

Abstract
The serine/threonine kinase protein kinase B (PKB)/ Akt is a critical regulator of insulin signaling, cell survival, and oncogenesis. The activation mechanisms of this key kinase are well characterized. In contrast, inactivation of PKB signaling by phosphatases is less well understood. To study the dynamics of PKB signaling in live cells, we generated a genetically encoded fluorescent reporter for PKB activity that reversibly responds to stimuli activating phosphatidylinositol 3-kinase. Specifically, phosphorylation of the reporter expressed in mammalian cells causes changes in fluorescence resonance energy transfer, allowing real-time imaging of phosphorylation catalyzed by PKB. Because of its reversibility, the reporter also allows termination of PKB signaling by phosphatases to be monitored. We found that PKB signaling in the cytosol was more rapid and more transient compared with that in the nucleus, suggesting the presence of differentially regulated phosphatase activity in these two compartments. Further more, targeting of the reporter to the plasma membrane, where PKB is activated, resulted in accelerated and prolonged response compared with the response in the cytosol, suggesting that release of PKB or its substrates from the membrane is required for desensitization of PKB signaling. These data reveal spatio-temporal gradients of both signal propagation and signal termination in PKB signaling.
Protein kinase B (PKB)1/Akt is a serine/threonine kinase that is the prominent mediator of pathways resulting in enhanced cell growth and cell survival. In 1991 it was identified as the transforming component of AKT8, a virus correlating with a high incidence of spontaneous lymphoma in mice, and named Akt (1). In the same year, two other groups isolated the same kinase via its homology to protein kinase A (PKA) and protein kinase C (PKC) and designated it Rac (related to the A and C kinases) (2) and PKB (3). There are three isozymes in mammals: Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ. While the kinase is referred to by these two names, PKB and Akt, herein it will be referred to as PKB.
PKB contains an NH2-terminal pleckstrin homology (PH) domain followed by a kinase domain and a short COOH-terminal regulatory tail containing an activating phosphorylation site (4). PKB is activated by recruitment to membranes following stimulation of growth factor receptors. Briefly, activated growth factor receptors lead to plasma membrane recruitment of phosphatidylinositol 3-kinase (PI 3-kinase), which leads to the production of phosphatidylinositol 3,4,5-trisphosphate (PIP3) at the plasma membrane. The PH domain of PKB binds to the newly formed PIP3 resulting in translocation of the kinase to the plasma membrane (5–8). Once at the membrane, PKB becomes activated via two sequential phosphorylation steps, first by its upstream kinase PDK-1 on Thr308 within its activation loop, and next via autophosphorylation at Ser473 within the COOH-terminal hydrophobic motif (9–12). PKB is maximally active when phosphorylated at both regulatory sites. Once phosphorylated, active PKB disengages from the plasma membrane and translocates through the cytosol to distinct subcellular compartments including the nucleus (13, 14). Some well characterized downstream substrates of PKB include Bad, the family of forkhead transcription factors, GSK3, and the NF-κB regulator IKK; phosphorylation of these substrates leads to an inhibition of apoptosis or directly promotes cell survival (15).
Although the mechanism of PKB activation has been well characterized, the molecular mechanisms of PKB desensitization and inactivation are poorly understood. We are interested in exploring the balance within the cell between PKB activity and phosphatase activities to elucidate mechanisms by which PKB signaling is regulated and terminated. The standard approach to monitoring PKB activity is through immunoblotting to detect phosphorylation at Thr308 and Ser473. This biochemical approach does not allow localized PKB activation to be monitored in live cells. The advent of genetically encoded fluorescent reporters provides a unique approach to elucidating the dynamic interplay between phosphorylation and dephosphorylation in live cells in real time (16, 17).
Genetically encoded fluorescent reporters generally consist of two different fluorescent proteins connected by a phosphoaminoacid-binding domain and a kinase substrate sequence (16–18). Phosphorylation of the substrate causes intramolecular complexation by the phosphoaminoacid-binding domain. The resulting conformational change alters fluorescence resonance energy transfer (FRET) between the fluorescent proteins, enabling one to visualize kinase signaling in live cells. Umezawa and colleagues (19) recently developed such a reporter for PKB, dubbed “Aktus,” but overexpression of PKB was required to see clear signals from Aktus.
Here we present BKAR (B kinase activity reporter), a novel PKB reporter. The predominant distinctions between BKAR and Aktus are the phosphoaminoacid-binding domain and its corresponding substrate sequence. We chose to utilize the FHA2 domain as the phosphoaminoacid-binding domain as we had previously utilized it in the context of CKAR (C kinase activity reporter) (18); its binding affinity to the phosphorylated substrate sequence was such that CKAR was sensitive to endogenous kinase signaling but also was reversible upon kinase inhibition. We show that BKAR responds reversibly to endogenous PKB activation, allowing detection of both activation and termination of signaling downstream of PKB. Through use of a plasma membrane-targeted BKAR, we show that phosphorylation of PKB substrates at the membrane occurs more rapidly than cytosolic substrates and that there is a further delay before nuclear substrates are phosphorylated. In addition, BKAR localized in the nucleus and plasma membrane is dephosphorylated slower than BKAR in the cytosol. These data reveal a spatio-temporal gradient of signal propagation and termination in PKB signaling.
EXPERIMENTAL PROCEDURES
Materials
PDGF-BB, phorbol 12,13-dibutyrate (PdBu), calyculin A, LY294002, and wortmannin were obtained from Calbiochem. PKB P-T308, PKB P-S473, and PKB antibodies were obtained from Cell Signaling Technology (Beverly, MA). Biomax MR film used for Western analyses was from Eastman Kodak Co. All other materials were reagent-grade.
Plasmid Constructs
BKAR was generated through substitution of a PKB substrate sequence, RKRDRLGTLGI, for the substrate sequence within pcDNA3-CKAR (18) by PCR. The phosphoacceptor threonine was mutated to an alanine to create BKAR-T/A following the QuikChange protocol (Stratagene). MyrPalm-BKAR was generated by PCR with a primer containing the 5′ 21 bp of the gene for Lyn kinase. The corresponding 7 amino acids are known to become myristoylated and palmitoylated. A fluorescent-tagged PKB-PH domain was generated by subcloning the DNA encoding monomeric red fluorescent protein (mRFP) (20) to that encoding the PH domain of PKB. For in vitro experiments, BKAR was subcloned into the bacterial expression vector pRSET B (Invitrogen).
Protein Purification
pRSET-BKAR or pRSET-BKAR-T/A was transformed into BL21(DE3) Escherichia coli. A single colony was grown for 2 days at room temperature. Pelleted cells were lysed by Dounce homogenization and French press in 20 mM HEPES, pH 7.5, containing 1 mM DTT, 300 nM phenylmethylsulfonyl fluoride, 10 μM bestatin, 500 nM benzamidine, and 500 ng/ml leupeptin. BKAR was purified from cleared lysates by nickel chelation chromatography using nickel-nitrilotriacetic acid-agarose (Qiagen). Following washes, BKAR was eluted in 20 mM HEPES, 1 mM DTT containing 200 mM imidazole. Imidazole was removed by dialysis. Protein yield was estimated by CFP absorption at 434 nm.
Kinase Assays and in Vitro Spectroscopy
For kinase assays, 1 μg of purified BKAR or BKAR-T/A was incubated with 100 ng of pure Akt1/ PKBα (ΔPH, S473D) (1739 units/mg, Upstate Biotechnology), PKA (4290 units/mg, S. Taylor’s laboratory) or PKC-βII (1240 units/mg) at 30 °C for the indicated times. Buffer compositions were as follows: 20 mM HEPES, 2 mM DTT, 200 μM ATP, 5 mM MgCl2 for PKB; 20 mM HEPES, 2 mM DTT, 200 μM ATP, 10 mM MgCl2 for PKA; and 20 mM HEPES, 2 mM DTT, 5 mM MgCl2, 100 μM ATP, 500 mM CaCl2, with 140 μM phosphatidlylserine, 3.8 μM diacylglycerol vesicles for PKC. For radioactive assays, [γ-32P]ATP was included in the reaction, BKAR and BKAR-T/A were isolated by SDS-PAGE, identified by Coomassie Blue staining, and excised, and 32P incorporation was measured by scintillation counting. For analysis by Western blotting, BKAR or BKAR-T/A was incubated with the designated kinase for 0 or 15 min and analyzed by SDS-PAGE and Western blotting with the phospho-(Ser/Thr) Akt/ PKB substrate antibody (Cell Signaling). Western blots were analyzed by chemiluminescence using SuperSignal (Pierce). Fluorescence emission scans were performed on BKAR phosphorylated by PKB (+ATP) or unphosphorylated (−ATP) by exciting at 434 nm to excite CFP.
Cell Transfection
NIH3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (Cellgro) containing 10% bovine calf serum and 1% penicillin/streptomycin at 37 °C in 5% CO2. COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium (Cellgro) containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in 5% CO2. Cells were plated onto sterilized glass coverslips in 35-mm dishes prior to transfection. Transient transfection was carried out using Effectene (Qiagen) for NIH3T3 cells or FuGENE 6 (Roche Diagnostics) for COS-7 cells. Cells were serum-starved within 24 h post-transfection and imaged within the following 24 h.
Cell Imaging
Cells were washed one time in Hanks’ balanced salt solution (HBSS, Cellgro) and imaged in HBSS in the dark at room temperature. Images were acquired on a Zeiss Axiovert microscope (Carl Zeiss Microimaging, Inc.) using a MicroMax digital camera (Roper-Princeton Instruments) controlled by MetaFluor software (Universal Imaging, Corp.). All optical filters were obtained from Chroma Technologies. Data were collected through a 10% neutral density filter. CFP and FRET images were obtained every 15 s through a 420/20-nm excitation filter, a 450-nm dichroic mirror, and a 475/40-nm or 535/25-nm emission filter for CFP and FRET, respectively. A YFP image was obtained as a control for photobleaching through a 495/10-nm excitation filter, a 505-nm dichroic mirror, and a 535/25-nm emission filter. RFP images were obtained through a 568/55-nm excitation filter, a 600-nm dichroic mirror, and a 653/95-nm emission filter. Excitation and emission filters were switched in filter wheels (Lambda 10–2, Sutter). Integration times were 200 ms for CFP and FRET and 50–100 ms for YFP.
Western Blotting
NIH3T3 cells were grown to near confluence in 60-mm dishes. Cells were washed once in HBSS and treated for the indicated times with 50 ng/ml PDGF in HBSS at room temperature. Cells were lysed and separated through a low speed spin to isolate the cytosolic fractions as described (21). One-third of each fraction was analyzed by SDS-PAGE and Western blotting to determine the relative amounts of PKB Thr308 and Ser473 phosphorylation over time. Western blots were analyzed by chemiluminescence.
RESULTS
Design of BKAR
BKAR, a B kinase activity reporter, is a genetically encoded fluorescent reporter designed to monitor PKB-mediated phosphorylation in live cells. Its modular structure is analogous to the previously described PKC activity reporter, CKAR, having only a small region (the substrate phosphorylation sequence) modified to make it a specific readout of PKB signaling (18). BKAR consists of the FRET pair mCFP (cyan fluorescent protein rendered monomeric by the mutation A206K) and mYFP2 (yellow fluorescent protein like-wise with A206K), bracketing an FHA2 phosphothreonine-binding domain and a consensus PKB phosphorylation sequence (22, 23). The consensus sequence, RKRDRLGTLGI, where the underlined T is the phospho-acceptor residue, was designed based on a combination of data describing optimal PKB phosphorylation consensus sequences (24) and information identifying which residues are critical for FHA2 domain binding (25). Analysis of this sequence using Scansite (scansite.mit.edu) suggested that it is optimal for PKB phosphorylation and suboptimal for phosphorylation by other basophilic kinases; PKA and PKC were predicted to be the next most likely kinases to phosphorylate the sequence. As with CKAR, unphosphorylated BKAR is in a conformation resulting in FRET from the donor molecule CFP to the acceptor YFP. Phosphorylation of the threonine within the substrate sequence triggers an intramolecular clamp with the FHA2, leading to a decrease in FRET (Fig. 1A).
Fig. 1. Design of BKAR.

A, BKAR consists of mCFP, the FHA2 domain of Rad53p, a consensus PKB phosphorylation sequence (red and mYFP. In the unphosphorylated state, mCFP and mYFP are in a proximity and orientation resulting in FRET. Once phosphorylated by PKB at the threonine within the substrate sequence (circled in red), the FHA2 domain binds the phosphorylated sequence resulting in a conformational change that alters the FRET ratio. B, time course of phosphorylation of His-tagged BKAR or BKAR-T/A purified from bacteria and incubated with constitutively active Akt1/PKBα (ΔPH, S473D) in an in vitro kinase assay. C, in vitro kinase assay monitoring reporter phosphorylation of BKAR or BKAR-T/A after 15 min of incubation with PKB, PKA, or PKC. Western blots show phosphorylation via the antiphospho-PKB substrate antibody (top panel) and total reporter present via the anti-GFP antibody (bottom panel). D, emission spectra of BKAR upon excitation of CFP after incubation with active PKB with (blue) or without (red) ATP. Emission spectra of BKAR following treatment with trypsin (green).
In Vitro Phosphorylation of BKAR by PKB
To determine whether BKAR can serve as a PKB substrate, BKAR was expressed in bacterial cells as a His-tagged fusion protein, purified, and subjected to in vitro phosphorylation by a constitutively active construct of PKB, Akt1/PKBα (ΔPH, S473D). Fig. 1B shows that pure PKB phosphorylated pure BKAR in vitro with a t1/2 of ~5 min under the conditions of the assay.Importantly, a construct of BKAR in which the phosphoacceptor threonine was mutated to alanine (BKAR-T/A) was not phosphorylated revealing that PKB specifically phosphorylates the threonine within the designed substrate sequence of BKAR. The stoichiometry of the phosphorylation was routinely 0.3 mol of phosphate per mol of reporter, suggesting possible aggregation or masking of some of the phosphorylation sites in vitro.
Because PKA and PKC were both identified by Scansite as the next most likely kinases to phosphorylate the consensus sequence in BKAR, we tested whether these enzymes phosphorylated the threonine in the consensus sequence. Phosphorylation at this site was detected with a phospho-specific antibody to the consensus PKB phosphorylation sequence. Fig. 1C shows that this antibody labeled BKAR, but not the BKAR-T/A construct, following an in vitro kinase assay using PKB as the kinase but not when PKA or PKC βII were used. Thus, neither PKA nor PKC recognize the phospho-acceptor site on BKAR in vitro. Curiously, analysis of total phosphorylation by monitoring 32P incorporation revealed that PKC βII (but not PKA) phosphorylated BKAR to a stoichiometry double that catalyzed by PKB. This phosphorylation also occurred on BKAR-T/A reflecting silent phosphorylations of the GFPs (data not shown).
Having demonstrated that pure PKB can recognize and phosphorylate BKAR on the threonine within its substrate sequence, we next addressed the effect of this phosphorylation on FRET between the donor/acceptor pairs on the reporter. Analysis of the emission spectrum of BKAR upon excitation at 434 nm (excitation of CFP) revealed a change in FRET upon phosphorylation. Specifically, 15 min of incubation with pure PKB resulted in an increase in CFP emission (around 475 nm) reflecting a loss in energy transfer to YFP, and an decrease in YFP emission reflecting a decrease in FRET. Furthermore, treatment of BKAR with trypsin to cleave the reporter and separate the amino-terminal CFP from the carboxyl-terminal YFP confirmed that the peak observed around 530 nm (YFP emission) was a result of FRET from CFP to YFP. Thus, phosphorylation of BKAR at the threonine within the substrate sequence induces a change in FRET. However, the critical test of the specificity of BKAR as a reporter to visualize kinase signaling is to examine its responses in living cells.
BKAR Reports PKB, but Not PKA or PKC, Activity in Live Cells
To examine the specificity of BKAR phosphorylation in live cells, NIH3T3 cells were transiently transfected with BKAR and then serum-starved overnight before imaging. Fig. 2A shows that BKAR responded to PDGF stimulation within a few minutes after drug delivery and this response reached its maximum level 15 min after PDGF treatment, as evidenced by an increased FRET ratio (cyan emission/yellow emission). Note that the data are plotted as the ratio of cyan fluorescence (which increases as FRET decreases) to yellow emission (which decreases as FRET decreases). To determine whether this change in the FRET ratio was due to phosphorylation at the threonine within the PKB consensus sequence in BKAR, we examined the response of BKAR-T/A in which the phosphoacceptor threonine had been mutated to an alanine. Serum-starved NIH3T3 cells expressing BKAR-T/A did not show any change in the FRET ratio in response to PDGF treatment. Thus, the observed change in the FRET ratio of BKAR results from phosphorylation at the designed PKB phosphorylation site.
Fig. 2. BKAR reversibly reports signaling by endogenous PKB.

A, serum-starved NIH3T3 cells overexpressing BKAR or BKART/A, in which the phospho-acceptor site was mutated to Ala, were treated with PDGF (50 ng/ml) and the ratio of cyan emission to yellow emission monitored with time. The inset shows pseudocolor images corresponding to the basal (pre-PDGF) and maximal (post-PDGF) FRET levels where blue/green represents unphosphorylated BKAR and red reflects phosphorylated BKAR. B, serum-starved NIH3T3 cells overexpressing BKAR were stimulated with 10 μM forskolin to activate PKA and imaged. C, serum-starved NIH3T3 cells overexpressing BKAR were stimulated with 200 nM PdBu and then 50 ng/ml PDGF. D, serum-starved NIH3T3 cells overexpressing BKAR were stimulated with 50 ng/ml PDGF for 25 min and then treated with 30 μM LY294002 and 300 nM wortmannin to inhibit PI 3-kinase. E, BKAR is a more sensitive PKB reporter than Aktus. Serum-starved COS-7 cells coexpressing mouse PKBα and BKAR or Aktus are treated with EGF (50 ng/ml) and then treated with the LY294002 (30 μM).
To further explore the specificity of BKAR in vivo, we tested whether activation of PKA or PKC altered the FRET response of the reporter. Treatment of serum-starved NIH3T3 cells expressing BKAR with 10 μM forskolin to activate PKA did not result in an increase in the FRET ratio, confirming in vitro data that PKA does not effectively phosphorylate BKAR (Fig. 2B). (Note the slight decrease in FRET ratio suggesting a change in basal BKAR phosphorylation, perhaps from activation of cyclic AMP-sensitive phosphatases). Importantly, treatment of cells with 200 nM PdBu, a potent activator of PKC, caused no significant change in the FRET ratio. Subsequent addition of PDGF resulted in a robust FRET ratio change (Fig. 2C). These data reveal that BKAR is not sensitive to either PKA or PKC activation.
We next addressed the reversibility of BKAR as a PKB activity reporter by examining the effect of inhibiting PI 3-kinase, and hence PKB, on the PDGF-stimulated phosphorylation of the reporter. Fig. 2D shows that PDGF stimulation resulted in an increase in the FRET ratio and this response began to level off typically around 10 min after PDGF treatment. Addition of LY294002 and wortmannin to inhibit PI 3-kinase caused a decrease in the FRET ratio (Fig. 2D), with the signal returning to baseline. This demonstrates that BKAR responds specifically to PI 3-kinase signaling, which is directly upstream of PKB signaling. Taken together, these data clearly indicate that BKAR specifically and reversibly reports PKB activity in vivo.
In Fig. 2E, we compared the sensitivity of BKAR to the described previously reporter, Aktus. We observed no FRET change from Aktus in response to endogenous PKB activation in starved NIH3T3 cells (data not shown) so we moved to a more robust PKB signaling system. EGF stimulation of COS-7 cells overexpressing PKBα and BKAR routinely results in a robust response that is reversed by addition of LY294002. Under the same conditions, cells expressing Aktus showed a barely detectable response to EGF (Fig. 2E). As reported previously (19), Aktus is suitable for examining the activity of localized and overexpressed PKB; in contrast, BKAR allows sensitive monitoring of endogenous activity.
PKB Signaling in the Cytosol and Nucleus
BKAR distributes throughout both the cytosol and the nucleus allowing analysis of PKB signaling from these two areas side by side in the same cell (Fig. 3A). Analyses of the FRET ratio changes from these two regions revealed differential timing of BKAR phosphorylation following growth factor stimulation. Specifically, the t1/2 for maximal response of BKAR in the cytosol was approximately three times faster than that of BKAR localized in the nucleus. This delay in nuclear PKB signaling likely reflects the time it takes for activated PKB to translocate into the nuclear compartment; however, the delay could also result from higher phosphatase activity in the nucleus compared with the cytosol. To discriminate between these two possibilities, cells were treated with calyculin A, a potent protein phosphatase-1 and protein phosphatase-2 inhibitor, prior to stimulation with PDGF. Fig. 3B shows that the delay in the nuclear response compared with the response in the cytosol was maintained following inhibition of phosphatase activity. Note that the magnitude of the response was enhanced more than 3-fold in the presence of calyculin, revealing robust phosphatase activity in these cells (compare maximal response in Fig. 3, A and B). Furthermore, when PDGF-stimulated cells were treated with LY294002 and wortmannin to inhibit PI 3-kinase, cytosolic BKAR was dephosphorylated much more rapidly than nuclear BKAR, revealing much more robust phosphatase activity toward BKAR in the cytosol compared with the nucleus (Fig. 3C). Thus, the delay in the nuclear response of BKAR does not arise from increased phosphatase activity in the nucleus. Rather, our data are consistent with the delay in nuclear signaling resulting from delay in the phosphorylation step.
Fig. 3. BKAR is distributed in the cytosol and nucleus.

A, distribution of BKAR expressed in serum-starved NIH3T3 cells imaged by monitoring CFP emission (top). Concurrent imaging of the FRET ratio from cytosolic and nuclear BKAR within the same cells reveals that cytosolic phosphorylation by PKB occurs more rapidly than nuclear phosphorylation by PKB following treatment with PDGF (50 ng/ml) (bottom). Data are presented as the emission of CFP over the emission of YFP normalized to that at t = 0. B, serum-starved NIH3T3 cells were first treated with calyculin A for 3 min and then treated with 50 ng/ml PDGF. Data are presented as described in Fig. 3A. C, concurrent imaging of the FRET ratio from cytosolic and nuclear BKAR in cells treated with 50 ng/ml PDGF and then treated with 30 μM LY294002 and 300 nM wortmannin. Data are presented as described for A. D, serum-starved NIH3T3 cells were treated with PDGF (50 ng/ml) for over 15 min and then treated with calyculin A (100 nM). E, Western analysis of endogenous PKB from serum-starved NIH3T3 cells treated with PDGF (50 ng/ml) at room temperature for the indicated times. Cytosolic fractions were isolated and the phosphorylation state of PKB was probed using antibodies against phospho-Thr308 and phospho-Ser473.
Cytosolic BKAR
Fig. 3A shows that cytosolic BKAR is considerably more sensitive to dephosphorylation than nuclear BKAR; extended imaging from these regions depicts the onset of a decline in the FRET ratio from cytosolic BKAR typically after 15 min of PDGF stimulation, whereas the nuclear BKAR response was sustained over this time. The decay of the cytosolic BKAR response could reflect an increase in phosphatase activity acting on PKB or on the reporter itself. Alternatively, the integrity of cytosolic BKAR could be affected following PKB phosphorylation. To examine these possibilities, starved NIH3T3 cells were treated with PDGF and the BKAR response was monitored until the FRET ratio had peaked and was beginning to decline. At this point, the cells were treated with the phosphatase inhibitor, calyculin A. Calyculin A treatment not only prevented further decline in the FRET ratio, it resulted in a robust increase in the BKAR response. This confirms that cytosolic BKAR is still intact and competent to report kinase activity; furthermore this indicates that PKB is still active in the cytosol after 15 min of PDGF treatment (Fig. 3D). Thus, the decline in BKAR phosphorylation following PDGF stimulation reflects phosphatase action.
To explore whether the dephosphorylation of BKAR reflects inactivation of PKB by phosphatases, or specific dephosphoryl-ation of the substrate, we examined the phosphorylation state of PKB following PDGF treatment. PKB is activated via phosphorylation first at its activation loop sequence (Thr308) and subsequently via autophosphorylation at its hydrophobic motif (Ser473). It is this dually phosphorylated PKB species that is most active. Starved NIH3T3 cells were treated with PDGF for the indicated times up to 1 h under the same conditions used for cell imaging. Cytosolic fractions were isolated, and the phosphorylation state of PKB was analyzed by Western blotting. Fig. 3E shows that PKB was rapidly phosphorylated following PDGF treatment, and this phosphorylation state was sustained for at least 1 h after growth factor stimulation. Thus, cytosolic PKB appears active (as measured by PKB phosphorylation at Thr308 and Ser473) during the time at which cytosolic BKAR displays a decline in the FRET ratio. Most simply, these experiments suggest that phosphatases are acting on BKAR and competing against PKB phosphorylation of the reporter 10–15 min after growth factor treatment.
PKB Signaling at the Membrane
PKB is first activated at the plasma membrane, where it is recruited and phosphorylated, following PI 3-kinase activation. Thus, we were interested in monitoring PKB activity at the membrane itself. To do this, we generated a membrane-targeted BKAR (MyrPalm-BKAR) in which the seven NH2-terminal residues (derived from Lyn kinase) encode for myristoylation and palmitoylation modifications of BKAR, thereby tethering it to the plasma membrane (Fig. 4A) (18, 22). Fig. 4B shows that the kinetics of the MyrPalm-BKAR response following PDGF stimulation were much more rapid than the kinetics described above for cytosolic BKAR. Typically, the t1/2 for maximal response of the membrane-tethered reporter was about three times faster than that observed for cytosolic BKAR. This phosphorylation was sustained for as long as cells were imaged (over 20 min) (data not shown) revealing that equilibrium is achieved between kinase and phosphatase function at the membrane. Addition of PI 3-kinase inhibitors reversed the phosphorylation, revealing, again, the dynamic interplay between kinase and phosphatase activities in PKB signaling. These data are consistent with PKB being first activated at the membrane and also reveal that substrate phosphorylation at this location is sustained relative to that which follows in the cytosol.
Fig. 4. Membrane targeting of BKAR reveals rapid PKB signaling.
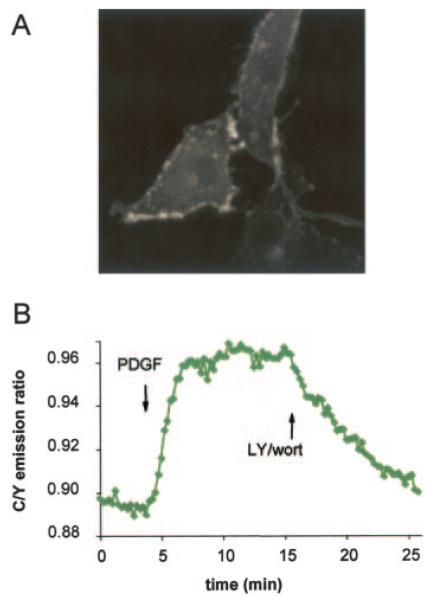
A, BKAR was targeted to the plasma membrane by fusion of the 7-amino acid NH2 terminus of Lyn to the amino terminus of BKAR, providing a myristoylation and palmitoylation tag. The distribution of membrane-targeted BKAR in serum-starved NIH3T3 cells is shown by monitoring CFP emission. B, serum-starved NIH3T3 cells expressing MyrPalm-BKAR were treated with PDGF (50 ng/ml) for 15 min and then treated with LY294002 (30 μM) and wortmannin (300 nM) and imaged.
Overexpression of PKB-PH Domain
We took advantage of BKAR to ask two questions: 1) what are the kinetics of PIP3 formation compared with Akt activation, and 2) does overexpression of PH domains interfere with endogenous PKB signaling? To this end, we concurrently imaged the translocation of the PH domain of PKBα fused to monomeric red fluorescent protein and PKB activity using cytosolic BKAR. Fig. 5 shows that PIP3 production, as measured by PH domain translocation, occurred with a t1/2 of ~0.5–1 min. These kinetics coincide with PKB activation at the plasma membrane (Fig. 4), and precede PKB activation in the cytosol, which has a t1/2 of ~3–5 min (Fig. 5). These kinetics of BKAR phosphorylation (cytosolic) were comparable with those observed in the absence of overexpressed PH domain (see Fig. 2). This reveals that overexpression of a PIP3-binding PH domain does not interfere with activation or signaling of PKB.
Fig. 5. Simultaneous imaging of PH domain translocation and PKB signaling.
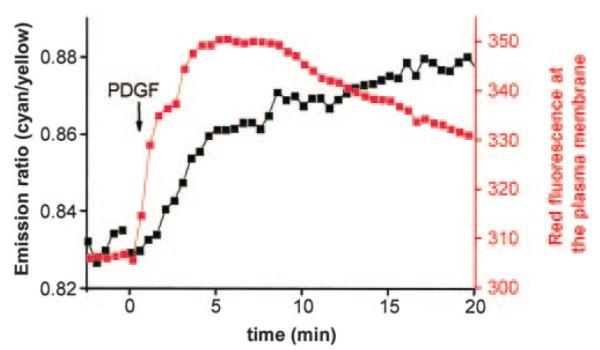
Serum-starved NIH3T3 overexpressing a monomeric RFP-tagged PKB-PH domain and BKAR were imaged in response to 50 ng/ml PDGF treatment. The intensity of the RFP signal at the plasma membrane (red) and the FRET ratio change from cytosolic BKAR (black) were assayed concurrently from the same cells and plotted.
DISCUSSION
In this study we generated BKAR, a novel reporter for endogenous PKB signaling in live cells. We show that BKAR is specific for PKB signaling and is reversible, allowing monitoring of both signal propagation and signal termination. Using this reporter, we examine the kinetics and spatial distribution of PKB signaling in serum-starved cells following growth factor treatment. Our data reveal a spatio-temporal gradient of PKB signaling that initiates at the membrane, propagates through the cytosol, and is last observed in the nucleus. Using a reporter for PIP3 production, we show that the rapid activation of PKB at the membrane coincides with the kinetics of PIP3 formation.
Propagation of PKB Signaling; Gradient of Phosphorylation
Monitoring BKAR at the membrane, in the cytosol and within the nucleus, reveals a temporal gradient of PKB signaling downstream of growth factor receptor stimulation. As depicted in Fig. 6, PKB is recruited to, and activated at, the plasma membrane following the production of PIP3. By targeting BKAR to the membrane, we observe very rapid kinetics of PKB signaling (t1/2 typically on the order of minutes) indicating that an active PKB species is generated and present at this location. This rapid activation coincides with the kinetics of PIP3 formation, as detected using a PH domain reporter for PIP3. Phosphorylation of BKAR localized in the cytosol is about three times slower. This could reflect the time required for PKB to disengage from the membrane and diffuse through the cytosol to phosphorylate BKAR. In addition, the slower kinetics of phosphorylation observed in the cytosol could result from higher phosphatase activity in this region compared with the phosphatase activity at the membrane. Phosphorylation of nuclear BKAR is typically another 3-fold slower, most likely reflecting a rate-limiting translocation of PKB into the nucleus. This lag does not result from increased phosphatase activity in the nucleus because it is also observed in the presence of phosphatase inhibitors; in addition, BKAR phosphorylation is sustained in the nucleus compared with the cytosol indicating reduced phosphatase activity in this compartment. As a genetically encoded reporter, BKAR can be targeted to a number of distinct intracellular regions where PKB is known to signal. As part of this study, we also show that overexpression of a PIP3 binding PH domain does not affect PKB signaling, indicating that the local concentration of PIP3 is saturating.
Fig. 6. Spatiotemporal gradient of PKB signaling.
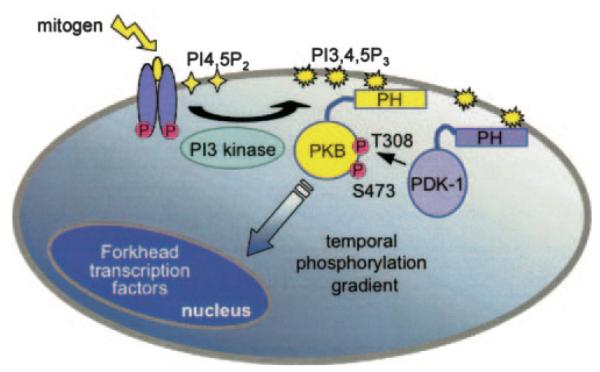
Stimulation of growth factor receptors leads to the recruitment of PI 3-kinase to the plasma membrane where it converts the phospholipid phosphatidylinositol 4,5-bisphosphate to PIP3. PKB translocates to the plasma membrane via its PH domain which binds to PI 3-kinase products (e.g. PIP3). At the membrane PDK-1 phosphorylates and activates PKB via phosphorylation at its activation loop site, Thr308. PKB subsequently autophosphorylates at its hydrophobic motif, Ser473; it is this dually phosphorylated PKB species that is most active. Once activated at the membrane, PKB phosphorylates substrates throughout the cell, including proteins such as the family of forkhead transcription factors within the nucleus. BKAR localized at the membrane, within the cytosol, or in the nucleus reveals a temporal gradient of PKB signals emanating from the point of PKB activation at the membrane, propagating through the cytosol, and terminating within the nucleus.
Termination of PKB Signaling
Protein phosphorylation is dependent upon the dynamics between active kinases and phosphatases within the cell. By monitoring BKAR, we observed differential dephosphorylation of the reporter that was dependent upon subcellular location. Membrane-targeted BKAR displayed rapid kinetics of activation with a prolonged phosphorylation that lasted as long as the cells were imaged (up to 20 min after growth factor treatment). PKB is activated at the membrane, thus the signaling kinase is concentrated in this region allowing it to efficiently compete with any phosphatase activity that might be present at the membrane. Cytosolic BKAR, on the other hand, exhibited a decay in its response following ~10 min of growth factor treatment. This decay was not a result of dephosphorylation of PKB but likely caused by phosphatases acting directly on BKAR itself; this suggests that PKB signaling in the cytosol may be more closely regulated by cellular phosphatases that act on PKB substrates. Last, nuclear BKAR responses remained elevated following over 25 min of growth factor treatment, similar to what was observed with the membrane-tethered BKAR. In the case of nuclear BKAR, however, the results are consistent with the presence of lower levels of the relevant phosphatase activity within the nuclear compartment.
Advantages of BKAR
Prior to the generation of BKAR, Umezawa and colleagues (19) developed a PKB reporter that they termed Aktus and which was used to detect activity of overexpressed PKB. In their studies, the authors targeted this reporter to mitochondria or golgi and assayed for PKB activity at these two compartments downstream of two distinct pathways. With Aktus, the authors found that there was differential phosphorylation depending both on the location of the reporter and the specific pathway activated; specifically, 17β estradiol was able to elicit a response from Aktus at both subcellular locations assayed, whereas insulin only elicited a response from the Golgi-targeted Aktus (19). Our new PKB reporter, BKAR, offers several advantages over Aktus. First, BKAR is much more sensitive to PKB signaling as it reports endogenous PKB activity in NIH3T3 cells; the cytosolic Aktus reporter elicited no response within these same cells (data not shown). In fact, even under conditions that elicit large responses from BKAR (imaging cells that overexpress PKB), we are not able to readily detect a FRET change from Aktus (Fig. 2E). Consistent with this, the authors state that responses from Aktus were best observed when the reporter was targeted to subcellular compartments where PKB signaling has been described previously, i.e. mitochondria and Golgi, rather than when it was diffuse within the cytosol (17, 19). Second, BKAR distributes throughout both the cytosol and the nucleus allowing for concurrent imaging from both areas within the same cell; untargeted Aktus is present solely in the cytosol. Last, BKAR responds reversibly to PKB activity allowing one to monitor the termination of PKB signaling in live cells, either through the deactivation of PKB itself or through the increased dephosphorylation of the reporter; we were unable to address the reversibility of Aktus given the low signal upon activation under our imaging conditions, and Sasaki et al. (19) did not show any attempts at reversal. Thus, while Aktus elegantly demonstrated the specificity of PKB signaling via its targeted versions, BKAR appears to be an improved reporter, offering more sensitivity in monitoring PKB signaling in live cells and allowing monitoring of both signal propagation and signal termination.
Acknowledgments
We thank Dr. Jonathan Violin for helpful advice, Dr. Susan Taylor’s laboratory for purified PKA, Qing Xiong for technical assistance, and Dr. Y. Umezawa for the Aktus construct.
Footnotes
This work was supported by National Institutes of Health Grants P01 DK54441 (to A. C. N.), NS27177 (to R. Y. T.), DK07233 (to M. T. K.), The Johns Hopkins University School of Medicine, and the W. M. Keck Center (to J. Z.).
This article was selected as a Paper of the Week.
- PKB
- protein kinase B
- PKA
- protein kinase A
- PKC
- protein kinase C
- BKAR
- B kinase activity reporter
- CFP
- cyan fluorescent protein
- CKAR
- C kinase activity reporter
- FRET
- fluorescence resonance energy transfer
- MyrPalm
- myristoylated and palmitoylated
- PdBu
- phorbol 12,13-dibutyrate
- PDGF
- platelet-derived growth factor
- PDK-1
- phosphoinositide-dependent kinase 1
- PH domain
- pleckstrin homology domain
- PI 3-kinase
- phosphatidylinositol 3-kinase
- PIP3
- phosphatidylinositol 3,4,5-trisphosphate
- RFP
- red fluorescent protein
- YFP
- yellow fluorescent protein
- DTT
- dithiothreitol
- HBSS
- Hanks’ balanced salt solution
The YFP variant with enhanced stability, citrine, was used (21).
REFERENCES
- 1.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 1.Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Proc. Natl. Acad. Sci. U. S. A. 1991;88:4171–4175. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffer PJ, Woodgett JR. Eur. J. Biochem. 1991;201:475–481. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- 4.Brazil DP, Hemmings BA. Trends Biochem. Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 5.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. J. Biol. Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 6.Franke TF, Kaplan DR, Cantley LC, Toker A. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 7.Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA. J. Biol. Chem. 1997;272:8474–8481. doi: 10.1074/jbc.272.13.8474. [DOI] [PubMed] [Google Scholar]
- 8.James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, Alessi DR. Biochem. J. 1996;315:709–713. doi: 10.1042/bj3150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alessi DR, Janes SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, Cohen P. Curr. Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 10.Stokoe D, Stephens L, Copeland T, Gaffney PRJ, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins P. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 11.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 12.Toker A, Newton AC. J. Biol. Chem. 2000;275:8271–8274. doi: 10.1074/jbc.275.12.8271. [DOI] [PubMed] [Google Scholar]
- 13.Brownawell AM, Kops GJ, Macara IG, Burgering BM. Mol. Cell. Biol. 2001;21:3534–3546. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meier R, Alessi DR, Cron P, Andjelkovic M, Hemmings BA. J. Biol. Chem. 1997;272:30491–30497. doi: 10.1074/jbc.272.48.30491. [DOI] [PubMed] [Google Scholar]
- 15.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Campbell RE, Ting AY, Tsien RY. Nat. Rev. Mol. Cell. Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 17.Sato M, Umezawa Y. Methods. 2004;32:451–455. doi: 10.1016/j.ymeth.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Violin JD, Zhang J, Tsien RY, Newton AC. J. Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki K, Sato M, Umezawa Y. J. Biol. Chem. 2003;278:30945–30951. doi: 10.1074/jbc.M212167200. [DOI] [PubMed] [Google Scholar]
- 20.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. Proc. Natl. Acad. Sci. U. S. A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim MA, Kikani CK, Wick MJ, Dong LQ. Proc. Natl. Acad. Sci. U. S. A. 2003;100:14006–14011. doi: 10.1073/pnas.2335486100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zacharias DA, Violin JD, Newton AC, Tsien RY. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 23.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. J. Biol. Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 24.Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. J. Biol. Chem. 2000;275:36108–36115. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- 25.Durocher D, Taylor IA, Sarbassova D, Haire LF, Westcott SL, Jackson SP, Smerdon SJ, Yaffe MB. Mol. Cell. 2000;6:1169–1182. doi: 10.1016/s1097-2765(00)00114-3. [DOI] [PubMed] [Google Scholar]