES pH 7.8, 50 mM NaCl, 5mM MgCl2). Buffer solutions were filtered prior to experiments using 0.02 μm filters (Anatop 25, Whatman, Maidstone, UK) and heated for 10 minutes to 65°C. Protein solutions were incubated at ambient temperature for 20 and 50 minutes. Samples were diluted to 20 nM PKR for deposition and 20 μL was then immediately applied to the surface of freshly cleaved mica and rinsed with water after 30 seconds. The surface was blotted, dried under nitrogen, and imaged immediately. Surface bound conformations of proteins are fixed after removal of the surrounding buffer solution and do thus not further change during the experiment. Visual analysis of the topography of PKR molecules and protein-dsRNA complexes was carried out using DI imaging software (Veeco). Volume analysis of protein peaks in the AFM images was achieved using Image SXM 169-3x software (Steve Barrett, University of Liverpool, UK), as previously described (39, 40). Between 300 and 1000 molecules of PKR from AFM images went into the statistical analysis.
Results
Fluorescence changes of mant nucleotides upon binding to PKR
In equilibrium experiments, which employed WT PKR, the nonhydrolyzable ATP analogs mant-AMPPNP or unlabeled AMPPNP were used to avoid complications from PKR-catalyzed autophosphorylation reactions. In control experiments, we confirmed that both mant-AMPPNP and AMPPNP are not substrates for PKR (data not shown). We then examined changes in mant nucleotide fluorescence upon binding to PKR. Upon direct excitation of the mant fluorophore at 360 nm, the fluorescence emission of mant-AMPPNP at 440 nm increased about 1.5-fold upon addition of a large molar excess of WT unphosphorylated PKR or to the inactive K296R mutant. This enhancement could be reversed by competing off the mant-labeled nucleotide with excess unlabeled AMPPNP or ATP (results not shown). In contrast to previous reports, these data suggest that ATP is able to bind inactive PKR. Because the mant absorption band at 360 nm strongly overlaps with PKR tryptophan emission at 346 nm, we examined whether mant-nucleotide binding could be detected via fluorescence resonance energy transfer. As shown in Figure 1A, mant-ATP fluorescence emission is observed upon excitation at 295 nm and there is a ~10% intensity increase upon addition of 1 μM PKR. The fluorescence increase is reversed by adding an excess of ATP, indicating that the enhancement is due to resonance energy transfer from the PKR tryptophans to the bound mant. Using the same concentrations of mant-nucleotide and PKR, upon direct excitation of mant at 360 nm there is no increase in mant emission upon addition of PKR (Figure 1B), confirming that the observed enhancement in Figure 1A is due to fluorescence energy transfer. A greater extent of tryptophan→ mant energy transfer is often observed upon binding of mant-nucleotides to ATP binding sites (e.g. references (35, 36). Although PKR contains three tryptophans within the kinase domain, two of which are within ~ 15–20 Å of the ATP binding site, the mant moiety is perpendicular to the tryptophan indole rings in a model of mant-ATP bound to PKR, which is unfavorable for energy transfer (Brown, R. and Lemaire, P.A., unpublished observations). Finally, we examined changes in mant - nucleotide emission anisotropy upon binding to PKR. Figure 2 shows that the emission anisotropy of mant-AMPPNP exhibits a large increase from ~ 0.02 to ~ 0.17 upon addition of a large excess of PKR.
Figure 1.

Fluorescence spectra of mant-ATP in the absence and presence of wild-type PKR. A: mant-ATP (—), PKR (.....), mant-ATP + PKR ( , blue) and mant-ATP +PKR + unlabeled ATP (
, blue) and mant-ATP +PKR + unlabeled ATP ( , red). Spectra were recorded at an excitation wavelength of 295 nm with excitation and emission bandwidths of 2 nm. B: Spectra of mant-ATP (—), and mant-ATP + PKR (, red). Spectra were recorded at excitation wavelength of 360 nm with excitation and emission bandwidths of 2 nm. Concentrations: mant-ATP, 122 μM; PKR, 1 μM; ATP, 2 mM.
, red). Spectra were recorded at an excitation wavelength of 295 nm with excitation and emission bandwidths of 2 nm. B: Spectra of mant-ATP (—), and mant-ATP + PKR (, red). Spectra were recorded at excitation wavelength of 360 nm with excitation and emission bandwidths of 2 nm. Concentrations: mant-ATP, 122 μM; PKR, 1 μM; ATP, 2 mM.
Figure 2.

Binding of mant-AMPPNP to PKR measured by forward anisotropy titration. The fluorescence anisotropy of 20 μM mant-AMPPNP was measured at increasing concentrations of PKR at an excitation wavelength of 360 nm and emission wavelength of 443 nm with spectral bandwiths of 2 nm and 4 nm for excitation and emission respectively. Fluorescence intensity at each polarizer orientation were integrated for 1 s and measurements were averaged until the maximum standard deviation drops below 0.1%. Each data point represents the average of 3–4 measurements. The solid line represents a fit of the data to a 1:1 binding model to obtain Kd = 33.8± 0.7μM, rL = 0.0213± 0.0003 and rEL = 0.1914± 0.0010.
Equilibrium binding of ATP to PKR
Based on the large anisotropy changes upon binding of mant-nucleotides to PKR we have chosen to use this method for detailed characterization of equilibrium binding parameters. Binding constants can reliably be obtained from anisotropy titrations performed using either variable enzyme or ligand concentrations. Figure 2 shows a titration of a fixed concentration of mant-AMPPNP with increasing concentrations of unphosphorylated, wild type PKR; we refer to this format as a “forward titration”. In contrast to previous reports (13, 14), we find that ATP analogs bind readily to unactivated PKR. Analysis of the binding isotherm using equation 2 yields a Kd of 33.8 ± 0.7 μM assuming a 1:1 binding stoichiometry (Table 1). If the stoichiometry is treated as an adjustable parameter, a best-fit value of 0.95 binding sites/PKR monomer is obtained with a similar Kd of 31.3 μM. Thus, binding of mant-AMPPNP occurs with the expected 1:1 stoichiometry, indicating that our PKR preparations are fully competent for nucleotide binding. In subsequent analyses, the stoichiometries were fixed at 1:1 to improve the precision of the Kd determinations.
Table 1.
Equilibrium dissociation constants for interaction of nucleotides with PKR.a
| Nucleotide | Enzyme Form | |||
|---|---|---|---|---|
| Unphosphorylated | Phosphorylated | K296R Mutant | Unphos. + 40mer dsRNA | |
| mant-AMPPNP | 33.8 ± 0.7b 34.8 ± 3.6c |
34.0 ± 0.7b | 11.5 ± 0.3b | 102.3 ± 3.6c |
| mant-ATP | - | 24.6 ± 1.7c | 10.3 ± 0.8b | - |
| ATP | - | 24.4 ± 0.1d | 20.2 ± 1.7d | - |
| AMPPNP | 97.7 ± 1.7d | 34.0 ± 0.1d | 22.8 ± 2.0d | 160.5 ± 1.4d |
| ADP | 76.5 ± 1.1d | 11.6 ± 0.7d | 12.5 ± 0.3d | 67.0 ± 0.7d |
Kd values in units of μM.
Forward anisotropy titration.
Reverse anisotropy titration.
Competition titration using mant-AMPPNP.
Although PKR exists in a monomer-dimer equilibrium (29), at the maximum enzyme concentration employed in the titration in Figure 2 only about 20% dimer is present. However, to avoid potential complications associated with variable extents of PKR dimerization during the course of a titration we have devised an alternative reverse anisotropy titration where the enzyme concentration is fixed and the ligand concentration is varied. Figure 3 shows a “reverse titration” using the same enzyme and ligand combination employed in figure 2. The average anisotropy, <r>, decreases with increasing [mant-AMPPNP] due to dilution of the bound ligand with excess free ligand and the shape of this curve is determined by the dissociation constant. The reverse titration data were fit to a simple 1:1 binding isotherm using equation 2, giving a value of Kd = 34.8 ± 3.6μM, which closely agrees with the value obtained from the forward titration.
Figure 3.

Binding of mant-AMPPNP to PKR measured by reverse fluorescence anisotropy. mant-AMPPNP was titrated against 40 μM PKR and the fluorescence anisotropy was measured at an excitation wavelength of 360 nm and emission wavelength of 443 nm with spectral bandwiths of 2 nm and 4 nm for excitation and emission respectively. Fluorescence intensity at each polarizer orientation were integrated for 1 s and measurements were averaged until the maximum standard deviation drops below 0.1%. Each data point represents the average of 3–4 measurements. The solid line represents a fit of the data to a 1:1 binding model to obtain Kd = 34.8± 3.6 μM, rL = 0.0407± 0.0016 and rEL = 0.2356 ± 0.0096.
The reverse anisotropy titration assay was used to compare the Kd values for mant-AMPPNP and mant-ATP binding to different PKR forms. Table 1 shows that the dissociation constant for mant-AMPPNP binding to in vitro phosphorylated PKR is, within error, no different from the unactivated enzyme. The Kd for a complex of unphosphorylated PKR with an activating 40 bp dsRNA is increased about three-fold relative to the wild type enzyme. Thus, there appears to be no strong correlation between activation of PKR and enhanced nucleotide binding affinity; indeed, dsRNA binding slightly weakens nucleotide binding. Because PKR undergoes dsRNA-independent autophosphorylation at the higher protein concentrations used in the fluorescence measurements (29) it is not possible to directly characterize binding of mant-ATP to wild type, unphosphorylated enzyme without complications from the catalytic reaction. Thus, we have employed a catalytically inactive K296R mutant. In protein kinases this lysine binds the α and β phosphates of ATP and is required for phosphoryl transfer but not ATP binding (41, 42). Mant-AMPPNP binds to the K296R mutant with a Kd of 11.5 ± 0.3 μM and a similar dissociation constant is observed for mant-ATP. For the phosphorylated enzyme, mant-ATP binds slightly stronger than mant-AMPPNP. Thus, the substitution of the β-γ bridging O by N does not substantially affect ATP binding.
Competitive anisotropy titrations were performed to measure the binding affinity of unlabeled nucleotides. Figure 4 shows that unlabeled ATP effectively competes with mant-ATP for binding to phosphorylated PKR. Fitting the titration using the previously-measured Kd for the labeled nucleotide gives a Kd = 24.4 ± 0.1μM for unlabeled ATP; thus, the mant moiety does not perturb equilibrium binding of ATP to phosphorylated PKR. Similarly, mant does not alter the affinity of AMPPNP binding to phosphorylated PKR (Table 1). However, mant enhances the affinity of AMPPNP binding to WT, K296R and the PKR-dsRNA complex by three-fold, and two-fold, and ~ 1.5-fold, respectively. Finally, competition titrations were used to assess binding of the reaction product ADP. ADP binds about twofold more strongly than ATP to the phosphorylated and K296R enzymes and about two-fold more strongly than AMPPNP to the PKR-dsRNA complex. However, the affinity of ADP and AMPPNP are approximately equal for unphosphorylated PKR.
Figure 4.

Binding of ATP to phosphorylated PKR measured by competition fluorescence anisotropy. In this case, ATP was titrated against 39.5 μM of phosphorylated PKR (measured by UV absorption at 280 nm) with 11.3 μM mant-ATP and the fluorescence anisotropy measured with excitation and emission at 360 and 443 nm respectively. Each data point represents and average of 3–4 repeated measurements at the same ATP concentration. The Kd of mant-ATP binding to phosphorylated PKR measured by reverse anisotropy titration and used in the fitting function was 24.6 μM.
Kinetics of nucleotide binding to PKR
We have characterized the kinetics of nucleotide binding to PKR by stopped-flow fluorescence measurements to define the binding mechanism and to determine whether the association or dissociation rates are sensitive to the enzyme activation state. Under pseudo first-order conditions where the ligand is in excess, the most sensitive kinetic measurements are obtained by monitoring mant emission stimulated by tryptophan →mant energy transfer. Figure 5A shows that binding of 50 μM mant-ATP to unphosphorylated PKR gives rise to a large rapid fluorescence increase complete within 20 ms along with a slow, low amplitude increase. The data fit well to a two exponential model with kobs, 1= 514 ± 9 s−1 (96% amplitude) and kobs,2 of 39 ± 9 s−1 (4% amplitude). The fluorescence amplitude of the kinetic transient extrapolated to t=0 corresponds to the fluorescence of the free ligand, indicating that there are no undetected fast phases within the instrument dead time (data not shown).
Figure 5.

Kinetics of mant-ATP binding to unphosphorylated wild-type PKR measured by stopped-flow fluorescence spectroscopy. A: Time-dependent changes in fluorescence of 50 μM mant-ATP in the presence of 1 μM unphosphorylated PKR. The mixture was excited at 295 nm and fluorescence emission above 400 nm measured using a cutoff filter. The plot represents average of 12 runs with each run made of 1000 data points taken over 10 s in a log time interval. The solid line is the two exponential fits to the data. The first phase contributed 96% to the total amplitude with the observed rate constant (kobs,1) of 514 ± 9 s−1 while the second phase contributed 4% to the total amplitude with kobs,2 of 39 ± 9 s−1. B: The effect of mant-ATP concentration on kobs. The solid line is a linear fit to the data from two experiments. kobs,1 (●) increases linearly as the concentration of mant-ATP and a least squares fit yields kobs,1:slope = 6.25 ± 0.35 μM−1·s−1 and kobs,1:intercept = 206 ± 11 s−1·kobs,2 (□) is essentially independent of mant-ATP concentration with an average value of 48.9 ± 9.9 s−1.
Figure 5B shows that the rate of the fast phase increases linearly with ligand concentration but the rate of the slow phase is essentially concentration-independent. The slope and intercept values for kobs,1 and the average value of kobs,2 are displayed in table 2. The relative amplitudes of the two phases are dependent on mant-ATP concentration: the contribution of kobs,1 ranges from 87% at 5 μM to 96% at 50 μM ligand (see Table S1).
Table 2.
Kinetics of mant-ATP binding to PKR.
| Enzyme Form | Association Kinetics | Dissociation Kineticsa | |||
|---|---|---|---|---|---|
| kobs, 1 : Slope (μM−1·s−1)b | kobs, 1 :Intercept (s−1)c | kobs, 2: Average (s−1)d | kobs, 1 (s−1) | kobs, 2 (s−1) | |
| Unphosphorylated | 6.25 ± 0.35 | 206 ± 11 | 48.9 ± 9.9 | 175 ± 2 | 43.1 ± 5.5 |
| Phosphorylated | 4.75 ± 0.40 | 239 ± 14 | 43.2 ± 4.9 | 214 ± 5 | 56.0 ± 0.8 |
| K296R | 5.56 ± 0.38 | 91 ± 13 | 39.7 ± 5.7 | 134 ± 4 | 18.6 ± 0.1 |
| Unphos. + 40mer dsRNA | 5.36 ± 0.50 | 232 ± 16 | 36.5 ± 8.1 | 175 ± 1 | 23.2 ± 5.5 |
Dissociation kinetics measured upon addition of 2 mM unlabeled ATP.
Slope from linear fit of kobs,1 vs. [mant-ATP].
Y-intercept from linear fit of kobs,1 vs. [mant-ATP].
Average value of kobs,2 over the entire range of [mant-ATP].
Because preparations of mant-ATP contain a mixture of 2′- and 3′-isomers, we also examined the binding kinetics of a single isomer nucleotide, 3′-mant-2′deoxy ATP. As with mant-ATP, 3′-mant-2′deoxy ATP binds to both WT and K296R PKR with biphasic association kinetics, indicating that this reaction pattern is not due to ligand heterogeneity (data not shown).
Binding of mant-ATP to other PKR forms also gives rise to a biexponential fluorescence increase with a concentration-dependent fast phase and a concentration-independent slow phase. Table 2 compare the kinetic parameters. Overall, the binding kinetics are not strongly affected by phosphorylation, binding to dsRNA or the K296R mutation. The largest change occurs for K296R where the value of kobs,1 :intercept is decreased about two-fold relative to unphosphorylated PKR. Also, in all cases the relative amplitude of the fast phase increases with increasing ligand concentration (see Table S1). For the phosphorylated and dsRNA-bound forms, the relative amplitudes are similar to the unphosphorylated enzyme; however, for the K296R mutant the fraction of the fast phase varies more strongly, increasing from 44% to 91% between 10 and 50 μM mant-AMPPNP.
The dissociation kinetics for mant-ATP were directly measured by trapping experiments where the PKR-mant-ATP complex was mixed with a large molar excess of unlabeled ATP. Figure 6 shows that the dissociation of mant-ATP from unphosphorylated PKR occurs with a dominant fast phase with kobs,1 of 175 ± 2 s−1 (94% amplitude) and a slow, low amplitude phase with kobs,2 of 43.1 ± 5.5 s−1. Biphasic dissociation kinetics were observed by the other PKR forms (table 2). As in the case of the association kinetics the observed rate constants for dissociation are fairly similar for the different PKR forms. The most notable effect is a decrease in both kobs,1 and kobs,2 for the K296R mutant. More dramatic changes are observed in the relative amplitudes of the two phases: for phosphorylated PKR the fast phase decreases to 49% and for the K296R mutant it drops to 13%.
Figure 6.

Kinetics of mant-ATP dissociation from unphosphorylated wild-type PKR measured by stopped-flow fluorescence spectroscopy. Time-dependent changes in fluorescence of 40 μM mant-ATP in the presence of 1 μM unphosphorylated PKR competed off with 2 mM ATP. The mixture was excited at 295 nm and fluorescence emission above 400 nm measured using a cutoff filter. The plot represents average of 21 runs with each run made of 1000 data points taken over 10 s in a log time interval. The solid line is the two exponential fits to the data. The first phase contributed 94% to the total amplitude with rate constant (kobs,1) of 175 ± 2 s−1 while the second phase contributed 6% to the total amplitude with kobs,2 of 43.1 ± 5.5 s−1.
The presence of two rate constants for the binding of mant-ATP indicates a minimal two step kinetic mechanism involving bimolecular association and isomerization reactions. In scheme 1, the nucleotide first binds to PKR to form EL, which then undergoes a conformational change to E′L.
Scheme 1.
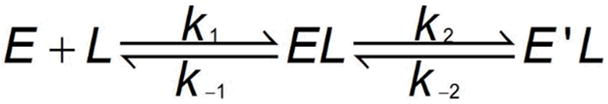
Alternatively, the free enzyme may undergo a conformational change (E →E′) prior to binding nucleotide (Scheme 2).
Scheme 2.
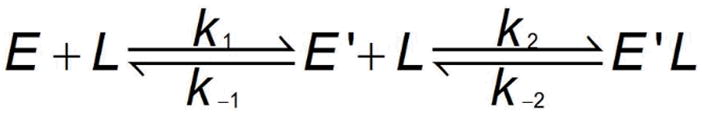
Although these two mechanisms cannot be distinguished based solely on the measured rate constants (43), only scheme 1 is fully consistent with the kinetic data. In Scheme 2, E′L is the only species that gives rise to fluorescence energy transfer and in the dissociation experiments its decay would be monoexponential with a rate constant of k−2. However, a biexponential fluorescence decrease is observed experimentally (Figure 6). In contrast, Scheme 1 predicts biexponential decay kinetics for the dissociation experiment. For the association reaction, both schemes predict that formation of E′L proceeds with a lag phase associated with population of an intermediate (either EL or E′). However no lag phase is observed and both kinetic phases of the association reaction have positive amplitude, indicating that scheme 2 is incorrect. Scheme 1 is consistent with the data provided that both EL and E′L contribute to the fluorescence increase, giving rise to an overall fluorescence change with two positive kinetic phases.
For scheme 1, the values of fast and slow phases can be expressed in terms of the elementary rate constants using the square root approximation (43) to give
| (5) |
| (6) |
Thus, as observed in Figure 5B, kobs,1 should exhibit a linear substrate concentration dependence, where the slope is given by k1 and the intercept by k−1+k2+k−2. Although kobs,2 is predicted to show a hyperbolic concentration dependence, this dependence may not be apparent in systems where k2 and k−2 are small compared to k1[S] and k−1. In the limit of a single step mechanism, where the isomerization reaction is not present, k2= k−2= 0 and only a single exponential phase is expected with kobs= k1[S] + k−1. For the unphosphorylated enzyme, if we neglect the slow kinetic phase and calculate Kd = k−1/k1 based on the intercept and slope of kobs,1 we obtain a value Kd= 33.0 μM, which agrees well with the equilibrium values of Kd =34–35 μM for mant-AMPPNP (Table 1). This agreement presumably is associated with the small contribution of the slow phase and low population of the E′L state for this enzyme form.
Figure 7 shows a global fit of the wild type PKR binding kinetics to scheme 1 over the range of 5 - 30 μM mant-ATP, making the simplifying assumption that EL and E′L contribute equally to the observed fluorescence change. The good fit provides further support for scheme 1 and gives best values of k1=7.33 ± 0.07 μM−1s−1, k−1 = 137± 2 s−1, k2 =1.8± 0.7 s−1 and k−2 =13.0 ± 10.7 s−1. The parameters obtained from the global analysis are reasonably consistent with the value of k1 obtained from the linear substrate concentration dependence of kobs,1 and with the trapping experiments, where the fast phase is equal to k−1 and the slow phase corresponds to k−2. Although the standard errors for k2 and k−2 are large, it is clear that ligand binding and dissociation are significantly faster than the subsequent isomerization reaction.
Figure 7.

Simulation of the kinetics of mant-ATP binding to unphosphorylated PKR: [EL] (– –), [E′L] × 8 (.....), [EL] + [E′L] (—). The kinetics were simulated based on a global fit of the experimental data to scheme 1 over the range of 5 – 30 μM mant-ATP, assuming that that EL and E′L contribute equally. The adjustable parameters are the four rate constants and an overall scaling factor, giving best fit values of k1=7.33 ± 0.07 μM−1s−1, k−1 = 137± 2 s−1, k2 =1.8± 0.7 s−1 and k−2 =13.0 ± 10.7 s−1.
Atomic force microscopy
In AFM images of latent PKR (Figure 8A), we observe molecules with clearly resolved features. Based on volume analysis of the individual particles, we identify the molecules as monomers of the protein (data not shown). A large number of proteins have been imaged using AFM in air on mica surfaces (40, 44–49). In these images, proteins generally appear globular with little or no disordered regions, although multiple domains and multiple subunits have been resolved in a few cases [D.A. Erie, unpublished data; (45, 50)]. The degree of structure apparent in the images of the PKR monomers is striking for a small (62 kDa) protein.
Figure 8.
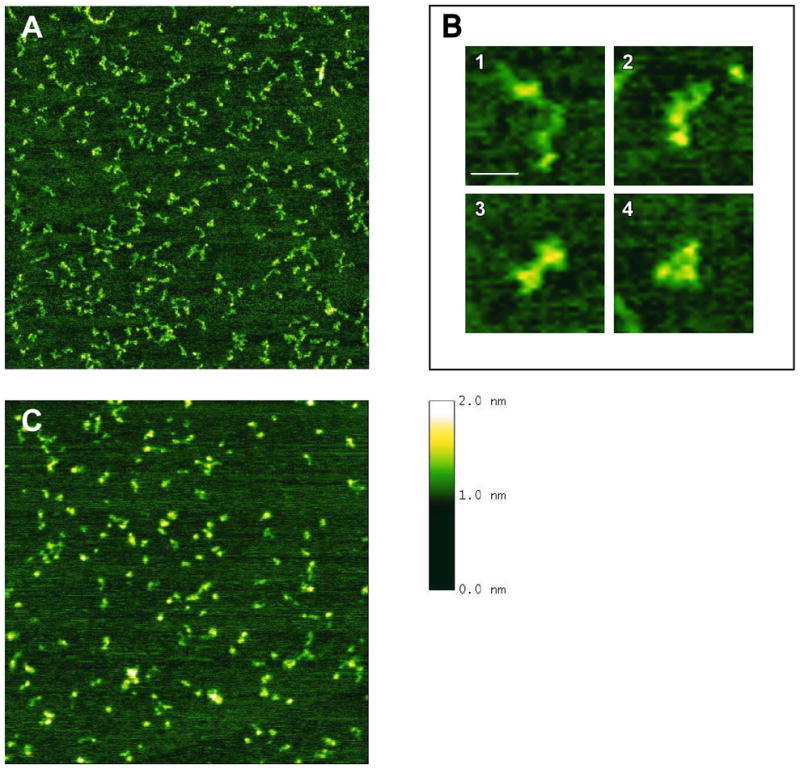
AFM images of (A) unphosphorylated PKR and (C) phosphorylated PKR. Images are 1 μm × 1 μm with a height scale of 2 nm. In (A), different molecular conformations can be found. Examples of these are shown enlarged in (B): almost entirely extended conformation (1), shapes with three resolvable segments (2), shapes showing only two resolved segments and a broader linking region (3), and molecules in which the individual segments are very close together (4). The scale bar in (B1) is 20 nm. Height scale of all images is 2 nm as indicated next to image (C).
The AFM images the PKR monomers reveal up to three distinguishable structured regions linked by bridge-like stretches. In some cases, the linker regions are structured (for example Figure 8B-4); however, this structured conformation appears unstable because in most molecules the linker adopts an unfolded, extended form. We have classified the different observed conformations into four groups, as shown in Figure 8B. Some molecules feature a compact arrangement (Figure 8B-4), while others appear almost completely extended (Figure 8B-1). Statistical analysis of the different conformations shows that the four shape categories are represented with approximately equal frequencies in the images (data not shown), which is consistent with a high linker flexibility.
Addition of ATP had no effect on the molecular conformations of PKR seen in AFM (data not shown). Because these experiments were carried out at low protein concentrations, PKR did not undergo dsRNA-autophosphorylation during AFM analysis (29). Phosphorylated PKR (Figure 8C) exhibited volumes consistent with PKR monomers and dimers (and higher oligomeric states) in agreement with previous studies finding enhanced dimerization of phosphorylated PKR (29). Although the phosphorylated protein frequently showed globular, condensed shapes, many of the molecules still displayed open extended shapes similar to those seen for unphosphorylated PKR, suggesting that the kinase domain is accessible to substrate in both the unphosphorylated and the phosphorylated protein.
Discussion
A key finding from our equilibrium and kinetic studies of nucleotide binding to PKR is that ATP is capable of binding to unphosphorylated enzyme and the dissociation constants and kinetics are largely unaffected by phosphorylation or binding to dsRNA. Although slight changes in PKR affinity and binding kinetics are observed, these differences are not likely to be physiologically significant, since the millimolar intracellular concentration of ATP is about two orders of magnitude higher than the average Kd. These results contrast with earlier studies indicating that photocrosslinking of either ATP or 8-azido ATP to PKR requires dsRNA (13, 14) or prior autophosphorylation (14). The origin of these discrepancies is not clear but may relate to differences in methodology or sample preparation. In particular, the current measurements were performed using direct fluorescence binding methods with purified enzyme. Control experiments demonstrate that the mant fluorophore does not strongly perturb the affinity or kinetics.
Based on the nucleotide binding data, we cannot exclude the possibility that dsRNA binding or phosphorylation alter accessibility of the peptide substrate binding region. No correlation between substrate recognition and dsRNA binding has been described. However, it has been reported that eIF2α binding to a PKR kinase domain construct requires phosphorylation at threonine 446 within the activation loop whereas Km for the nonspecific substrate histone H2A is unaffected by PKR phosphorylation (34). In contrast, earlier studies using full length PKR demonstrated that eIF2α can bind to unphosphorylated PKR (51). Although the activities of many eukaryotic protein kinases are enhanced by phosphorylation events within the activation loop, in most cases, peptide substrate or nucleotide binding are not controlled by phosphorylation; instead, the dominant effect is to enhance the phosphoryl transfer rate (33, 52). Further studies are required to determine whether PKR follows this dominant paradigm.
Although mutation of lysine 296 renders PKR completely inactive (53), the K296R mutant is capable of binding adenosine nucleotides. This lysine is conserved in most kinases where it interacts with the α and β phosphates of ATP. Mutants of this lysine in other kinases, including lck (42), erk2 (41) and PKA (54), are also capable of binding ATP but are catalytically impaired. In PKR, the K296R mutation decreases adenosine nucleotide Kd values by 3–6 fold but, in erk2 the analogous lysine → arginine mutation slightly increases the Km for ATP, suggesting that the lysine – ATP interaction does not contribute strongly to the overall binding affinity.
The observation of biphasic kinetics in the stopped-flow measurements of ATP binding to PKR supports a minimal two-step model involving bimolecular association and isomerization steps. In general, these steps can occur in either order; however, only scheme 1, where binding precedes the isomerization reaction, is consistent with the biphasic fluorescence decrease observed in dissociation experiments and the presence of two positive phases in the association measurements. Furthermore, kinetic transients obtained at multiple ligand concentrations can be globally fit to scheme 1. Qualitatively similar kinetics are observed in all PKR forms, indicating that the overall reaction mechanism is not altered by phosphorylation, dsRNA binding or the K296R mutation. Transient kinetic measurement have led to a similar kinetic model for nucleotide binding to protein kinase A involving ligand binding and subsequent isomerization; however, an additional kinetic phase was observed and assigned to an additional isomerization step (35). Based on kinetic data alone the isomerization step cannot be definitively assigned to a specific conformational change or to a step in the catalytic cycle. The ATP bound form of the PKR kinase domain has a slightly more open conformation of the two lobes of the catalytic core than the unliganded kinase (8), suggesting that PKR undergoes a conformational change upon nucleotide binding. The slow isomerization may be associated with this conformational change.
The AFM images of latent PKR reveal monomeric molecules that are extended and segmented. These highly asymmetric structures are fully consistent with the previously reported high frictional ratio f/f0 of 1.62 (29) and the large radius of gyration for PKR (19). Structured domains are resolved in the AFM images and are linked by flexible, bridge-like regions. Resolution of this domain structure indicates that latent PKR exists in an open conformation consistent with unhindered binding of ATP to the kinase domain. The range of different PKR conformations observed on a surface using AFM indicates that highly flexible regions link the individual segments, allowing these domains to land on the surface in different orientations and at different distances from each other. In some cases, the linker regions appear to be structured; however, this structured conformation is very unstable since the majority of these regions adopt an unfolded, extended conformation. The high flexibility of these regions is consistent with NMR evidence that the 20 amino acid linker between the dsRBMs is unstructured in solution (7). The conformation of the ~ 100 residue segment lying between the dsRBD and kinase domain is unknown but may contain additional flexible regions. Although globular, condensed shapes are frequently observed in the AFM images of phosphorylated PKR, many molecules exhibit domain structure. Thus, phosphorylated PKR can adopt an open shape similar to the latent enzyme, consistent with an insignificant effect of the protein phosphorylation state on ATP binding affinity.
The present nucleotide binding and AFM data do not support the autoinhibition model for PKR activation and indicate an open conformation for the resting enzyme. Equilibrium and kinetic measurements of nucleotide binding demonstrate that the ATP binding site is fully accessible in the unphosphorylated enzyme in the absence of dsRNA. AFM data indicate that the dsRBD is not stably associated with the kinase domain. PKR can be activated by dimerization at high protein concentrations in the absence of dsRNA (29), arguing against a specific involvement of the dsRNA binding domains in enzymatic activation. In addition, fusion of a heterologous dimerization domain with the PKR catalytic domain enhances autophosphorylation and eIF2α kinase function in vivo, suggesting that dimerization is sufficient to induce activation in the absence of exogenous activator (9). Heparin, a known activator, is capable of activating PKR deletion mutants lacking the dsRNA binding domain (55, 56), providing further evidence that the dsRNA binding domain is not directly associated with enzymatic activity. Thus, PKR activation by dsRNA and heparin may simply be associated with increasing the local enzyme concentration and enhancing functional dimerization by binding PKR to a polymeric substrate containing multiple binding sites.
Acknowledgments
We thank Carolyn Teschke for use of the fluorescence stopped flow instrument, Eric Anderson for help in carrying out the kinetic measurements and Ray Brown for modeling of mant-ATP into the PKR kinase active site. This work was supported by grant number AI-53615 from the NIH (J.L.C.) and by the American Cancer Society and NIH grant number GM-54136 (D.A.E.).
Abbreviations
- AFM
atomic force microscopy
- AMPPNP
adenylyl imidodiphosphate
- dsRBD
dsRNA binding domain
- dsRNA
double-stranded RNA
- mant
N-methylanthraniloyl
- WT
wild type
Footnotes
Supporting Information
Table S1: Relative contribution of the fast phase for association of mant-AMPNP with PKR. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Clemens MJ. PKR--a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol. 1997;29:945–949. doi: 10.1016/s1357-2725(96)00169-0. [DOI] [PubMed] [Google Scholar]
- 2.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 3.Williams BR. Signal integration via PKR. Sci STKE. 2001;2001:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- 4.Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 5.Barber GN. Host defense, viruses and apoptosis. Cell Death Differ. 2001;8:113–126. doi: 10.1038/sj.cdd.4400823. [DOI] [PubMed] [Google Scholar]
- 6.Tian B, Bevilacqua PC, Diegelman-Parente A, Mathews MB. The double-stranded RNA binding motif: Interference and much more. Nature Rev Mol Cell Biol. 2004;5:1013–1023. doi: 10.1038/nrm1528. [DOI] [PubMed] [Google Scholar]
- 7.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 9.Vattem KM, Staschke KA, Wek RC. Mechanism of activation of the double-stranded-RNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2) Eur J Biochem. 2001;268:3674–3684. doi: 10.1046/j.1432-1327.2001.02273.x. [DOI] [PubMed] [Google Scholar]
- 10.Nanduri S, Rahman F, Williams BRG, Qin J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000;19:5567–5574. doi: 10.1093/emboj/19.20.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu S, Kaufman RJ. trans-Autophosphorylation by the isolated kinase domain is not sufficient for dimerization or activation of the dsRNA-activated protein kinase PKR. Biochemistry. 2004;43:11027–34. doi: 10.1021/bi0360105. [DOI] [PubMed] [Google Scholar]
- 12.Wu S, Kaufman RJ. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem. 1997;272:1291–6. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- 13.Bischoff JR, Samuel CE. Mechanism of interferon action. The interferon-induced phosphoprotein P1 possesses a double-stranded RNA-dependent ATP-binding site. J Biol Chem. 1985;260:8237–8239. [PubMed] [Google Scholar]
- 14.Galabru J, Hovanessian A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 15.Rivas C, Gil J, Esteban M. Identification of functional domains of the interferon-induced enzyme PKR in cells lacking endogenous PKR. J Interferon Cytokine Res. 1999;19:1229–1236. doi: 10.1089/107999099312885. [DOI] [PubMed] [Google Scholar]
- 16.Vattem KM, Staschke KA, Zhu S, Wek RC. Inhibitory sequences in the N-terminus of the double-stranded-RNA-dependent protein kinase, PKR, are important for regulating phosphorylation of eukaryotic initiation factor 2alpha (eIF2alpha) Eur J Biochem. 2001;268:1143–1153. doi: 10.1046/j.1432-1327.2001.01979.x. [DOI] [PubMed] [Google Scholar]
- 17.Saelens X, Kalai M, Vandenabeele P. Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-alpha phosphorylation. J Biol Chem. 2001;276:41620–41628. doi: 10.1074/jbc.M103674200. [DOI] [PubMed] [Google Scholar]
- 18.Zhu S, Romano PR, Wek RC. Ribosome targeting of PKR is mediated by two double-stranded RNA- binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2. J Biol Chem. 1997;272:14434–14441. doi: 10.1074/jbc.272.22.14434. [DOI] [PubMed] [Google Scholar]
- 19.Carpick BW, Graziano V, Schneider D, Maitra RK, Lee X, Williams BRG. Characterization of the solution complex between the interferon-induced double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J Biol Chem. 1997;272:9510–9516. doi: 10.1074/jbc.272.14.9510. [DOI] [PubMed] [Google Scholar]
- 20.Manche L, Green SR, Schmedt C, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kostura M, Mathews MB. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol Cell Biol. 1989;9:1576–1586. doi: 10.1128/mcb.9.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor DR, Lee SB, Romano PR, Marshak DR, Hinnebusch AG, Esteban M, Mathews MB. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol Cell Biol. 1996;16:6295–6302. doi: 10.1128/mcb.16.11.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor DR, Tian B, Romano PR, Hinnebusch AG, Lai MM, Mathews MB. Hepatitis C virus envelope protein E2 does not inhibit PKR by simple competition with autophosphorylation sites in the RNA-binding domain. J Virol. 2001;75:1265–1273. doi: 10.1128/JVI.75.3.1265-1273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, Hinnebusch AG. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol Cell Biol. 1998;18:2282–2297. doi: 10.1128/mcb.18.4.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem. 2001;276:24946–24958. doi: 10.1074/jbc.M102108200. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Herring CJ, Romano PR, Szczepanowska J, Brzeska H, Hinnebusch AG, Qin J. Identification of phosphorylation sites in proteins separated by polyacrylamide gel electrophoresis. Anal Chem. 1998;70:2050–2059. doi: 10.1021/ac971207m. [DOI] [PubMed] [Google Scholar]
- 27.Su Q, Wang S, Baltzis D, Qu LK, Wong AH, Koromilas AE. Tyrosine phosphorylation acts as a molecular switch to full-scale activation of the eIF2{alpha} RNA-dependent protein kinase. Proc Natl Acad Sci U S A. 2006;103:63–68. doi: 10.1073/pnas.0508207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galabru J, Katze MG, Robert N, Hovanessian AG. The binding of double-stranded RNA and adenovirus VAI RNA to the interferon-induced protein kinase. Eur J Biochem. 1989;178:581–589. doi: 10.1111/j.1432-1033.1989.tb14485.x. [DOI] [PubMed] [Google Scholar]
- 29.Lemaire PA, Lary J, Cole JL. Mechanism of PKR activation: dimerization and kinase activation in the absence of double-stranded RNA. J Mol Biol. 2005;345:81–90. doi: 10.1016/j.jmb.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 30.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 31.Johnson LN, Noble MEM, Owen DJ. Active and inactive protein kinases: structural basis of regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 32.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 33.Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chem Rev. 2001;101:2271–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- 34.Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 35.Ni Q, Shaffer J, Adams JA. Insights into nucleotide binding in protein kinase A using fluorescent adenosine derivatives. Protein Sci. 2000;9:1818–1827. doi: 10.1110/ps.9.9.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talavera MA, De La Cruz EM. Equilibrium and kinetic analysis of nucleotide binding to the DEAD-box RNA helicase DbpA. Biochemistry. 2005;44:959–970. doi: 10.1021/bi048253i. [DOI] [PubMed] [Google Scholar]
- 37.Barshop BA, Wrenn RF, Frieden C. Analysis of numerical methods for computer simulation of kinetic processes: development of KINSIM--a flexible, portable system. Anal Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 38.Anderson KS, Sikorski JA, Johnson KA. A tetrahedral intermediate in the EPSP synthase reaction observed by rapid quench kinetics. Biochemistry. 1988;27:7395–406. doi: 10.1021/bi00419a034. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Wang H, Erie DA. Quantitative characterization of biomolecular assemblies and interactions using atomic force microscopy. Methods. 2003;29:175–187. doi: 10.1016/s1046-2023(02)00308-0. [DOI] [PubMed] [Google Scholar]
- 40.Ratcliff GC, Erie DA. A novel single-molecule study to determine protein--protein association constants. J Am Chem Soc. 2001;123:5632–5635. doi: 10.1021/ja005750n. [DOI] [PubMed] [Google Scholar]
- 41.Robinson MJ, Harkins PC, Zhang J, Baer R, Haycock JW, Cobb MH, Goldsmith EJ. Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry. 1996;35:5641–5646. doi: 10.1021/bi952723e. [DOI] [PubMed] [Google Scholar]
- 42.Carrera AC, Alexandrov K, Roberts TM. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc Natl Acad Sci U S A. 1993;90:442–446. doi: 10.1073/pnas.90.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson KA. Transient state kinetic analysis of enzyme reaction pathways. In: Sigman DS, editor. The Enzymes. Academic Press; San Diego: 1992. pp. 1–61. [Google Scholar]
- 44.Dame RT, Wyman C, Goosen N. Insights into the regulation of transcription by scanning force microscopy. J Microsc. 2003;212:244–253. doi: 10.1111/j.1365-2818.2003.01243.x. [DOI] [PubMed] [Google Scholar]
- 45.Janicijevic A, Ristic D, Wyman C. The molecular machines of DNA repair: scanning force microscopy analysis of their architecture. J Microsc. 2003;212:264–272. doi: 10.1111/j.1365-2818.2003.01254.x. [DOI] [PubMed] [Google Scholar]
- 46.Xue Y, Ratcliff GC, Wang H, Davis-Searles PR, Gray MD, Erie DA, Redinbo MR. A minimal exonuclease domain of WRN forms a hexamer on DNA and possesses both 3′-5′ exonuclease and 5′-protruding strand endonuclease activities. Biochemistry. 2002;41:2901–2912. doi: 10.1021/bi0157161. [DOI] [PubMed] [Google Scholar]
- 47.Thomson NH, Smith BL, Almqvist N, Schmitt L, Kashlev M, Kool ET, Hansma PK. Oriented, active Escherichia coli RNA polymerase: an atomic force microscope study. Biophys J. 1999;76:1024–1033. doi: 10.1016/S0006-3495(99)77267-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasas S, Thomson NH, Smith BL, Hansma HG, Zhu X, Guthold M, Bustamante C, Kool ET, Kashlev M, Hansma PK. Escherichia coli RNA polymerase activity observed using atomic force microscopy. Biochemistry. 1997;36:461–468. doi: 10.1021/bi9624402. [DOI] [PubMed] [Google Scholar]
- 49.Wyman C, Rombel I, North AK, Bustamante C, Kustu S. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science. 1997;275:1658–1661. doi: 10.1126/science.275.5306.1658. [DOI] [PubMed] [Google Scholar]
- 50.Shiomi Y, Usukura J, Masamura Y, Takeyasu K, Nakayama Y, Obuse C, Yoshikawa H, Tsurimoto T. ATP-dependent structural change of the eukaryotic clamp-loader protein, replication factor C. Proc Natl Acad Sci U S A. 2000;97:14127–14132. doi: 10.1073/pnas.97.26.14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai R, Williams BR. Mutations in the double-stranded RNA-activated protein kinase insert region that uncouple catalysis from eIF2alpha binding. J Biol Chem. 1998;273:11274–11280. doi: 10.1074/jbc.273.18.11274. [DOI] [PubMed] [Google Scholar]
- 52.Adams JA. Activation loop phosphorylation and catalysis in protein kinases: is there functional evidence for the autoinhibitor model? Biochemistry. 2003;42:601–607. doi: 10.1021/bi020617o. [DOI] [PubMed] [Google Scholar]
- 53.Katze MG, Wambach M, Wong ML, Garfinkel M, Meurs E, Chong K, Williams BR, Hovanessian AG, Barber GN. Functional expression and RNA binding analysis of the interferon-induced, double-stranded RNA-activated, 68,000-Mr protein kinase in a cell-free system. Mol Cell Biol. 1991;11:5497–5505. doi: 10.1128/mcb.11.11.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iyer GH, Garrod S, Woods VL, Jr, Taylor SS. Catalytic independent functions of a protein kinase as revealed by a kinase-dead mutant: study of the Lys72His mutant of cAMP-dependent kinase. J Mol Biol. 2005;351:1110–1122. doi: 10.1016/j.jmb.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 55.Patel RC, Stanton P, Sen GC. Role of the amino-terminal residues of the interferon-induced protein kinase in its activation by double-stranded RNA and heparin. J Biol Chem. 1994;269:18593–18598. [PubMed] [Google Scholar]
- 56.Patel RC, Sen GC. Requirement of PKR dimerization mediated by specific hydrophobic residues for its activation by double-stranded RNA and its antigrowth effects in yeast. Mol Cell Biol. 1998;18:7009–7019. doi: 10.1128/mcb.18.12.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]