

Abstract
Sulindac is a commonly used nonsteroidal anti-inflammatory drug. This study tested the hypothesis that sulindac-mediated drug–drug interactions and/or hepatotoxicity may be caused, in part, by inhibition of proteins responsible for the hepatic transport of drugs and/or bile acids by sulindac and/or sulindac metabolites [sulindac sulfone (S-sulfone) and sulindac sulfide (S-sulfide)]. The uptake and excretion of model substrates, [3H]taurocholate (TC), [3H]estradiol 17-β-glucuronide (E217G), and nitrofurantoin (NF), were investigated in rat and human suspended and sandwich-cultured hepatocytes (SCH). In suspended rat hepatocytes, S-sulfone and S-sulfide inhibited Na+-dependent TC initial uptake (IC50 of 24.9 ± 6.4 and 12.5 ± 1.8 μM, respectively) and Na+-independent E217G initial uptake (IC50 of 12.1 ± 1.6 and 6.3 ± 0.3 μM, respectively). In rat SCH, sulindac metabolites (100 μM) decreased the in vitro biliary clearance (Clbiliary) of TC, E217G, and NF by 38 to 83%, 81 to 97%, and 33 to 57%, respectively; S-sulfone and S-sulfide also decreased the TC and NF biliary excretion index by 39 to 55%. In suspended human hepatocytes, S-sulfone and S-sulfide inhibited Na+-dependent TC initial uptake (IC50 of 42.2 and 3.1 μM, respectively); S-sulfide also inhibited the TC Clbiliary in human SCH. Sulindac/metabolites markedly inhibited hepatic uptake and biliary excretion of E217G by 51 to 100% in human SCH. In conclusion, sulindac and metabolites are potent inhibitors of the uptake and biliary clearance of bile acids in rat and human hepatocytes and also inhibit substrates of rat breast cancer resistance protein, rat and human organic anion-transporting polypeptides, and human multidrug resistance-associated protein 2. Inhibition of multiple hepatic transport proteins by sulindac/metabolites may play an important role in clinically significant sulindac-mediated drug–drug interactions and/or liver injury.
Sulindac is a nonsteroidal anti-inflammatory drug (NSAID) indicated for the relief of signs and symptoms of arthritic conditions, including osteoarthritis and rheumatoid arthritis. Sulindac is reduced by the aldehyde oxidase system to sulindac sulfide (S-sulfide) (Fig. 1), which has analgesic and anti-inflammatory properties. S-sulfide is converted by flavin-containing monooxygenase 3 back to sulindac and further to sulindac sulfone (S-sulfone), which has been recognized as a promising antiproliferative agent in colon cancer (Tatsumi et al., 1983; Davies and Morris, 1993; Hamman et al., 2000). Among NSAIDs, sulindac has been associated most often with hepatotoxicity (Aithal and Day, 2007), particularly with cholestatic hepatitis, and to a lesser extent with hepatocellular damage (Tarazi et al., 1993). Despite the high incidence of liver injury, the mechanisms underlying sulindac-mediated hepatotoxicity remain to be elucidated.
Fig. 1.

Chemical structures of sulindac (center), S-sulfone (right), and S-sulfide (left).
Inhibition of hepatic transport proteins has been proposed as one mechanism of drug-induced liver injury (DILI). Inhibition of the canalicular bile salt export pump (BSEP) in rats (ABCB11) by hepatotoxic drugs such as troglitazone and bosentan may lead to elevated hepatic concentrations of detergent-like bile acids, which can disrupt cellular function (Fattinger et al., 2001; Funk et al., 2001). Other hepatic transport proteins also are involved in bile acid transport, acting in concert to maintain homeostasis of bile acids. In humans, bile acids are taken up primarily by Na+-dependent taurocholate cotransporting polypeptide (NTCP; SLC10A1) and Na+-independent organic anion-transporting polypeptides (OATPs; SLCOs). Once taken up by hepatocytes, bile acids are excreted into the canalicular lumen primarily by BSEP. Alternatively, bile acids can be excreted into sinusoidal blood by basolateral hepatic transport proteins such as the multidrug resistance-associated protein 3 (MRP3; ABCC) 3, MRP4, and organic solute transporter α/β (Kosters and Karpen, 2008).
Some interactions between sulindac and hepatic transport proteins have been demonstrated previously. Sulindac inhibited organic anion transporter (OAT) 1-mediated [14C]para-aminohippurate uptake (IC50 ∼36 μM) and OAT3-mediated [3H]estrone sulfate uptake (IC50 ∼3 μM); sulindac also was the most potent inhibitor among tested NSAIDs of organic cation transporters 1 and 2 in stably transfected cells (Khamdang et al., 2002). Sulindac inhibited [3H]methotrexate transport in MRP2- and MRP4-overexpressing membrane vesicles (IC50 ∼38 and ∼2 μM, respectively) (El-Sheikh et al., 2007). MRP2 functions to excrete compounds from the hepatocyte into bile. In addition, S-sulfide was shown to be a potent inhibitor of MRP4-mediated leukotriene B4 and C4 transport in MRP4-overexpressing membrane vesicles (Rius et al., 2003). However, no studies demonstrating inhibition of transport by sulindac/metabolites in rat or human hepatocytes have been reported.
Sulindac is metabolized extensively by the liver in rats and humans, and plasma concentrations of sulindac metabolites are sustained because of extensive enterohepatic recycling (Dujovne et al., 1983). After intravenous administration of sulindac (10 mg/kg) in rats, the plasma area under the concentration-time curve (AUC) for S-sulfone was ∼2.5-fold higher than that of sulindac, whereas the AUC for S-sulfide was only ∼40% of that for sulindac (Duggan et al., 1978); Clbiliary of sulindac and S-sulfone were comparable, whereas S-sulfide Clbiliary was only 40% of that for sulindac. In humans, after oral administration of sulindac, peak plasma total concentrations (Cmax) for sulindac, S-sulfone, and S-sulfide were 10 to 34, 4 to 19, and 3 to 33 μM, respectively (Davies and Morris, 1993); the apparent Clbiliary of sulindac and S-sulfone, measured by an occludable T-tube that was placed in the common bile duct of patients after elective gallbladder surgery, was ∼25- and ∼18-fold greater, respectively, than that of S-sulfide (Dobrinska et al., 1983). Several groups have reported a proportional relationship between S-sulfide concentrations and the incidence of hepatotoxicity, emphasizing the potential for metabolites, in addition to the parent drug, to cause hepatotoxicity (Duggan et al., 1978; Laffi et al., 1986).
The present study was designed to examine the interaction of sulindac and metabolites with hepatic transport proteins in primary rat and human hepatocytes. To examine the effect of sulindac and metabolites on the function of hepatic transport proteins, taurocholate was used as a model substrate for Ntcp/NTCP and Bsep/BSEP, and estradiol 17-β-glucuronide (E217G) was used as a model substrate for Oatps/OATPs and Mrp2/MRP2 in rat and human hepatocytes, respectively. In addition, nitrofurantoin (NF) was used to examine the interaction between sulindac/metabolites and Bcrp.
Materials and Methods
Chemicals and Reagents.
[3H]TC, [3H]E217G, and [14C]inulin were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Dulbecco's modified Eagle's medium (DMEM) and minimum essential medium nonessential amino acids were purchased from Invitrogen (Carlsbad, CA). BioCoat culture plates, Matrigel extracellular matrix, and insulin/transferrin/selenium culture supplement were purchased from BD Biosciences Discovery Labware (Bedford, MA). NF, TC, E217G, sulindac, S-sulfone, S-sulfide, penicillin-streptomycin solution, dexamethasone, Hanks' balanced salt solution (HBSS), modified HBSS (HBSS without Ca2+ and Mg+), and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals and reagents were of analytical grade and were readily available from commercial sources.
Culture of Primary Rat and Human Hepatocytes in a Sandwich Configuration.
Primary rat hepatocytes were cultured as described previously (Lee et al., 2010). In brief, hepatocytes were seeded at a density of 1.75 × 106 cells/well onto six-well BioCoat plates. Approximately 24 h later, hepatocytes were overlaid with 2 ml of 0.25 mg/ml BD Matrigel basement membrane matrix in DMEM containing 1% (v/v) insulin/transferrin/selenium culture supplement, 0.1 μM dexamethasone, 2 mM l-glutamine, 1% (v/v) minimum essential medium nonessential amino acids, 100 units penicillin G sodium/ml, and 100 μg streptomycin sulfate/ml. CellzDirect (Durham, NC) kindly provided human hepatocytes cultured in six-well plates, seeded at a density of 1.5 × 106 cells/well, and overlaid with Matrigel. Hepatocytes were cultured for 4 days (rat) or 7 to 8 days (human) to allow extensive formation of canalicular networks between cells before experimentation; medium was changed daily.
Transport Studies Using Rat and Human Sandwich-Cultured Hepatocytes.
The method to determine substrate accumulation in sandwich-cultured hepatocytes (SCH) has been described previously (Liu et al., 1999). In brief, SCH were washed twice with 2 ml of warm HBSS containing Ca2+ (standard; cells + bile) or Ca2+-free HBSS (cells), followed by incubation in the same buffer for 10 min at 37°C. Subsequently, cells were incubated at 37°C for 10 min with 1.5 ml of standard HBSS containing NF (5 μM), [3H]TC (1 μM; 100 nCi/ml), or [3H]E217G (1 μM; 100 nCi/ml) in the presence of sulindac or its metabolites (0–100 μM). After 10 min, cells were washed three times with ice-cold standard HBSS. Cells were lysed with either 1 ml of methanol/water (70:30; v/v) (NF studies) or 0.5% (v/v) Triton X-100 in phosphate-buffered saline (TC and E217G studies), and plates were shaken for at least 20 min. Samples of cells + bile and cells were quantified by HPLC/MS/MS for NF and by liquid scintillation spectroscopy (Packard Tricarb; PerkinElmer Life and Analytical Sciences) for [3H]TC or [3H]E217G, and transport function was normalized to the protein content of the hepatocytes by using a BCA protein assay (Pierce Chemical, Rockford, IL). The biliary excretion index (BEI; %) and in vitro biliary clearance (Clbiliary; ml/min/kg) were calculated by using B-CLEAR technology (Qualyst, Inc., Research Triangle Park, NC) based on the following equations:
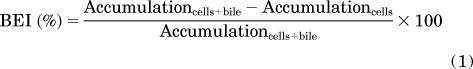 |
 |
where AUCmedium represents the area under the substrate concentration-time curve, which was determined by multiplying the initial substrate concentration in the incubation medium by the incubation time (10 min). Clbiliary values were converted to milliliter per minute per kilogram based on previously reported values for protein content in liver tissue (200 mg/g liver weight for rats; 160 mg/g liver weight for humans) and liver weight (40 g/kg body weight for rats; 25.7 g/kg body weight for humans) (Davies and Morris, 1993; Wilson et al., 2003).
HPLC/MS/MS Analysis of NF.
NF accumulation in cells + bile and cells was quantified by HPLC/MS/MS as described previously (Yue et al., 2009). In brief, proteins were precipitated in samples obtained from rat SCH by adding 1 ml of methanol/water (70:30, v/v), and samples were sonicated for 20 s. After centrifugation at 4°C (12,000g) for 3 min, the supernatant (20 μl) was diluted with 100 μl of methanol and water containing internal standard (ethyl warfarin). A Shimadzu (Columbia, MD) solvent delivery system and a LEAP Technologies (Carrboro, NC) HTC Pal thermostated autosampler connected to an Applied Biosystems (Foster City, CA) API 4000 triple quadruple mass spectrometer with a Turbo Spray ion source were used for analysis. NF and internal standards were eluted from an Aquasil C18 column (50 × 2.1 mm, dp = 5 μm; Thermo Fisher Scientific, Waltham, MA) using a mobile phase gradient (hold at 100% 10 mM ammonium acetate aqueous solution from 0 to 0.75 min; to 40% methanol from 0.75 to 1.39 min; to 90% methanol at 3.3 min; hold at 90% methanol until 4 min; equilibrated with 100% 10 mM ammonium acetate aqueous solution at 4 min; flow rate, 0.75 ml/min; 0.75 to 1.39 min directed to the mass spectrometer) and were detected in negative ion mode by using multiple reaction monitoring: NF, 236.8 → 151.8 m/z; ethyl warfarin, 320.8 → 160.9 m/z. Eight-point calibration curves (2–1000 nM) were applied, and all points on the curves were back-calculated to within ± 15% of the nominal value.
Transport Studies Using Suspended Primary Rat and Human Hepatocytes.
The initial uptake of [3H]TC and [3H]E217G was determined in freshly isolated hepatocytes, as described previously (Leslie et al., 2007). In brief, hepatocytes were washed twice with ice-cold standard HBSS containing 10 mM Tris and 5 mM glucose (Na+-containing condition) or Na+-free choline buffer. Hepatocytes were suspended at 1 × 106 cells/ml in the same buffer and used immediately in experiments. Hepatocytes (2 ml) in 16 × 100-mm test tubes were incubated at 37°C in an orbital shaking water bath for 3 min. Vehicle control (0.1% DMSO), S-sulfone, or S-sulfide (0.1–100 μM) was added, followed by [3H]TC (1 μM; 60 nCi/ml) or [3H]E217G (1 μM; 60 nCi/ml). At 30 s (TC) or 90 s (E217G), samples (200 μl) were placed into 0.4-ml polyethylene tubes prefilled with 3 M KOH (50 μl) layered with silicone oil/mineral oil (82:18, v/v; 100 μl) and immediately centrifuged. Radioactivity in the cell pellet and supernatant was quantified by liquid scintillation counting and corrected for the adherent fluid volume, which was determined by the incubation of cells with [14C]inulin (Brouwer et al., 1987). At the end of each experiment, an aliquot (10 μl) of suspended cells was used to determine protein concentrations by the BCA protein assay.
Determination of Inhibitory Potency of Sulindac Metabolites on Na+-Dependent [3H]TC Uptake or Na+-Independent [3H]E217G Uptake.
The data obtained from substrate transport studies ([3H]TC and [3H]E217G) in suspended hepatocytes were plotted against the inhibitor concentration (S-sulfone and S-sulfide). Based on standard model selection criteria including residual distribution, Akaike's information criterion, and visual inspection, the sigmoidal inhibitory Emax model was selected to estimate the concentration for 50% inhibition of uptake (IC50) by using WinNonlin 5.0.1 (Pharsight, Mountain View, CA).
where E is the rate of substrate uptake, Emax is the maximum rate of substrate uptake, C is the inhibitor concentration, and r is the curve shape factor.
Statistical Analysis.
Data are expressed as mean and the associated S.D. or S.E.M., as indicated. Statistical differences were determined by one-way analysis of variance (ANOVA) with Bonferroni's post hoc test using SigmaStat 2.03 (Aspire Software International, Ashburn, VA). Differences were considered significant at p < 0.05.
Results
Effect of Sulindac and Metabolites on TC Hepatobiliary Disposition in Rat SCH.
[3H]TC was used as a model substrate to examine the impact of sulindac and metabolites on the hepatic uptake and biliary excretion of bile acids in rat SCH (Fig. 2). The accumulation of TC (1 μM, 100 nCi/ml, 10 min) in cells + bile in the presence of 100 μM S-sulfide was significantly decreased compared with vehicle control. TC BEI in the presence of 1 and 10 μM sulindac, S-sulfone, and S-sulfide was not statistically different from those values in vehicle control. TC BEI was decreased significantly by 100 μM S-sulfone (42% decrease) and S-sulfide (55% decrease), but TC BEI in the presence of 100 μM sulindac was not different from those values in vehicle control. Sulindac (10 μM; 53% decrease), S-sulfone (100 μM; 76% decrease), and S-sulfide (100 μM; 84% decrease) significantly inhibited TC Clbiliary.
Fig. 2.

Effect of sulindac and metabolites on hepatobiliary disposition of TC in rat SCH. Accumulation of [3H]TC (1 μM, 100 nCi/ml, 10 min) in cells + bile (black bars) and cells (white bars) was measured in the presence of increasing concentrations (1–100 μM) of sulindac (A), S-sulfone (B), or S-sulfide (C) in rat SCH. BEI and in vitro Clbiliary of TC were calculated as described under Materials and Methods. Data are presented as the mean ± S.E.M (n = 3–4 livers in triplicate); ∗, statistically different from vehicle control (CTL) by one-way ANOVA with Bonferroni's post hoc test.
Effect of Sulindac and Metabolites on E217G Hepatobiliary Disposition in Rat SCH.
[3H]E217G was used as a model substrate to examine the impact of sulindac and metabolites on Oatp-mediated uptake and Mrp2-mediated biliary excretion in rat SCH (Fig. 3). Accumulation of E217G (1 μM, 100 nCi/ml, 10 min) in cells + bile and cells was decreased significantly in the presence of sulindac (100 μM), S-sulfone (10 and 100 μM), and S-sulfide (10 and 100 μM). E217G BEI was not affected by sulindac, S-sulfone, or S-sulfide (1–100 μM), whereas E217G Clbiliary was decreased significantly by S-sulfone (1, 10, and 100 μM) and S-sulfide (100 μM), but not by sulindac.
Fig. 3.

Effect of sulindac and metabolites on hepatobiliary disposition of E217G in rat SCH. Accumulation of [3H]E217G (1 μM, 100 nCi/ml, 10 min) in cells + bile (black bars) and cells (white bars) was measured in the presence of increasing concentrations (1–100 μM) of sulindac (A), S-sulfone (B), or S-sulfide (C) in rat SCH. BEI and in vitro Clbiliary of E217G were calculated as described under Materials and Methods. Data are presented as the mean ± S.E.M (n = 3–4 livers in triplicate); ∗, statistically different from vehicle control (CTL) by one-way ANOVA with Bonferroni's post hoc test.
Effect of Sulindac and Metabolites on NF Hepatobiliary Disposition in Rat SCH.
NF, a specific Bcrp substrate in rat SCH (Yue et al., 2009), was used as a model substrate to examine the impact of sulindac and metabolites on Bcrp-mediated biliary excretion in rat SCH (Fig. 4). NF accumulation (5 μM, 10 min) in cells + bile was not significantly different in the presence of 100 μM sulindac, S-sulfone, or S-sulfide, but cellular accumulation was increased significantly in the presence of S-sulfide. NF BEI was decreased by 30, 39, and 57% in the presence of 100 μM sulindac, S-sulfone, and S-sulfide, respectively, compared with vehicle control. Clbiliary of NF was decreased, with more potent inhibition by S-sulfide (50% decrease), followed by sulindac (33% decrease) and S-sulfone (33% decrease).
Fig. 4.

Effect of sulindac and metabolites on hepatobiliary disposition of NF in rat SCH. Accumulation of NF (5 μM, 10 min) in cells + bile (black bars) and cells (white bars) was measured in the presence of 100 μM sulindac, S-sulfone, or S-sulfide in rat SCH. BEI and in vitro Clbiliary of NF were calculated as described under Materials and Methods. Data are presented as the mean ± S.E.M (n = 3 livers in triplicate); ∗, statistically different from vehicle control (CTL) by one-way ANOVA with Bonferroni's post hoc test.
Influence of S-Sulfone and S-Sulfide on Na+-Dependent Initial Uptake of TC and Na+-Independent Initial Uptake of E217G in Suspended Rat Hepatocytes.
To further examine the mechanisms of inhibition of hepatic transport proteins by sulindac metabolites, the inhibitory potency of S-sulfone and S-sulfide on the initial uptake of TC in the absence and presence of Na+ was determined in suspended rat hepatocytes (Fig. 5, A and C, respectively). Initial uptake was determined at 30 s for TC (1 μM, 60 nCi/ml) based on preliminary studies (data not shown). The IC50 values for inhibition of Na+-dependent TC initial uptake by S-sulfone and S-sulfide were 24.9 ± 6.4 μM (Fig. 5B) and 12.5 ± 1.8 μM (Fig. 5D), respectively. E217G initial uptake was determined under Na+-free conditions (Oatp-mediated) to exclude Na+-dependent E217G uptake by Ntcp (Kouzuki et al., 1999). Na+-independent E217G initial uptake (1 μM, 60 nCi/ml, 90 s) was 114.8 ± 33.1 pmol/mg protein/min, which was comparable with previous data reported by Kemp et al. (2005). The IC50 values for inhibition of Na+-independent E217G initial uptake by S-sulfone and S-sulfide were 12.1 ± 1.6 μM (Fig. 6 A) and 6.3 ± 0.3 μM (Fig. 6B), respectively.
Fig. 5.

Inhibition of Na+-dependent TC initial uptake in suspended rat hepatocytes by S-sulfone (A and B) and S-sulfide (C and D). Na+-dependent [3H]TC initial uptake (1 μM, 60 nCi/ml, 30 s) was determined by subtracting [3H]TC uptake in Na+-free buffer (○) from [3H]TC uptake in Na+-containing buffer (●) in the presence of increasing concentrations of S-sulfone (0.5–100 μM; A) and S-sulfide (0.5–100 μM; C). Symbols and error bars represent the mean ± S.D. of n = 1–3 livers in duplicate. The dotted curve represents the best fit of the sigmoidal inhibitory Emax model to the data by using WinNonlin (B and D); the curve shape factor (r) was estimated as 0.31 ± 0.04 and 0.81 ± 0.09 for S-sulfone and S-sulfide, respectively.
Fig. 6.

Inhibition of Na+-independent E217G initial uptake in suspended rat hepatocytes by S-sulfone (A) and S-sulfide (B). Na+-independent [3H]E217G initial uptake (1 μM, 60 nCi/ml, 90 s) was determined in Na+-free buffer in the presence of increasing concentrations of S-sulfone (0.5–100 μM) and S-sulfide (0.5–100 μM). Symbols and error bars represent the mean ± S.D. of n = 3 livers in duplicate. The dotted curve represents the best fit of the sigmoidal inhibitory Emax model to the data by using WinNonlin; the curve shape factor (r) was estimated as 0.8 ± 0.2 and 1 ± 0.2 for S-sulfone and S-sulfide, respectively.
Effect of Sulindac and Metabolites on TC Hepatobiliary Disposition in Human SCH.
In human SCH, [3H]TC accumulation (1 μM, 100 nCi/ml, 10 min) in cells + bile in the presence of 10 and 100 μM sulindac and S-sulfone was comparable with vehicle control, but S-sulfide at 10 and 100 μM inhibited TC accumulation by 52 and 87%, respectively (Fig. 7). TC BEI was not decreased by sulindac, S-sulfone, or S-sulfide, but 10 and 100 μM S-sulfide decreased the Clbiliary of TC by 50 and 88%, respectively.
Fig. 7.

Effect of sulindac and metabolites on hepatobiliary disposition of TC in human SCH. Accumulation of [3H]TC (1 μM, 100 nCi/ml, 10 min) in cells + bile (black bars) and cells (white bars) was measured in the presence of 10 and 100 μM sulindac, S-sulfone, or S-sulfide in human SCH. BEI and in vitro Clbiliary of TC were calculated as described under Materials and Methods. Bars and error bars denote mean ± one-half the range from n = 2 livers in duplicate.
Influence of S-Sulfone and S-Sulfide on Na+-Dependent TC Initial Uptake in Suspended Human Hepatocytes.
Based on previously published data demonstrating that TC uptake was linear for at least 2 min in freshly isolated human hepatocytes (Shitara et al., 2003), the inhibitory potency of S-sulfone and S-sulfide on the initial uptake of [3H]TC (1 μM, 60 nCi/ml) in the absence and presence of Na+ was determined at 30 s in suspended human hepatocytes (Fig. 8, A and C, respectively). The IC50 for the inhibition of Na+-dependent TC initial uptake by S-sulfone and S-sulfide was 42.2 μM (Fig. 8B) and 3.1 μM (Fig. 8D), respectively.
Fig. 8.

Inhibition of Na+-dependent TC initial uptake in suspended human hepatocytes by S-sulfone (A and B) and S-sulfide (C and D). Na+-dependent [3H]TC initial uptake (1 μM, 60 nCi/ml, 30 s) was determined by subtracting [3H]TC uptake in Na+-free buffer (open symbols; n = 2 livers) from those values in Na+-containing buffer (closed symbols; n = 2 livers) in the presence of increasing concentrations of S-sulfone (0.5–300 μM; A) and S-sulfide (0.5–100 μM; C). X and + denote individual data from n = 2 livers in duplicate (B and D). The dotted curve represents the best fit of the sigmoidal inhibitory Emax model to the data by using WinNonlin; the curve shape factor (r) was estimated as 0.6 and 0.9 for S-sulfone and S-sulfide, respectively.
Effect of Sulindac and Metabolites on E217G Hepatobiliary Disposition in Human SCH.
In human SCH, [3H]E217G accumulation (1 μM, 100 nCi/ml, 10 min) in cells + bile and cells was markedly decreased with 100 μM sulindac and metabolites (68–85% decrease) (Fig. 9). Contrary to rat SCH, E217G BEI was decreased in the presence of sulindac (74% decrease), S-sulfone (100% decrease), and S-sulfide (51% decrease). The Clbiliary of E217G was decreased more than 85% by sulindac and metabolites in human SCH.
Fig. 9.

Effect of sulindac and metabolites on hepatobiliary disposition of E217G in human SCH. Accumulation of [3H]E217G (1 μM, 100 nCi/ml, 10 min) in cells + bile (solid bars) and cells (empty bars) was measured in the presence of 100 μM sulindac, S-sulfone, or S-sulfide in human SCH. BEI and in vitro Clbiliary of E217G were calculated as described under Materials and Methods. Bars and error bars denote mean ± half the range from n = 2 livers in duplicate.
Discussion
Among the NSAIDs, sulindac has been associated with the highest incidence of DILI. One potential mechanism of DILI may involve interference with hepatic transport protein function. The present study was designed to examine interactions between sulindac/metabolites and hepatic transport proteins by using TC, E217G, and NF as model substrates, respectively, for Ntcp-/Bsep-, Oatp-/Mrp2-, and Bcrp-mediated transport, respectively, in primary rat and human hepatocytes. BEI and Clbiliary from SCH were used as indices of hepatobiliary disposition of each model substrate; BEI is a measure of net canalicular efflux, whereas Clbiliary encompasses both uptake and efflux.
Both the BEI and Clbiliary of TC in rat SCH were decreased significantly by S-sulfone and S-sulfide (100 μM) (Fig. 2, B and C). Moreover, both metabolites inhibited Na+-dependent TC initial uptake in suspended rat hepatocytes (IC50 of 24.9 and 12.5 μM, respectively). Collectively, these data indicate that S-sulfone and S-sulfide inhibit Ntcp and Bsep, as depicted in Fig. 10. The balance between bile acid uptake and excretion in hepatocytes may influence the occurrence of DILI. Indeed, the preferential inhibition of rat Ntcp compared with human NTCP by bosentan may protect rats from hepatotoxicity by preventing further uptake of bile acids from blood when hepatocyte bile acid concentrations are increased because of bosentan-mediated Bsep/BSEP inhibition (Leslie et al., 2007).
Fig. 10.

A diagram of the proposed inhibition of transport proteins by sulindac and metabolites in hepatocytes. Sulindac and metabolites inhibited multiple hepatic transport proteins including Ntcp/NTCP, Oatps/OATPs, Bsep, Bcrp, and MRP2 in rat and/or human hepatocytes. Inhibition of transport proteins by sulindac and metabolites are depicted by thick lines; dashed lines indicate the marginal inhibition profiles of transport proteins by sulindac and metabolites. Impaired function of multiple hepatic transporters by sulindac/metabolites may disturb bile acid homeostasis and affect hepatobiliary disposition of coadministered drugs. Thus, when sulindac is coadministered with other drugs/xenobiotics, the possibility of drug interactions should be considered. N.D., not determined; N.S., not significant.
In rats, E217G is taken up by multiple Oatps, whereas biliary excretion of E217G is governed by Mrp2. The decreased hepatocellular accumulation of E217G in the presence of S-sulfone and S-sulfide in rat SCH, combined with the inhibition of E217G Clbiliary but not BEI, suggested that sulindac metabolites inhibited Oatp-mediated uptake but not Mrp2-mediated biliary excretion of E217G (Fig. 3, B and C). Subsequent investigations in suspended rat hepatocytes revealed that S-sulfone and S-sulfide were potent inhibitors (IC50 of 12.1 and 6.3 μM, respectively) of E217G initial uptake, consistent with the inhibition of Oatp 1a1 and 1a4 (Sugiyama et al., 2001).
NF was chosen as a model substrate for Bcrp-mediated transport based on recent data demonstrating that NF is a specific Bcrp substrate in rat SCH (Yue et al., 2009). Several clinical studies have reported that genetic polymorphisms in BCRP (ABCG2) can influence drug disposition. For example, the ABCG2 C421A polymorphism is associated with decreased protein expression and function in vitro (Imai et al., 2002) and increased plasma concentrations of several BCRP substrates, including rosuvastatin, diflomotecan, and sulfasalazine (Morisaki et al., 2005; Zhang et al., 2006; Yamasaki et al., 2008). In the present study, sulindac/metabolites significantly decreased NF BEI and Clbiliary in rat SCH, consistent with the inhibition of Bcrp (Fig. 4), suggesting the potential for BCRP-mediated interactions when sulindac is administered with other drugs/xenobiotics. Investigations are underway to determine whether sulindac and metabolites alter human BCRP-mediated transport.
TC was tested further in human SCH to examine whether sulindac/metabolites also inhibited the hepatic transport of TC. In human SCH, TC accumulation in cells + bile and the TC Clbiliary were decreased markedly by S-sulfide without affecting TC BEI. These findings may reflect, at least in part, S-sulfide-mediated inhibition of NTCP. The IC50 (3.1 μM) of Na+-dependent TC initial uptake by S-sulfide determined in human hepatocyte suspensions, which do not contain serum, was near the unbound plasma concentration range of S-sulfide (∼0.14–1.5 μM) based on reported Cmax values of 3 to 33 μM and an unbound fraction (fu) of 4.6% after oral administration of a therapeutic dose of sulindac (Davies and Morris, 1993). However, the IC50 (42.2 μM) of Na+-dependent TC initial uptake by S-sulfone was ∼100-fold greater than the unbound plasma concentration range (∼0.1–0.4 μM) of S-sulfone (Cmax, 4–19 μM; fu, 2.1%). Although the IC50 for inhibition of Na+-dependent TC initial uptake by S-sulfone was higher than unbound plasma concentrations, it is probable that the initial inhibitor concentrations in the portal vein when sulindac is administered orally are higher than unbound plasma concentrations, because ∼85% of total hepatic blood flow is composed of portal venous blood, which contains drug absorbed directly from the gastrointestinal tract (Ito et al., 1998). In addition, it is plausible that the extensive enterohepatic recycling of sulindac and metabolites may result in prolonged inhibition of hepatic transport proteins caused by much higher concentrations in the portal circulation (Dujovne et al., 1983).
The Food and Drug Administration has recommended an in vivo evaluation of drug interactions when the estimated [I]/Ki ratio is more than 0.1, where [I] represents the mean steady-state Cmax value for total drug (bound plus unbound) after administration of the highest proposed clinical dose (Huang et al., 2008). In theory, the IC50 values determined in the present study would be identical to Ki values, regardless of the mechanism of inhibition, based on previous data demonstrating the relationships between the Ki and IC50 values (Cheng and Prusoff, 1973); that is, the Ki value is equal to the IC50 value under conditions of either noncompetitive or uncompetitive inhibition, whereas under competitive inhibition conditions, the IC50 value will be identical to Ki(1 + S/Km), where S is the substrate concentration and Km is the substrate concentration corresponding to one-half of the maximal velocity of the reaction. Because the TC concentration (1 μM) used for the IC50 determination in the present study was lower than the previously reported Km value for the uptake of TC in rat (18 μM) and human (4.8 μM) hepatocytes, the Ki values would be equal to or less than the IC50 value (Kouzuki et al., 1998; Shitara et al., 2003). With this assumption, the [I]/Ki ratio determined by using data from human hepatocytes ranged from 0.9 to 10.6 for S-sulfide and 0.1 to 0.4 for S-sulfone, based on Cmax values of 3 to 33 and 4 to 19 μM, respectively (Davies and Morris, 1993). These findings suggest that S-sulfide and S-sulfone may interact with NTCP-mediated transport of bile acids and/or drugs in humans.
In human SCH, the hepatic uptake of E217G was decreased markedly in the presence of 100 μM sulindac and metabolites, suggesting a potential interaction between sulindac/metabolites and other drugs that are transported by OATPs (OATP1B1 and OATP1B3). Because of the limited availability of human liver tissue, the IC50 of E217G initial uptake by sulindac/metabolites was not determined. It is noteworthy that, contrary to rat SCH, sulindac and its metabolites markedly decreased the biliary excretion index of E217G (a substrate for MRP2) in human SCH, consistent with previous data demonstrating inhibition of MRP2-mediated methotrexate transport by sulindac in MRP2-overexpressing membrane vesicles (El-Sheikh et al., 2007). This discrepancy may be caused by species differences in canalicular transport mechanisms between rats and humans, which requires further characterization (Li et al., 2009).
In conclusion, the present studies demonstrated that multiple hepatic transport proteins (Ntcp/NTCP, Oatp/OATP, Bsep, Bcrp, or MRP2) are inhibited by sulindac and metabolites. Impaired hepatic transport of bile acids by sulindac/metabolites may disturb bile acid homeostasis, resulting in sulindac-mediated liver injury. Furthermore, interactions between sulindac/metabolites and drugs that are substrates for these transport proteins may result in clinically significant interactions. For example, Furst et al. (1990) demonstrated that oral administration of sulindac decreased methotrexate clearance in patients with rheumatoid arthritis. Considering that methotrexate is taken up by OATP1B1 and OATP1B3 and excreted by MRP2 in the human liver, the interaction between sulindac and methotrexate may be caused by the sulindac-mediated inhibition of these transport proteins, as depicted in Fig. 10. The possibility of hepatic transporter-mediated interactions should be considered when sulindac is coadministered with other drugs/xenobiotics.
This work was supported by the National Institutes of Health [Grant R01-GM41935] and pilot funding by the North Carolina Translational and Clinical Sciences Institute provided by the National Institutes of Health National Center for Research Resources, [Award UL1RR025747].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.165852.
- S-sulfone
- sulindac sulfone
- S-sulfide
- sulindac sulfide
- NSAID
- nonsteroidal anti-inflammatory drug
- TC
- taurocholate
- E217G
- estradiol 17-|gb-glucuronide
- NF
- nitrofurantoin
- SCH
- sandwich-cultured hepatocytes
- BSEP
- bile salt export pump
- NTCP
- Na+-dependent taurocholate cotransporting polypeptide
- OAT
- organic anion transporter
- OATP
- organic anion-transporting polypeptide
- MRP
- multidrug resistance-associated protein
- DILI
- drug-induced liver injury
- BEI
- biliary excretion index
- ANOVA
- analysis of variance
- HBSS
- Hanks' balanced salt solution
- DMEM
- Dulbecco's modified Eagle's medium
- AUC
- area under the concentration-time curve
- BCRP
- breast cancer resistance protein
- Clbiliary
- biliary clearance
- HPLC
- high-performance liquid chromatography
- MS/MS
- tandem mass spectroscopy
- Cmax
- peak plasma total concentration.
References
- Aithal GP, Day CP. (2007) Nonsteroidal anti-inflammatory drug-induced hepatotoxicity. Clin Liver Dis 11:563–575, vi–vii [DOI] [PubMed] [Google Scholar]
- Brouwer KLR, Durham S, Vore M. (1987) Multiple carriers for uptake of [3H]estradiol-17 β(β-d-glucuronide) in isolated rat hepatocytes. Mol Pharmacol 32:519–523 [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108 [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095 [DOI] [PubMed] [Google Scholar]
- Dobrinska MR, Furst DE, Spiegel T, Vincek WC, Tompkins R, Duggan DE, Davies RO, Paulus HE. (1983) Biliary secretion of sulindac and metabolites in man. Biopharm Drug Dispos 4:347–358 [DOI] [PubMed] [Google Scholar]
- Duggan DE, Hooke KF, Noll RM, Hucker HB, Van Arman CG. (1978) Comparative disposition of sulindac and metabolites in five species. Biochem Pharmacol 27:2311–2320 [DOI] [PubMed] [Google Scholar]
- Dujovne CA, Pitterman A, Vincek WC, Dobrinska MR, Davies RO, Duggan DE. (1983) Enterohepatic circulation of sulindac and metabolites. Clin Pharmacol Ther 33:172–177 [DOI] [PubMed] [Google Scholar]
- El-Sheikh AA, van den Heuvel JJ, Koenderink JB, Russel FG. (2007) Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J Pharmacol Exp Ther 320:229–235 [DOI] [PubMed] [Google Scholar]
- Fattinger K, Funk C, Pantze M, Weber C, Reichen J, Stieger B, Meier PJ. (2001) The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 69:223–231 [DOI] [PubMed] [Google Scholar]
- Funk C, Pantze M, Jehle L, Ponelle C, Scheuermann G, Lazendic M, Gasser R. (2001) Troglitazone-induced intrahepatic cholestasis by an interference with the hepatobiliary export of bile acids in male and female rats. Correlation with the gender difference in troglitazone sulfate formation and the inhibition of the canalicular bile salt export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology 167:83–98 [DOI] [PubMed] [Google Scholar]
- Furst DE, Herman RA, Koehnke R, Ericksen N, Hash L, Riggs CE, Porras A, Veng-Pedersen P. (1990) Effect of aspirin and sulindac on methotrexate clearance. J Pharm Sci 79:782–786 [DOI] [PubMed] [Google Scholar]
- Hamman MA, Haehner-Daniels BD, Wrighton SA, Rettie AE, Hall SD. (2000) Stereoselective sulfoxidation of sulindac sulfide by flavin-containing monooxygenases. Comparison of human liver and kidney microsomes and mammalian enzymes. Biochem Pharmacol 60:7–17 [DOI] [PubMed] [Google Scholar]
- Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, Habet SA, Baweja RK, Burckart GJ, et al. (2008) New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol 48:662–670 [DOI] [PubMed] [Google Scholar]
- Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T, Miki Y, Sugimoto Y. (2002) C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther 1:611–616 [PubMed] [Google Scholar]
- Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. (1998) Prediction of pharmacokinetic alterations caused by drug–drug interactions: metabolic interaction in the liver. Pharmacol Rev 50:387–412 [PubMed] [Google Scholar]
- Kemp DC, Zamek-Gliszczynski MJ, Brouwer KLR. (2005) Xenobiotics inhibit hepatic uptake and biliary excretion of taurocholate in rat hepatocytes. Toxicol Sci 83:207–214 [DOI] [PubMed] [Google Scholar]
- Khamdang S, Takeda M, Noshiro R, Narikawa S, Enomoto A, Anzai N, Piyachaturawat P, Endou H. (2002) Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J Pharmacol Exp Ther 303:534–539 [DOI] [PubMed] [Google Scholar]
- Kosters A, Karpen SJ. (2008) Bile acid transporters in health and disease. Xenobiotica 38:1043–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzuki H, Suzuki H, Ito K, Ohashi R, Sugiyama Y. (1998) Contribution of sodium taurocholate cotransporting polypeptide to the uptake of its possible substrates into rat hepatocytes. J Pharmacol Exp Ther 286:1043–1050 [PubMed] [Google Scholar]
- Kouzuki H, Suzuki H, Ito K, Ohashi R, Sugiyama Y. (1999) Contribution of organic anion transporting polypeptide to uptake of its possible substrates into rat hepatocytes. J Pharmacol Exp Ther 288:627–634 [PubMed] [Google Scholar]
- Laffi G, La Villa G, Pinzani M, Ciabattoni G, Patrignani P, Mannelli M, Cominelli F, Gentilini P. (1986) Altered renal and platelet arachidonic acid metabolism in cirrhosis. Gastroenterology 90:274–282 [DOI] [PubMed] [Google Scholar]
- Lee JK, Marion TL, Abe K, Lim C, Pollock GM, Brouwer KLR. (2010) Hepatobiliary disposition of troglitazone and metabolites in rat and human sandwich-cultured hepatocytes: use of Monte Carlo simulations to assess the impact of changes in biliary excretion on troglitazone sulfate accumulation. J Pharmacol Exp Ther 332:26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EM, Watkins PB, Kim RB, Brouwer KLR. (2007) Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1) by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther 321:1170–1178 [DOI] [PubMed] [Google Scholar]
- Li N, Bi YA, Duignan DB, Lai Y. (2009) Quantitative expression profile of hepatobiliary transporters in sandwich cultured rat and human hepatocytes. Mol Pharm 6:1180–1189 [DOI] [PubMed] [Google Scholar]
- Liu X, LeCluyse EL, Brouwer KR, Lightfoot RM, Lee JI, Brouwer KLR. (1999) Use of Ca2+ modulation to evaluate biliary excretion in sandwich-cultured rat hepatocytes. J Pharmacol Exp Ther 289:1592–1599 [PubMed] [Google Scholar]
- Morisaki K, Robey RW, Ozvegy-Laczka C, Honjo Y, Polgar O, Steadman K, Sarkadi B, Bates SE. (2005) Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol 56:161–172 [DOI] [PubMed] [Google Scholar]
- Rius M, Nies AT, Hummel-Eisenbeiss J, Jedlitschky G, Keppler D. (2003) Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology 38:374–384 [DOI] [PubMed] [Google Scholar]
- Shitara Y, Li AP, Kato Y, Lu C, Ito K, Itoh T, Sugiyama Y. (2003) Function of uptake transporters for taurocholate and estradiol 17β-d-glucuronide in cryopreserved human hepatocytes. Drug Metab Pharmacokinet 18:33–41 [DOI] [PubMed] [Google Scholar]
- Sugiyama D, Kusuhara H, Shitara Y, Abe T, Meier PJ, Sekine T, Endou H, Suzuki H, Sugiyama Y. (2001) Characterization of the efflux transport of 17β-estradiol-d-17β-glucuronide from the brain across the blood-brain barrier. J Pharmacol Exp Ther 298:316–322 [PubMed] [Google Scholar]
- Tarazi EM, Harter JG, Zimmerman HJ, Ishak KG, Eaton RA. (1993) Sulindac-associated hepatic injury: analysis of 91 cases reported to the Food and Drug Administration. Gastroenterology 104:569–574 [DOI] [PubMed] [Google Scholar]
- Tatsumi K, Kitamura S, Yamada H. (1983) Sulfoxide reductase activity of liver aldehyde oxidase. Biochim Biophys Acta 747:86–92 [DOI] [PubMed] [Google Scholar]
- Wilson ZE, Rostami-Hodjegan A, Burn JL, Tooley A, Boyle J, Ellis SW, Tucker GT. (2003) Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br J Clin Pharmacol 56:433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki Y, Ieiri I, Kusuhara H, Sasaki T, Kimura M, Tabuchi H, Ando Y, Irie S, Ware J, Nakai Y, et al. (2008) Pharmacogenetic characterization of sulfasalazine disposition based on NAT2 and ABCG2 (BCRP) gene polymorphisms in humans. Clin Pharmacol Ther 84:95–103 [DOI] [PubMed] [Google Scholar]
- Yue W, Abe K, Brouwer KLR. (2009) Knocking down breast cancer resistance protein (Bcrp) by adenoviral vector-mediated RNA interference (RNAi) in sandwich-cultured rat hepatocytes: a novel tool to assess the contribution of Bcrp to drug biliary excretion. Mol Pharm 6:134–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yu BN, He YJ, Fan L, Li Q, Liu ZQ, Wang A, Liu YL, Tan ZR, Fen J, et al. (2006) Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta 373:99–103 [DOI] [PubMed] [Google Scholar]