

Abstract
Approximately10% of patients with amyotrophic lateral sclerosis (ALS) have familial ALS (FALS), and 20% of FALS is caused by mutant Cu/Zn superoxide dismutase type 1 (MTSOD1). Previous studies have convincingly demonstrated that MTSOD1 expression in other cell types besides motor neurons (MNs) contributes to disease in MTSOD1 FALS transgenic mice. Using Cre/LoxP methods, we knocked down G85R SOD1 mRNA by 66% in all cell types in 3-month-old FALS transgenic mice, delaying disease onset and lengthening disease duration. Surprisingly, the effect on onset and early disease duration was similar to that seen with ~25% knockdown prenatally in G85R SOD1 mRNA restricted to MNs and some interneurons in FALS transgenic mice. These results demonstrate no clear cumulative effect on disease onset or early disease duration from knocking down G85R SOD1 in other cell types in addition to MNs/interneurons; the findings bring up the possibility that MTSOD1 has a pathogenic effect early in life that our later knockdown did not affect. Despite the more limited amelioration of disease than expected, the effect of the knockdown on disease supports the value of this approach in FALS patients and asymptomatic individuals with SOD1 mutations.
Keywords: familial amyotrophic lateral sclerosis (FALS), ALS, superoxide dismutase type 1 (SOD1), motor neuron, neurodegeneration
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the selective loss of motor neurons (MNs). Approximately 10% of ALS cases are FALS, and ~20% of FALS cases are caused by mutant Cu/Zn superoxide dismutase type 1 (MTSOD1) (reviewed in (Cleveland & Rothstein 2001)). There is convincing evidence that MTSOD1 kills MNs because of a toxicity rather than a deficiency in dismutase activity. Of note, MTSOD1s that have full superoxide dismutase activity (such as G37R and G93A) as well as no activity (such as G85R) cause FALS. However, the basis for the toxicity of MTSOD1 remains unclear. Of interest, recent data suggest that sporadic ALS may also involve abnormalities in the SOD1 protein or its activity (Ezzi et al. 2007, Gruzman et al. 2007). Therefore, investigations of MTSOD1 may not only clarify our understanding of the pathogenesis and treatment of FALS, but also increase our understanding and treatment of the sporadic disease.
A number of mechanisms have been proposed by which MTSOD1 leads to MN cell death, including the following: misfolding of the MT protein; interference with axonal transport or cytoplasmic trafficking; altered oxidative activity of the mutant enzyme; a defect in the EAAT2 glutamate transporter leading to excitotoxicity (Cleveland & Rothstein 2001). Investigators have emphasized the importance of the misfolding of the MTSOD1 enzyme in disease pathogenesis, hypothesizing that misfolded MTSOD1 causes disease by sequestering proteins that normally bind SOD1 and are important for the function and viability of MNs. A role for misfolding of MT proteins, with sequestration of cell type-specific proteins, has also been proposed as key to the pathogenesis of a number of other neurodegenerative diseases.
ALS is a desperate disease with no effective treatment. Most patients will die 3-5 years after the onset. This prognosis is even more grim in the case of FALS caused by certain MTSOD1s. For example, patients with the A4V SOD1 mutation, the most common mutation in North America, usually die in ~ a year following the onset of symptoms (Rosen et al. 1994). One potential treatment of MTSOD1-induced FALS is to knock down the expression of MTSOD1. Data from a number of studies, including the following, suggest that this ameliorates disease in MTSOD1 FALS transgenic mice: immunization with MTSOD1 and administration of monoclonal antibodies specifically directed against MTSOD1 prolonged survival (Urushitani et al. 2007); delivery of antisense oligonucleotides to SOD1 slowed disease progression (Smith et al. 2006); delivery of siRNA directed against MTSOD1 delayed disease onset (Ralph et al. 2005); knockdown of MTSOD1 in a particular cell type by means of Cre/LoxP methods delayed onset and/or extended the duration of disease (Boillee et al. 2006a, Boillee et al. 2006b, Yamanaka et al. 2008, Wang et al. 2009b).
Studies using a regulatable system to produce a conditional model of Huntington’s disease found that blockage of expression led to a disappearance of inclusions and amelioration of the behavioral phenotype, suggesting that disease pathology and symptoms require a continuous influx of the mutant protein (Yamamoto et al. 2000). In the present study, we used a regulatable system to shut-off G85R SOD1 expression in all cells in a conditional model of G85R SOD1-induced FALS.
Materials and methods
Preparation of G85Rflox/Cre-ER™ double transgenic mice
G85Rflox mice (Wang et al. 2009a), which carry G85R genomic sequence flanked by LoxP sites, were crossed with Cre-ER™ mice (Jackson Lab) (Hayashi & McMahon 2002), which carry a tamoxifen-inducible Cre driven by the chicken beta actin promoter/enhancer coupled with the CMV immediate-early enhancer. G85Rflox/Cre-ER™ double transgenic mice were identified by PCR using previously published primers for detection of the human SOD1 gene (Wang et al. 2009a), and the following primers for detection of the Cre transgene: forward primer: 5′-GCG GTC TGG CAG TAA AAA CTA TC-3′ and reverse primer: 5′-GTG AAA CAG CAT TGC TGT CAC TT-3′.
Tamoxifen treatment and G85R knockdown
In order to induce Cre recombinase, 1mg of tamoxifen (Sigma) /10g body weight was administered intraperitoneally to 3-month-old double transgenic mice (~7 months before disease onset) over 5 consecutive days (Doerflinger et al. 2003). To confirm tamoxifen-inducibility of Cre in Cre-ER™ mice, Cre-ER™ mice were crossed with Cre-reporter mice (Rosa26-LoxSTOPLox-LacZ mice) which activate expression of β-galactosidase following Cre-mediated DNA recombination (Soriano 1999) (Jackson Labs). One month after tamoxifen treatment, the mice were sacrificed; β-galactosidase staining was carried out on the spinal cord as previously described (Lu et al. 2003).
In order to determine the amount of knockdown of human SOD1 mRNA in MNs, we performed laser capture microdissection (LCM) followed by real time polymerase chain reaction (qPCR). Approximately 1000 MNs were collected from the cervical spinal cord of each of 4 mice from each of the following three groups: tamoxifen-treated G85Rflox/Cre-ER™ mice, untreated G85Rflox/Cre-ER™ mice and G85Rflox mice. qPCR was performed as previously described (Wang et al. 2009b). We also assessed the amount of knockdown of human SOD1 mRNA in the whole spinal cord by dissecting the L4, 5 segment of the lumbar spinal cord followed by qPCR. These studies were performed on tissues from the same 4 mice of each group that was used for the MN qPCR described above. Human SOD1 protein levels were assessed on Western blots of homogenates of the remainder of the lumbar spinal cord from the same mice. For the protein studies, spinal cords were homogenized in a buffer containing 50mM HEPES, 1mM EDTA, 100mM iodoacetamide and 2.5% sodium dodecyl sulfate. After centrifugation, the supernatants were subjected to SDS-PAGE and then immunoblotted as previously described (Wang et al. 2009a), using a human specific anti-SOD1 antibody (Deng et al. 2006) and anti-β-tubulin antibody (to make certain that similar amounts of protein in each sample were applied) (Developmental Studies Hybridoma Bank). The amount of knockdown of the human SOD1 protein was determined by quantitating band intensities through densitometric analysis using Image J (National Institute of Mental Health, Bethesda, MD, USA).
Assessment of disease phenotype
Clinical assessment
Mice were weighed every two days and clinically assessed as previously described (Boillee et al. 2006b): the onset of disease was defined as peak weight before a decline; early phase of disease was the period from peak weight until loss of 10% of maximal weight; late phase of disease was the time from 10% loss in weight until death (when a mouse was unable to right itself within 20 seconds after being put on its back). In experiments monitoring disease parameters and survival, the tamoxifen-treated G85Rflox/Cre-ER™ mice were compared with G85Rflox mice as well untreated G85Rflox/Cre-ER™ mice.
Pathology
The three groups of mice that were evaluated clinically were also compared with respect to pathology using previously described methods (Wang et al. 2002, Wang et al. 2008, Wang et al. 2009b).
Immunohistochemical evaluation
SOD1 aggregation was assessed by immunohistochemical staining as previously described (Wang et al. 2008, Wang et al. 2009a), using a rabbit antibody that recognizes the carboxyl end of mouse and human SOD1(Deng et al. 2006). Nissl staining and glial fibrillary acidic protein (GFAP) antibody staining have been described (Wang et al. 2009a, Wang et al. 2009b).
Results
The preparation and tamoxifen treatment of G85Rflox/Cre-ER™ double transgenic mice
In order to knock down G85R SOD1, we first generated G85Rflox/Cre-ER™ double transgenic mice by crossing G85Rflox transgenic mice (Wang et al. 2009a) with Cre-ER™ mice (Hayashi & McMahon 2002) (Jackson Lab), which carry a tamoxifen-inducible Cre driven by the chicken beta actin promoter/enhancer coupled with the CMV immediate-early enhancer (Fig. 1). Previous studies had assessed the expression pattern and effectiveness of the Cre-ER™ conditional system by tamoxifen treating mice that carried Cre-ER™ as well as a reporter gene (Rosa26-LoxSTOPLox-LacZ (Soriano 1999)) that activates expression of β-galactosidase following Cre-mediated DNA recombination; these studies showed β-galactosidase staining in a broad range of cell types in many tissues (Hayashi & McMahon 2002). We extended these studies, and showed that tamoxifen-treated Rosa26-LoxSTOPLox-LacZ/Cre-ER™ double transgenic mice demonstrate β-galactosidase staining in multiple cell types in the anterior (Fig. 2B) and posterior (Fig. 2D) horn of the spinal cord; β-galactosidase staining of MNs is apparent in sections of the anterior horn (Fig. 2B). No β-galactosidase staining was seen in untreated Rosa26-LoxSTOPLox-LacZ/Cre-ER™ transgenic mice (Fig. 2A, C).
Figure 1.

Diagram depicting the generation of G85Rflox/Cre-ER™ mice and knockdown of G85R following tamoxifen treatment.
Figure 2.

Conditional expression of Cre in Cre-ER™ mice. Cre-ER™ mice were crossed with Rosa26-LoxSTOPLox-LacZ mice (which carry a reporter gene that activates expression of β-galactosidase following Cre-mediated DNA recombination). Rosa26-LoxSTOPLox-LacZ/Cre-ER™ mice were treated with tamoxifen at 3 months of age and sacrificed one month later. A section from the spinal cord anterior horn of an untreated (A) and tamoxifen-treated (B) Rosa26-LoxSTOPLox-LacZ/Cre-ER™ mouse was stained with β-galactosidase and then counterstained with eosin. The spinal cord anterior horn of tamoxifen-treated mice shows β-galactosidase staining of MNs as well as other cells. A section of the spinal cord posterior horn of an untreated (C) and tamoxifen-treated (D) Rosa26-LoxSTOPLox-LacZ/Cre-ER™ mouse was stained with β-galactosidase and counterstained with eosin. The posterior horn of tamoxifen-treated mice shows β-galactosidase staining of multiple cell types. Scale bar = 50μm.
In order to quantitate the knockdown of G85R SOD1 mRNA in MNs, we used LCM followed qPCR and compared the amount of G85R SOD1 mRNA in MNs of the cervical spinal cord from 4 G85Rflox/Cre-ER™ mice that were treated with tamoxifen at 3 months of age and sacrificed at 4 months of age with 4 untreated G85Rflox/Cre-ER™ and 4 G85Rflox transgenic mice (Fig. 3A). There was no significant decrease in the G85R SOD1 mRNA level when untreated G85Rflox/Cre-ER™ mice were compared to G85Rflox mice (P=0.17), demonstrating that there was no significant leakiness of Cre in untreated mice. We found a decrease of 59.5% (P=0.008) in G85R mRNA levels in the spinal cord MNs of tamoxifen-treated G85Rflox/Cre-ER™ mice compared to G85Rflox mice (Fig. 3A). In addition, we examined the knockdown of MT SOD1 mRNA as well as protein level in all cell types of the lumbar spinal cord from the same mice that were used for the MN qPCR (Fig. 3B, C). Again, there was no significant decrease in the G85R SOD1 mRNA level when untreated G85Rflox/Cre-ER™ mice were compared to G85Rflox mice (P=0.88) (Fig. 3B). There was a decrease of 65.9% (P<0.01) in G85R mRNA levels in the spinal cord of tamoxifen-treated G85Rflox/Cre-ER™ mice compared to G85Rflox mice (Fig. 3B). Quantitation of the knockdown of human SOD1 protein levels was determined by Western blots of homogenates of the lumbar spinal cord from the same mice (Fig. 3C). There was no significant decrease in the G85R protein level when untreated G85Rflox/Cre-ER™ mice were compared to G85Rflox mice. There was a decrease of human SOD1 protein of 57.7% (P<0.01) in 4 tamoxifen-treated G85Rflox/Cre-ER™ mice at 4 months of age when compared with 4 G85Rflox transgenic mice. A representative Western blot is shown in Fig. 3D.
Figure 3.

Knockdown of G85R SOD1 mRNA and protein in tamoxifen-treated G85Rflox/Cre-ER™ mice. G85R SOD1 mRNA in MNs of the cervical spinal cord (A) and the L4, 5 segments of the lumbar spinal cord (B) was quantitated by qPCR, and G85R SOD1 protein levels (C) were quantitated by densitometry of band intensities on Western blots of spinal cord homogenates. The SOD1 mRNA and protein analyses were performed on spinal cords from 4 mice from each of the following groups: tamoxifen-treated G85Rflox/Cre-ER™ mice, untreated G85Rflox/Cre-ER™ mice, and G85Rflox mice. The G85R SOD1 mRNA level and the G85R SOD1 protein level in G85Rflox mice were assigned a value of 100. The mean ± standard deviation is shown. *P < 0.05. (D) Representative Western blot of lumbar spinal cord homogenates immunostained with anti-human SOD1 antibody and anti-β-tubulin antibody. Non-Tg = non-transgenic mouse.
A knockdown of expression of G85R in all cell types in G85Rflox/Cre-ER™ transgenic mice delays the onset and extends disease duration of G85Rflox mice
Untreated and tamoxifen-treated G85Rflox/Cre-ER™ mice were compared with G85Rflox mice using previously defined clinical parameters (see Materials and Methods) (Boillee et al. 2006b). The onset of clinical disease and the duration of the early and late phases of disease were compared; progressive paralysis appeared during the late phase of disease. No difference was found in any of these clinical parameters when untreated G85Rflox/Cre-ER™ mice were compared to G85Rflox mice, indicating that there was no effect of the uninduced Cre-ER™ transgene on disease On the other hand, the mean onset of disease for tamoxifen-treated G85Rflox/Cre-ER™ mice was significantly delayed compared to disease onset in G85Rflox mice (350.3±45.2 days vs. 305.1±26.2 days, P = 0.007, n=12) (Fig. 4A). In addition, there was a prominent prolongation in the duration of the early phase of disease in tamoxifen-treated G85Rflox/Cre-ER™ mice vs. G85Rflox mice (28.7±8.1 days vs. 16.2±3.1 days, P <0 .001, n=12) (Fig. 4B, D), as well as the late phase of disease (20.8±6.7 days vs. 13.2±1.7 days, P <0 .001, n=12) (Fig. 4E). As expected, survival of tamoxifen-treated G85Rflox/Cre-ER™ mice was significantly greater than G85Rflox mice (399.8±56.7 days vs. 334.4±25.8 days, P = 0.002, n=12) (Fig. 4C).
Figure 4.

Clinical disease in tamoxifen-treated G85Rflox/Cre-ER™ mice, untreated G85Rflox/Cre-ER™ mice, and G85Rflox mice. (A) Disease onset. (B, D) Early phase of disease. (C) Survival. (E) Late phase of disease. The mean ± standard deviation is shown in (D) and (E). *P < 0.05 when tamoxifen-treated G85Rflox/Cre-ER™ mice are compared with G85Rflox mice. n=12 mice for each of the three groups.
A knockdown of expression of G85R in all cell types in G85Rflox/Cre-ER™ transgenic mice delays the pathology and immunohistochemical abnormalities of G85Rflox mice
In order to compare the pathology of the different groups of mice, we examined anterior horns of the lumbar spinal cord from tamoxifen-treated G85Rflox/Cre-ER™ mice, untreated G85Rflox/Cre-ER™ mice, and G85Rflox mice at ~335 days (Fig. 5). Untreated G85Rflox/Cre-ER™ and G85Rflox mice demonstrated a significant loss of MNs by Nissl staining (Fig. 5A), vigorous astrocytosis by GFAP staining (Fig. 5B), extensive microgliosis by Iba1-staining (Fig. 5C), and abundant SOD1 aggregation (in cells with MN morphology and in cell processes) (Fig. 5D). This pathology was not apparent in control non-transgenic mice and not as prominent in tamoxifen-treated G85Rflox/Cre-ER™ mice (except at the ~400 day time point shown in Fig. 6). These results indicate that the pathological and immunohistochemical hallmarks of disease were significantly delayed in tamoxifen-treated G85Rflox/Cre-ER™ mice.
Figure 5.

Pathological and immunohistochemical findings at ~335 days in the spinal cord anterior horn of non-transgenic wild type (WT) mice, G85Rflox mice, untreated G85Rflox/Cre-ER™ mice, and tamoxifen-treated G85Rflox/Cre-ER™ mice. Scale bar = 50μm.
Figure 6.
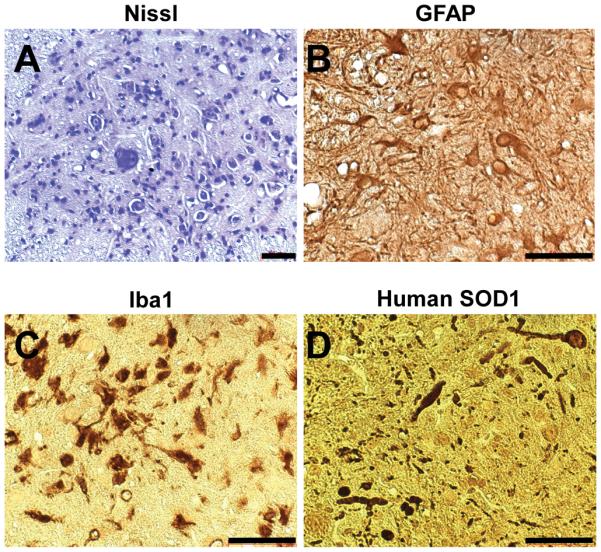
Pathological and immunohistochemical findings at ~400 days in the spinal cord anterior horn of tamoxifen-treated G85Rflox/Cre-ER™ mice. Scale bar = 50μm.
Discussion
Neurodegenerative diseases are challenging problems in biology and medicine as well as major contributors to disability and disease, with an enormous cost to society. The identification of mutated genes associated with particular neurodegenerative diseases and the generation of experimental models of these diseases have been key in directing new treatment approaches. Despite these efforts, there are still only symptomatic treatments available.
ALS remains a desperate disease because of its grim prognosis and the lack of treatment options. The identification of MTSOD1 as a cause of FALS provided an opportunity to better understand the reasons for MN death in a neurodegenerative disease as well as an experimental model to use to test treatments. There are a number of features of MTSOD1-induced experimental FALS that suggest that disease is a result of toxicity of the MT enzyme (reviewed in (Cleveland & Rothstein 2001)): some MTSOD1s have full dismutase activity (e.g., G37R); MTSOD1 transgenic mice develop ALS although the mice have full SOD1 activity; SOD1 knockout mice fail to develop disease. For this reason, knocking down the expression of MTSOD1 has been a treatment goal. This therapeutic approach has also gained support from noting that the time of disease onset in MTSOD1 transgenic mice is related to the number of copies of the MTSOD1 transgene (Dal Canto & Gurney 1997).
Several studies have explored the therapeutic implications of knocking down SOD1 expression in FALS experimental models, including the following: (i) A one month intraventricular infusion of an anti-sense oligonucleotide complementary to rat and human SOD1 mRNA into a G93A transgenic rat begun at 65 days of age (~30 days prior to the usual disease onset) knocked down G93A mRNA in the cervical cord ~40% (Smith et al. 2006). Although there was no change in disease onset or the early phase of disease, disease duration was extended by 37%, prolonging survival by ~8%. (ii) Immunization of G37R transgenic mice at age 8 weeks, 11 weeks, 14 weeks, and 6 months with bacterially expressed and purified demetallated G37R SOD1 lengthened disease onset by ~6% and survival by ~8% (Urushitani et al. 2007); the pathology showed that there was a relative preservation of MNs in the immunized mice compared to those that were not injected. The vaccination led to a “significantly lower” amount of MTSOD1 in the spinal cord; however, the extent of the decrease was not further detailed. In addition, a 28-day intraventricular infusion of (a high copy number) G93A transgenic mice with anti-G93A SOD1 antibody starting at day 85 (5 days before the onset of symptoms) led to a prolongation of survival by 4%; the degree of knockdown was not detailed. (iii) Intramuscular injection of siRNA delivered by a virus vector at 7 days of age into G93A transgenic mice resulted in a 40% reduction in SOD1 protein levels in the ventral horn, and a remarkable lengthening of disease onset by 115% and survival by 77% (Ralph et al. 2005); however, as noted by Smith et al. (Smith et al. 2006), there was actually an acceleration of disease duration by 30%, presumably because the intramuscular injection failed to knockdown MTSOD1 in non-neuronal cells. (iv) Using Cre/LoxP methods, investigators have knocked down MTSOD1 in specific neural cell types in the CNS of G37R (Boillee et al. 2006a, Boillee et al. 2006b, Yamanaka et al. 2008) and G85R (Wang et al. 2009b) FALS transgenic mice; these cell type-specific knockdowns have resulted in an amelioration of disease, affecting disease onset and/or disease duration.
There are several differences between the present study and previously published MTSOD1 knockdown studies summarized above. First, the present study involved a knockdown in all cell types of G85R SOD1, which has no superoxide dismutase enzymatic activity, whereas previous studies have involved a knockdown in specific cells and/or tissues of MTSOD1s that have full superoxide dismutase enzymatic activity. Second, the knockdown in the present study was a very substantial one, with ~66% knockdown of G85R SOD1 mRNA and ~58% knockdown of G85R SOD1 protein in all cells of the spinal cord; this knockdown included ~60% knockdown of G85R SOD1 mRNA in MNs, and with probably a similar knockdown in all cells of the body considering the promiscuous promoter driving Cre. Furthermore, the knockdown was started ~7 months prior to the expected disease onset of G85Rflox mice. The knockdown in the present study led to a significant lengthening of disease onset (by 14.8%), the early phase of disease (by 77.1%), the late phase of disease (by 57.6%), and overall survival (by 19.6%) compared to G85Rflox mice. We suspect that a previously published study using antisense oligonucleotides to knockdown MTSOD1 failed to change disease onset or early disease (Smith et al. 2006) because of the late timing of the knockdown (~30 days prior to the usual disease onset). Although the results of the present study showed a significant amelioration of disease, the effect was more limited than expected considering the results from previously published studies of MTSOD1 knockdowns restricted to particular cell types (see below) (Wang et al. 2009b) and considering the substantial degree of the knockdown in the present study.
We questioned how the results of G85R SOD1 knockdown in all cell types and tissues (in tamoxifen-treated G85Rflox/Cre-ER™ mice) differed from our previous results obtained with separate knockdowns of G85R SOD1 restricted to specific cell types (Wang et al. 2009b). Results from our previously published studies, like other studies involving knockdown of G37R SOD1 (Boillee et al. 2006a, Boillee et al. 2006b, Yamanaka et al. 2008), showed that expression of MTSOD1 in other cell types besides MNs contributes to disease in MTSOD1 transgenic mice, i.e., there is a non-cell autonomous degeneration. These studies found that a G85R SOD1 knockdown restricted to MNs/interneurons (in G85Rflox/Lhx3:Cre mice) delayed the onset and prolonged the early phase of disease of G85Rflox mice, while a knockdown of G85R SOD1 restricted to microglia/macrophages had no effect on onset (perhaps because microgliosis is minimal prior to disease onset), but increased early and late disease duration of G85Rflox mice (Wang et al. 2009b) (Table). In the present study, tamoxifen-treated G85Rflox/Cre-ER™ mice had an almost identical disease onset, duration of early disease, and survival as seen following a knockdown restricted just to MNs (in G85Rflox/Lhx3:Cre mice), with little apparent additional contribution from a knockdown of G85R SOD1 in other cells, including microglia (Wang et al. 2009b) (Table). For example, although our previous study showed that there was an extension of early disease duration by 87.5% following a knockdown restricted to MNs/interneurons (in G85Rflox/Lhx3:Cre mice) and by 58.8% following a knockdown restricted to microglia/macrophages (in G85Rflox/CD11b:Cre mice), results from the present study showed that a knockdown in all cell types in tamoxifen-treated G85Rflox/Cre-ER™ mice led to an extension of early disease duration of only 77.1%. Of note, the similar disease onset and early disease in tamoxifen-treated G85Rflox/Cre-ER™ mice and G85Rflox/Lhx3:Cre mice occurred despite ~66% knockdown of G85R SOD1 mRNA in all cell types (including ~60% knockdown of G85R SOD1 mRNA in MNs) in tamoxifen-treated G85Rflox/Cre-ER™ mice compared to only ~25% knockdown restricted to MNs/interneurons in G85Rflox/Lhx3:Cre mice. These results suggest that: (i) expression of G85R in MNs plays a driving role in determining disease onset and duration; (ii) knocking down G85R in all cell types has little additional cumulative effect on disease onset and duration of early disease from knocking down G85R just in MNs.
Table.
Change in clinical disease after G85R knockdown
| Transgenic | Onset | Early phase |
Late phase |
Disease duration |
Survival |
|---|---|---|---|---|---|
| G85Rflox/Cre-ER™ | 14.8% | 77.1% | 57.6% | 68.6% | 19.6% |
| G85Rflox/Lhx3:Cre | 14.6% | 87.5% | NS | 46.6% | 16.9% |
| G85Rflox/CD11b:Cre | NS | 58.8% | 37.5% | 43.2% | 12.5% |
Note: The numbers represent the per cent increase in the time to disease onset and increase in the duration of the early phase of disease, the late phase of disease, disease duration and survival compared to G85Rflox mice. The G85Rflox/Cre-ER™ mice were tamoxifen-treated. Data related to the knockdown in G85Rflox/Lhx:Cre mice (in which Cre is expressed in spinal and hindbrain MNs and a population of interneurons in the spinal cord) and G85Rflox/CD11b:Cre mice (in which Cre is expressed in microglia and activated macrophages) have been previously published (Wang et al. 2009b). NS=not statistically significant.
Why did a knockdown of G85R SOD1 in all cell types in 3-month-old tamoxifen-treated G85Rflox/Cre-ER™ mice transgenic mice result in a similar disease onset and early disease as that seen following a knockdown prenatally in G85Rflox/Lhx3:Cre mice that was restricted just to MNs? One possible explanation is that G85R SOD1 is pathogenic early in life in the transgenic mice, so that the later knockdown in all cell types did not have the same impact as it would have had if it had occurred earlier (as in the case of the knockdown in MNs in the G85Rflox/Lhx3:Cre mice). Of note, other neurodegenerative diseases have demonstrated effects of the pathogenic mutant gene early in life (Serra et al. 2006, Zabel et al. 2009). We are presently investigating whether an earlier knockdown in tamoxifen-treated G85Rflox/Cre-ER™ mice will have a more significant effect on disease. We are also carrying out a study to look at the effects of a later knockdown on disease, which would be more clinically relevant. An alternative explanation for the similar disease onset and early disease duration seen in tamoxifen-treated G85Rflox/Cre-ER™ mice and G85Rflox/Lhx3:Cre mice despite the different degree of knockdown of G85R SOD1 is that a variable degree of knockdown in MNs/interneurons has a similar but limited effect on disease parameters, perhaps because even very small amounts of G85R SOD1 have a prominent tendency to form pathogenic oligomers (Prudencio et al.), initiating a cascade of events in MNs/interneurons that is difficult to stop.
Although some of the clinical parameters of disease in tamoxifen-treated G85Rflox/Cre-ER™ mice and G85Rflox/Lhx3:Cre mice were similar, the prolonged duration of the late phase of disease seen in tamoxifen-treated G85Rflox/Cre-ER™ mice was not present in G85Rflox/Lhx3:Cre mice, i.e., a knockdown in non-MN/interneuron cell types influenced the duration of late disease in tamoxifen-treated G85Rflox/Cre-ER™ mice (demonstrating a non-cell autonomous degeneration). We presume that microglia/activated macrophages partly or completely influenced the duration of late disease in tamoxifen-treated G85Rflox/Cre-ER™ mice since previous results showed that knockdown of G85R in microglia/activated macrophages (in G85Rflox/CD11b:Cre mice (Wang et al. 2009b)) prolonged late disease (Table).
In a previously published study (Yamamoto et al. 2000), knockdown of mutant huntingtin in a regulatable conditional mouse model of Huntington’s disease led to a dramatic reduction in huntingtin inclusions over time with an improvement in motor function. Although the knockdown in G85R SOD1 in the present study ameliorated the disease, there was still evidence of progressive paralysis along with progression of the neuropathology and G85R SOD1 aggregation. The varying results suggest that a knockdown of disease-associated mutant protein may have different effects in different neurodegenerative diseases. Despite the continuing progression of G85R SOD1-induced FALS in the face of a knockdown in G85R SOD1, the present results support the value of this therapeutic approach and also provide a rationale for considering a knockdown in asymptomatic individuals who carry a mutation in SOD1.
ACKNOWLEGMENTS
This work was supported by the Muscular Dystrophy Association (#4346 to RPR), the National Institutes of Health (NS049333-01 to RPR), and the ALS Association (#1211 to RPR).
REFERENCES
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. A low expressor line of transgenic mice carrying a mutant human Cu,Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol. 1997;93:537–550. doi: 10.1007/s004010050650. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. Epub 2006 Apr 7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis. 2003;35:63–72. doi: 10.1002/gene.10154. [DOI] [PubMed] [Google Scholar]
- Ezzi SA, Urushitani M, Julien JP. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem. 2007;102:170–178. doi: 10.1111/j.1471-4159.2007.04531.x. Epub 2007 Mar 2029. [DOI] [PubMed] [Google Scholar]
- Gruzman A, Wood WL, Alpert E, et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104:12524–12529. doi: 10.1073/pnas.0705044104. Epub 12007 Jul 12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Lu YY, Wang LJ, Muramatsu S, et al. Intramuscular injection of AAV-GDNF results in sustained expression of transgenic GDNF, and its delivery to spinal motoneurons by retrograde transport. Neurosci Res. 2003;45:33–40. doi: 10.1016/s0168-0102(02)00195-5. [DOI] [PubMed] [Google Scholar]
- Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Variation in aggregation propensities among ALS-associated variants of SOD1: Correlation to human disease. 2009 doi: 10.1093/hmg/ddp260. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph GS, Radcliffe PA, Day DM, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. Epub 2005 Mar 2013. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Bowling AC, Patterson D, et al. A frequent ala 4 to val superoxide dismutase-1 mutation is associated with a rapidly progressive familial amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3:981–987. doi: 10.1093/hmg/3.6.981. [DOI] [PubMed] [Google Scholar]
- Serra HG, Duvick L, Zu T, et al. RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127:697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. Epub 2006 Jul 2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104:2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Deng HX, Grisotti G, Zhai H, Siddique T, Roos RP. Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum Mol Genet. 2009a;18:1642–1651. doi: 10.1093/hmg/ddp085. Epub 2009 Feb 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Sharma K, Deng HX, Siddique T, Grisotti G, Liu E, Roos RP. Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol Dis. 2008;29:400–408. doi: 10.1016/j.nbd.2007.10.004. Epub 2007 Oct 2023. [DOI] [PubMed] [Google Scholar]
- Wang L, Sharma K, Grisotti G, Roos RP. The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol Dis. 2009b;35:234–240. doi: 10.1016/j.nbd.2009.05.002. Epub 2009 May 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LJ, Lu YY, Muramatsu S, et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci. 2002;22:6920–6928. doi: 10.1523/JNEUROSCI.22-16-06920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. Epub 2008 Feb 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel C, Mao L, Woodman B, et al. A large number of protein expression changes occur early in life and precede phenotype onset in a mouse model of Huntington disease. Mol Cell Proteomics. 2009;8:720–734. doi: 10.1074/mcp.M800277-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]