

Abstract
Gastrulation is the first major morphogenetic movement in development, and requires dynamic regulation of cell adhesion and the cytoskeleton. C. elegans gastrulation begins with the migration of the two endodermal precursors, Ea and Ep, from the surface of the embryo into the interior. Ea/Ep migration provides a relatively simple system to examine the intersection of cell adhesion, cell signaling, and cell movement. Ea/Ep ingression depends on correct cell fate specification and polarization, apical myosin accumulation, and Wnt activated actomyosin contraction that drives apical constriction and ingression (Lee et al., 2006; Nance et al., 2005). Here, we show that Ea/Ep ingression also requires the function of either HMR-1/cadherin or SAX-7/L1CAM. Both cadherin complex components and L1CAM are localized at all sites of cell-cell contact during gastrulation. Either system is sufficient for Ea/Ep ingression, but loss of both together leads to a failure of apical constriction and ingression. Similar results are seen with isolated blastomeres. Ea/Ep are properly specified and appear to display correct apical-basal polarity in sax-7(eq1); hmr-1(RNAi) embryos. Significantly, in sax-7(eq1); hmr-1(RNAi) embryos Ea and Ep fail to accumulate myosin (NMY-2::GFP) at their apical surfaces, but in either sax-7(eq1) or hmr-1(RNAi) embryos, apical myosin accumulation is comparable to wildtype. Thus, the cadherin and L1CAM adhesion systems are redundantly required for localized myosin accumulation, and hence for actomyosin contractility during gastrulation. We also show that sax-7 and hmr-1 function are redundantly required for Wnt-dependent spindle polarization during division of the ABar blastomere, indicating that these cell surface proteins redundantly regulate multiple developmental events in early embryos.
Keywords: L1CAM, cadherin, C. elegans, gastrulation, non-muscle myosin
INTRODUCTION
Cell-cell adhesion plays an early and essential role in embryogenesis. Multicellular animals depend on adhesive systems to hold their cells together, and dynamic regulation of adhesion is essential for cellular movements involved in embryonic patterning and morphogenesis. A major unanswered question in developmental biology is how various adhesion systems function together to regulate adhesion. Two adhesion systems of particular interest in development are L1CAMs and cadherins. L1CAMs bind extracellularly to other L1CAMs, to a variety of other integral membrane proteins, and to the ECM, and are known to function in nervous system development (reviewed in (Maness and Schachner, 2007)).
Cadherins are a key class of molecules involved in cell-cell adhesion, both during morphogenesis and later, because they regulate many developmental events including migration, polarity, cell shape and cell sorting (Gumbiner, 2005). Cadherins act by binding homotypically to cadherins on adjacent cells, linking cells to each other and to the actin cytoskeleton through catenins and other linker proteins (Gates and Peifer, 2005). Cadherins are also involved in complex feedback to the actin cytoskeleton via Rho family GTPases (reviewed in (Braga and Yap, 2005; Nelson, 2008)).
Gastrulation, which involves the internalization of the endodermal and mesodermal precursors, is the first major morphogenetic movement in metazoans. In most species, cadherins are essential both for the organization of the pregastrula embryo, and for gastrulation. For example, in zebrafish the E-cadherin homologue, halfbaked/cdh1, is required for epiboly of the blastoderm around the yolk cell (Babb et al., 2001; Kane et al., 1996; Kane et al., 2005). Organization of the blastula (Heasman et al., 1994) and germ-layer separation at the onset of gastrulation (Wacker et al., 2000) in Xenopus requires cadherins as well. In Drosophila, E-cadherin/shotgun is required for gastrulation and tracheal outgrowth (Tepass et al., 1996; Uemura et al., 1996). The expression pattern of E-cadherin/CAD-1 also suggests a dynamic role for cadherins in sea urchin development (Miller and McClay, 1997). In mice, E-cadherin is required before gastrulation for cell-cell adhesion during compaction and trophectoderm formation, but not for earlier cleavage events (De Vries et al., 2004; Larue et al., 1994; Riethmacher et al., 1995). Later, β-catenin is required for gastrulation in mouse embryos (Haegel et al., 1995).
The C. elegans embryo is an excellent in vivo system for examining the role of cell-cell adhesion during gastrulation, because major classes of cell-cell adhesion molecules are well conserved (Cox et al., 2004). Gastrulation and subsequent cell movements can be readily observed via live cell microscopy (Hardin and Lockwood, 2004). Stereotypic cell divisions and lineages allow for a complete description of cell-cell signaling and other interactions in early C. elegans development. In addition, cell-cell adhesion molecules are conserved in C. elegans, including the Ig superfamily protein L1CAM, cadherin-1, claudin family proteins, Crumbs and neurexin family proteins, and selectins (Cox et al., 2004), making C. elegans a good system for examining combinatorial effects of multiple adhesion complexes.
Early gastrulation events in C. elegans are similar to invagination movements in other animals in that apical-basal polarity is defined by PAR proteins and internalization of endodermal precursors involves apical constriction (Chisholm, 2006). The C. elegans endoderm is derived from a single blastomere, E, that is specified by a series of maternal and zygotic signaling events (reviewed in (Maduro, 2006)). Before gastrulation begins, E divides to generate the two endodermal precursor cells, Ea and Ep. Gastrulation begins at the 26–28 cell stage with the apical constriction and ingression of Ea/Ep. Nearly half of the cells in the embryo then ingress over a period of 130 minutes, including additional endodermal, mesodermal and germ-line precursors. The number of cells in the embryo increases to around 350 during this time (Sulston et al., 1983). Finally, the gastrulation cleft is sealed by neuroblasts that migrate together over the cleft (reviewed in (Chisholm and Hardin, 2005)).
Evidence thus far favors a model in which apical constriction drives Ea/Ep ingression, thereby displacing Ea/Ep internally. At the same time MSxx and P4 close over the ventral surface (Lee and Goldstein, 2003; Nance et al., 2003). When Ea/Ep are properly polarized, myosin concentrates on their apical surfaces (Nance et al., 2003). In polarized cells a Wnt signal leads to phosphorylation of myosin light chains, presumably driving actomyosin contractility, causing apical constriction and ingression (Lee et al., 2006). Surprisingly, the role of major cell-cell adhesion systems in C. elegans gastrulation remains unexplored.
In C. elegans there is a single conserved classical cadherin-catenin complex (CCC) consisting of the core components HMR-1/cadherin, HMP-2/β-catenin, and HMP-1/α-catenin. Unlike other species, C. elegans gastrulation does not usually require HMR-1/cadherin or other members of the CCC. The CCC is essential for the subsequent morphogenetic process of ventral enclosure (Costa et al., 1998; Raich et al., 1999), during which the embryonic epidermis wraps around the embryo and seals at the midline. Maternal loss of CCC components leads to failure to form nascent cell-cell adhesions at the ventral midline, whereas zygotic loss typically leads to arrest slightly later, during elongation. Localization of other junctional molecules, including VAB-9, a C. elegans BCMP1/claudin (Simske et al., 2003), depends on HMR-1. Thus, while the CCC is important for later morphogenesis, it is not essential for gastrulation. Another cell-cell adhesion molecule expressed in early C. elegans embryos is the sole L1CAM homologue, SAX-7 (formerly known as LAD-1 (Chen et al., 2001)). SAX-7 plays no obvious role in gastrulation or early morphogenesis, but sax-7 null mutants exhibit cell-cell positioning defects later in multiple tissues, as well as defects in pharyngeal shape in larvae (Axang et al., 2007; Sasakura et al., 2005; Wang et al., 2005).
Here we show that the CCC and SAX-7 are present at cell-cell contacts throughout early embryogenesis in C. elegans, and that HMR-1 and SAX-7 act synergistically to regulate blastomere compaction and a Wnt-dependent spindle polarization event in the early embryo. In addition, loss of both HMR-1 and SAX-7 leads to severe disruption of gastrulation. Ea/Ep appear properly polarized and are correctly specified, but fail to concentrate non-muscle myosin II at their apical surfaces and fail to apically constrict. Thus, HMR-1 and SAX-7 work redundantly to localize myosin, which in turn is required for apical constriction and ingression.
METHODS AND MATERIALS
Strains and Alleles
The Bristol strain N2 was used as wildtype. Strains were maintained and cultured as previously described (Brenner, 1974). Previous studies used a sax-7(eq1) strain that was outcrossed 8 times (Wang et al., 2005), which showed 40% embryonic lethality that was later found to be due to an interacting, tightly linked second-site mutation (L. Chen, personal communication and (Zhou et al., 2008)). Further outcrossing recombined away the mutant to generate the strain LH81 sax-7(eq1) IV, which has minimal embryonic lethality (gift of L. Chen). LH81 adults are subtly Unc with defects in positional maintenance of axons and neuronal cell bodies, as seen with other sax-7 alleles (L. Chen, personal communication) (Sasakura et al., 2005; Zallen et al., 1999). Additional mutant and reporter strains used include: KK866 [itIs153 (axEx1094[pie-1p::par-2::gfp), pRF4(+)] (Morton et al., 2002), SU348 [sax-7(eq1) IV]; itIs153 [axEx1094[pMW103, pie-1p::par-2::gfp, pRF4(+)]], JJ1579 [unc-119(ed3) III; zuIs77(par-6p::par-6::gfp), unc-119(+)] (gift of Jim Priess), SU367 [sax-7(eq1) IV; zuIs77(par-6p::par-6::gfp), unc-119(+)]; JJ1473 [unc-119(ed3) III; zuIs45 (nmy-2p::nmy-2::gfp, unc-119(+)) V] (Nance et al., 2003), SU344 [sax-7(eq1) IV; zuIs45(nmy-2::gfp, unc-119(+)) V], CB362 [unc-44(e362) IV], JR2274[wIS137(rol-6; end-3p::end-3[p202L]::gfp)] (Walston et al., 2004); SU375 [sax-7(eq1) IV; jcEx102 pMM446 (end-3p::end-3[p202L]::gfp), pRF4]. All strains were maintained and filmed at 20°C, except for KK866 and SU348, which were maintained at 22–24°C.
RNA Interference (RNAi)
All RNAi experiments were done by injection of double-stranded RNA at a concentration of 2 μg/μl into young adult hermaphrodites. Injected worms were allowed to lay embryos for at least 20 hours prior to collection of embryos from gravid hermaphrodites for imaging. RNAi efficacy was verified by comparison with published RNAi phenotypes available on Wormbase (http://www.wormbase.org). For cDNAs, PCR amplification using T3 and/or T7 primers produced RNA templates. Ambion Megascript T3 and T7 kits (Ambion, Austin, Texas) were used for in vitro transcription (as described in (Walston et al., 2004)). Transcription products were purified, mixed in equimolar concentrations and double stranded. Sources of cDNA templates include Y. Kohara (Gene Network Lab, NIG, Japan) who kindly provided the following cDNA clones: pry-1 yk1084e03, afd-1 yk1725g10, spc-1 yk1502e02, igcm-1 yk1008b11, hmp-1 yk437b8, hmp-2 yk1047e06 and yk77d10, sax-7 yk766b04, jac-1 yk131h12; and the Ahringer feeding library: sma-1 (R31.1), unc-70 (KK11C4.3), erm-1 (C01G8.5) (Fraser et al., 2000; Kamath et al., 2003). Double stranded hmr-1 was made using a vector, JW12, which contains the full-length hmr-1 cDNA cloned into the PCR blunt vector (Invitrogen; clone courtesy of J. Wang). Primers used to amplify hmr-1A residues 52-1973 were (T7/T3 sequences are in lowercase and the hmr-1 sequence is in uppercase): T7HMR5A (taatacgactatagGGGAGTTCTAAGAGGCTCTGGGTG) and T3HMR3A (aattaaccctcactaaagggCTTCCAGTGAGATACGGCCGC). While hmr-1 is notoriously difficult to knock down by feeding RNAi (Kennedy et al., 2004), injection of double stranded RNA leads to phenotypes that are consistent between preparations and reflect strong loss of function, based on published data (Costa et al., 1998). Results shown are from multiple different preps.
Nomarski and Fluorescent Imaging
Nomarski images were collected as previously described (Raich et al., 1999). Twenty-two optical sections with 1 μm spacing were collected at one-minute intervals. Fluorescent images were collected with a Perkin-Elmer UltraVIEW spinning disk confocal microscope as described (Sheffield et al., 2007). For staining fixed embryos, forty optical sections with 0.5 μm spacing were captured; for live images 10–20 optical sections with 1.0 μm spacing were captured at one-minute intervals using a Hamamatsu ORCA-ER CCD camera using Perkin Elmer UltraVIEW Imaging Suite software. Images were analyzed using custom ImageJ plugins (available at http://worms.zoology.wisc.edu/research/4d/4d.html). Supplemental NMY-2 moves demonstrate NMY-2 levels over a 15 minute interval between P4 birth and the time of Ea/Ep ingression. Movies were synchronously staged by placing MSxx nuclear reformation at timepoint eight. ImageJ plugins generated by T. Collins at the McMaster Biophotonics Facility were used to correct for photobleacing (available at http://www.macbiophotonics.com/).
Antibody Staining
Gravid hermaphrodites were cut open in distilled water and embryos were mounted on poly-L-lysine coated slides for freeze cracking (Simske et al., 2003). The following primary antibodies/antisera and dilutions were used: goat anti-HMP-2, 1:50, (Santa Cruz, (cT-20):sc-15520); rabbit anti-SAX-7, 1:300, gift of L. Chen (Chen et al., 2001), rabbit anti-HMR-1 (1:3), gift of J. Priess (Costa et al., 1998); rabbit anti-GFP, 1:500–1:1000, RDI); mouse anti-PAR-3 (Developmental Studies Hybridoma Bank; 1:2 dilution of monoclonal supernatant). Overnight primary antibody incubations were at 4°C. Secondary antibodies were used at a 1:50 dilution. Because both the anti-HMR-1 and anti-SAX-7 antibodies were generated in rabbits, we used anti-HMP-2 staining as a marker for the CCC in some experiments.
Ea/Ep Differentiation
Standard markers for gut differentiation, birefringent rhabditin granules, were observed by polarizing optics (Laufer et al., 1980). END-3::GFP expression was assessed in JR2274 and SU375 via anti-GFP immunostaining as above, but with primary antibody at 1:100 dilution.
Apical Constriction Measurements
The surface connecting the border of Ea-MSxx to Ea-Ep (for Ea) and the surface connecting Ea-Ep to Ep-P4 (for Ep) were measured by tracing images in ImageJ using a Wacom CTE-630 drawing pad, computing the number of pixels and converting to μm distance in ImageJ, similar to previous studies (Lee et al., 2006). Apical surface length for Ea and Ep was always measured for the focal plane in which the nucleus of the cell being measured was most sharply in focus. In some double mutants the cell positions in the posterior vary, such that D contacts Ep in the focal plane containing the sharply focused Ep nucleus and P4 drops to a separate focal plane, but P4 still contacts Ep. In these instances, the measurements were made using the focal plane in which the Ep-P4 border was visible. Using the same end points from the measurement mentioned above, the feret length was also measured for Ea and Ep. The ratio of feret length (F) to apical perimeter (P) was used to describe the “flatness” of the apical surface of Ea and Ep. “Flatness” was then normalized by comparing the experimental surface to a flat surface and a half circle, which corresponds to a perfectly rounded cell:
A flatness of 0 corresponds to a perfectly circular cell; a flatness of 1 corresponds to a perfectly flat apex.
Blastomere Isolations and Imaging
Embryos were devitellinized and blastomeres were cultured essentially as described (Edgar, 1995). The following changes were made to the protocol: watch glasses were treated with Rain-X, eggshells were dissolved in a 3:20 dilution of 4–6% sodium hypochlorite (Sigma) for two minutes; tetramisole, chymotrypsin and silicon oil were not used; the P1 blastomere was separated from AB by drawing embryos through a narrow-bore pipet. Blastomeres were filmed by placing them on a scored ring slide in EGM (Edgar, 1995), covered with a coverslip and sealed with Velap (heated 1:1:1, petroleum jelly: lanolin: paraffin wax). Ea/Ep movement analysis was similar to (Lee and Goldstein, 2003). Contact length and circularity measurements are described in Supplemental Fig. S1. Birefringent rhabditin granules were always observed in terminal images of isolated blastomeres.
Analysis of PAR-2, PAR-6 and NMY-2 Localization
PAR-2 and PAR-6 localization was scored in P4 stage embryos. For NMY-2::GFP localization, a box drawn at the apical surface was converted into kymographs of maximum pixel intensity over time using ImageJ. NMY-2::GFP levels were calculated using methods similar to (Lee et al., 2006). In brief, in a middle focal plane of Ea and Ep the total pixel intensity of the apical surface or at the Ea/Ep border was compared to the pixel intensity of the cytoplasm above background at 0, 5 and 10 minutes after the P4 nuclear envelope was re-established following P4 birth. Total pixel intensity was measured to avoid potential artifacts due to changes in the area of the apical membrane.
Data and Statistical Analysis
Normalization of data was performed using Microsoft Excel. Contingency table analysis (Fig. S4) and Analysis of Variance (ANOVA) with Tukey-Kramer posthoc analysis (Fig. 3C, D, 4B, C and 6B, C) were performed using Microsoft Excel spreadsheets and StatPlus:mac 2009 (AnalystSoft, Inc.). Circular statistics (Fig. 4D) were performed using routines written for Macintosh OS 9 or below by JH as described (Hardin, 1989). For details on these tests, see (Zar, 2010).
Fig. 3.
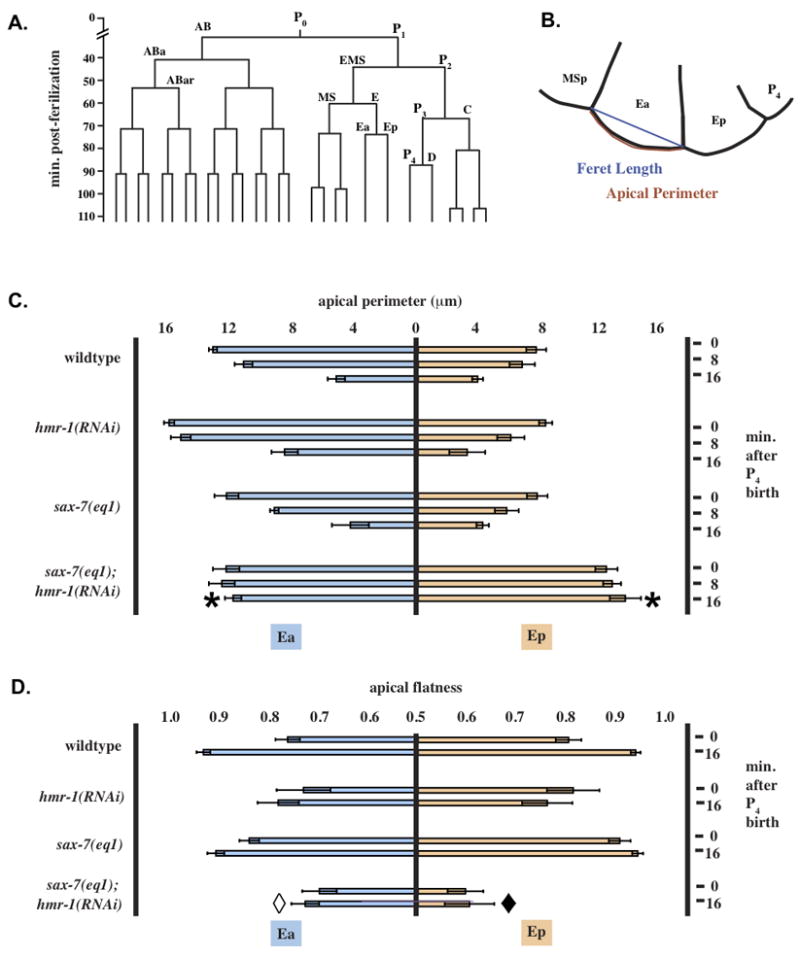
Ea and Ep do not apically constrict in intact sax-7(eq1); hmr-1(RNAi) embryos. (A) Early wild-type embryonic lineage referred to in the text (anterior daughter cells shown to the left). (B) Example of traces used to measure apical membrane length and feret length. (C) Apical membrane length of Ea/Ep at 0, 8 and 16 minutes after the P4 nuclear envelope was re-established following P4 birth. Error bars = 95% confidence intervals. *, change from t = 0 to t = 16 significantly different from other genotypes, p < 0.02, n = 5. Other genotypes not significantly different from one another. (D) Data shown represent normalized feret length/apical membrane length at 0 and 16 minutes after the P4 nuclear envelope was reestablished following P4 birth. Data were normalized as described Materials and Methods; a completely flat surface has a value of 1, while a half circle has a value of 0. Error bars are 95% confidence intervals. Open diamond, t = 16 value significantly different from wildtype, p < 0.02. Closed diamond, t = 16 value significantly different from wildtype and sax-7, p < 0.01, n = 5.
Fig. 4.

SAX-7 and the CCC cooperatively regulate blastomere compaction and displacement of P4 and MSxx. (A) Cell movements in P1 isolates over time. Ea/Ep cells are marked with white stars, the initial direction of movement of P4 was considered to be an indicator of the apical side of the blastomeres (black asterisks) and white arrows show directions of movements of MSxx and P4. Times shown are post-Ea/Ep birth. Ea and Ep divide prior to the 60 min. time point. Some cells are not visible due to their position along the Z-axis. (B) Length of contact area between Ea/Ep in blastomeres (see Supplemental Fig. S1 for method of measurement). Open diamond, significantly different from sax-7, p < 0.03; not significantly different from sax-7;hmr-1. *, significantly different from wildtype and sax-7, p < 0.0001; n = 5 for all genotypes. (C) Circularity of the Ep blastomere. Open diamond, significantly different from wildtype and sax-7, p < 0.01, n = 5 for each genotype; not significantly different from sax-7;hmr-1. #, not significantly different from wildtype. *, significantly different from wildtype and sax-7, p < 0.001; not significantly different from sax-7;hmr-1. (D) Changes in angular orientation of P4 and MSxx relative to Ea/Ep as a measure of apical constriction of Ea/Ep. MSxx angle in sax-7;hmr-1 significantly different, p < 0.05, n = 5 for each genotype.
Fig. 6.
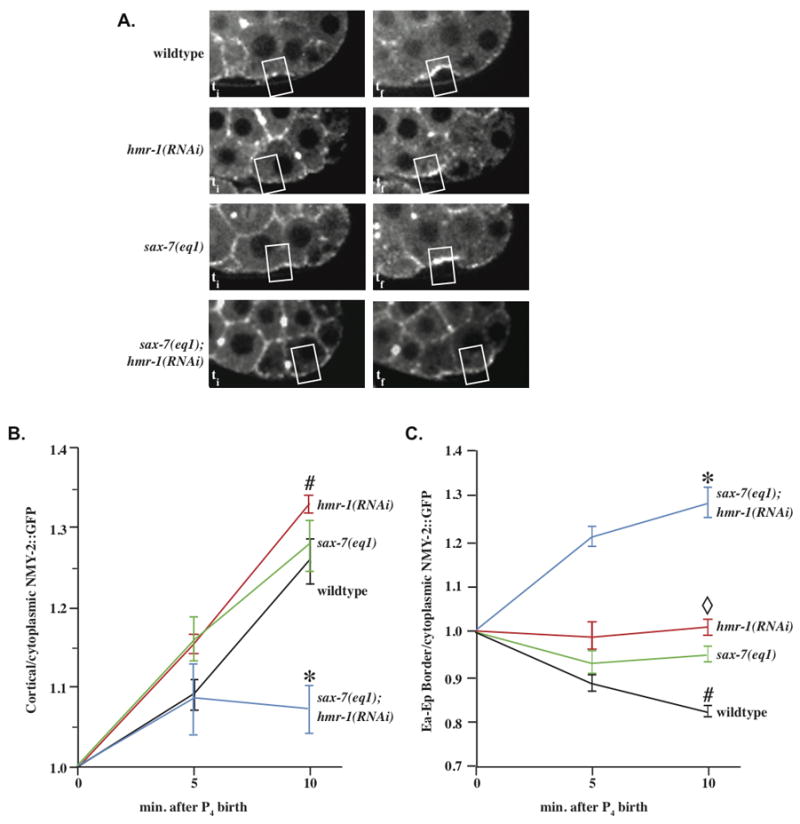
hmr-1(RNAi); sax-7(eq1) embryos fail to accumulate NMY-2 on the apical surfaces of Ea/Ep. (A) NMY-2::GFP levels at apical surface of Ea/Ep over time in wild-type and mutant embryos. The two left-hand panels show NMY-2 accumulation at two successive time points in wild-type and mutant embryos. The right column represents kymographs of the apical surface over time. (B) NMY-2::GFP levels at the apical surface of Ea/Ep over time in wild-type and mutant embryos. *, significantly different from other genotypes, p. < 0.02, n = 5 for each genotype. #, wildtype, hmr-1, and sax-7 not significantly different. (C) NMY-2::GFP levels at the Ea/Ep border over time in wild-type and mutant embryos. *, significantly different from other genotypes, p < = 0.01, n = 5 of each genotype. #, significantly different from hmr-1 and sax-7, p < 0.05. Open diamond, hmr-1 and sax-7 not significantly different.
RESULTS
The CCC and SAX-7/L1CAM localize to distinct submembrane domains
In early C. elegans embryos through the initiation of gastrulation, the CCC localizes to all sites of cell-cell contact ((Costa et al., 1998) and Fig. 1Ai, Bi, Bii). HMR-1/cadherin staining is similar to that of HMP-2/β-catenin (Fig. 1Ai, Bii); thus the CCC is found at cell-cell contacts in early embryos. SAX-7 is also at sites of cell-cell contact ((Chen et al., 2001) and Fig. 1Aii). Because the localization of the CCC and SAX-7 had not yet been examined simultaneously, we examined whether SAX-7 co-localized with the CCC. We observed that HMP-2 and SAX-7 sometimes co-localize but can also be present in distinct submembrane regions prior to or at the initiation of gastrulation (Fig. 1Aii).
Fig. 1.

The CCC and SAX-7 localize to regions of cell-cell contact. All embryos are wildtype unless indicated. (A) Anti-HMP-2 and anti-SAX-7 staining in a 26–28 cell embryo. (B) Anti-HMR-1 staining of (i) 4 cell, (ii) 26–28 cell. (C) Anti-HMR-1 staining of an embryo near the 26–28 cell stage and a Nomarski image of the same focal plane. (D) Anti-SAX-7 staining of (i) 4 cell, (ii) 26–28 cell embryos. (E) Anti-HMR-1 staining of (i) 4 cell, (ii) 26–28 cell sax-7(eq1) embryos. Scale in A smaller than other panels. Scale bars in this and all subsequent figures are 10 μm. Anterior is to the left in all figures. Nuclear signal in Dii and Eii is END-3::GFP.
To examine the function of hmr-1 in gastrulation, we knocked down maternal and zygotic expression in embryos by injection of dsRNA into hermaphrodite worms. hmr-1(RNAi) results in 100% embryonic lethality, primarily due to defects in ventral enclosure. We observed no HMR-1 in hmr-1(RNAi) pre-gastrulae (Fig. Ci, Cii). Thus, hmr-1(RNAi) is an effective way to knockdown hmr-1 in most tissues. SAX-7 localization in hmr-1(RNAi) embryos appears similar to that in wildtype (Fig. 1Di-ii). Thus, hmr-1(RNAi) does not affect SAX-7 localization. To examine the function of sax-7 during gastrulation, we used the eq1 allele, which contains a 2020 base pair deletion and is predicted to encode a protein missing the fifth extracellular fibronectin III domain, the transmembrane domain and the intracellular domains, and thus behaves as a genetic null (Wang et al., 2005). Our genetic experiments show that the eq1 allele is recessive (data not shown). Staining of sax-7(eq1) embryos for SAX-7 yields no signal (data not shown). Localization of HMR-1 in sax-7(eq1) embryos appears similar to wildtype (Fig. 1Ei-ii). Thus, HMR-1 and SAX-7 do not depend on one another for localization to sites of cell-cell contact.
Intestinal precursors Ea and Ep fail to ingress in sax-7(eq1); hmr-1(RNAi) embryos
Because loss of CCC components in C. elegans results in less severe phenotypes than seen in many other species, we asked whether SAX-7 might act redundantly with the CCC in early embryos. In wild-type C. elegans embryos, Ea/Ep initiate gastrulation by migrating from the ventral periphery to the center of the embryo, where they become surrounded by other cells, and only then do they begin the divisions that allow them to form the intestine (Fig. 2, Movie 1). At this time in development the outer surface of the embryo is considered apical and cells are arranged with their basal surfaces surrounding a small blastocoel (Nance and Priess, 2002). Ingression involves apical constriction of Ea/Ep, which pulls MSxx and P4 over the apical surface of Ea/Ep (Lee and Goldstein, 2003). Early gastrulation defects never occur in hmr-1(RNAi) or sax-7(eq1) embryos (Fig. 2, Table 1). However, in sax-7(eq1); hmr-1(RNAi) embryos Ea/Ep do not initiate gastrulation before dividing and instead divide on the surface (Fig. 2, Movie 2). Despite this failure to ingress, Ea/Ep remain in continuous contact with each other. Although co-RNAi against two genes gives somewhat weaker knockdown than targeting each gene separately (Gonczy et al., 2000), early gastrulation defects still occur in sax-7(RNAi); hmr-1(RNAi) embryos. These defects are less severe: although Ea and Ep remain in continual contact, one of the pair may ingress far enough to be surrounded by other cells at the time of division while the other is exposed to the apical surface of the embryo (Table 1).
Fig. 2.

Intestinal precursors Ea/Ep fail to ingress in sax-7(eq1); hmr-1(RNAi) embryos. Images are from Nomarski time-lapse movies of the indicated genotypes. E cells are pseudocolored blue in all frames.
Table 1.
Ea and Ep defects following CCC and SAX-7 loss of function
| Embryo | % with any Ea/Ep defect | % Ingression failure | n | |||
|---|---|---|---|---|---|---|
| Ea alone | Ep alone | Both Ea and Ep | % Embryos Surviving | |||
| Wildtype | 0 | 0 | 0 | 0 | 100 | 19 |
| hmr-1(RNAi) | 0 | 0 | 0 | 0 | 0 | 20 |
| sax-7(eq1) | 0 | 0 | 0 | 0 | 100 | 21 |
| sax-7(eq1); hmr-1(RNAi) | 100 | 0 | 0 | 100 | 0 | 39 |
| sax-7(RNAi); hmr-1(RNAi) | 96 | 25 | 8 | 63 | 0 | 24 |
| hmp-1(RNAi) | 0 | 0 | 0 | 0 | 0 | 34 |
| hmp-2(RNAi) | 0 | 0 | 0 | 0 | 0 | 32 |
| sax-7(eq1); hmp-1(RNAi) | 90 | 16 | 23 | 52 | 0 | 31 |
| sax-7(eq1); hmp-2(RNAi) | 91 | 11 | 29 | 51 | 0 | 35 |
| sax-7(eq1); jac-1(RNAi) | 0 | 0 | 0 | 0 | 100 | 45 |
Cadherins can mediate some functions without a link to catenins. For example, human leukemia tissue culture cells require exogenous E-cadherin to aggregate, but the aggregation still occurs in the absence of the E-cadherin cytoplasmic domain (Ozawa and Kemler, 1998). Thus, we examined whether HMR-1 provides sufficient function for early gastrulation in sax-7(eq1) by knocking down other members of the CCC. Although early gastrulation defects are not seen following hmp-1/α-catenin (RNAi) or hmp-2/β-catenin (RNAi) alone, such defects occurred in sax-7(eq1); hmp-1(RNAi) and in sax-7(eq1); hmp-2(RNAi) embryos (Table 1); however, these defects were less severe than in sax-7(eq1); hmr-1(RNAi) embryos and instead resembled double RNAi against hmr-1 and sax-7. Knockdown of the C. elegans p120 catenin, jac-1, did not result in gastrulation defects in the sax-7(eq1) background. The lack of a phenotype with jac-1(RNAi) is not surprising, since in C. elegans (Pettitt et al., 2003) and Drosophila (Myster et al., 2003) p120ctn is not essential, as it appears to be in vertebrates (Fang et al., 2004; Peifer and Yap, 2003; Reynolds, 2007). Thus, SAX-7 and the core CCC components function redundantly in the initiation of gastrulation in C. elegans. Since RNAi knockdown of gene function is more effective against hmp-2 and hmp-1 than against hmr-1 (our unpublished observations), the more severe phenotype seen with hmr-1(RNAi) suggests that HMR-1 carries out some adhesive functions independent of the rest of the CCC during gastrulation.
Ea and Ep differentiate properly but fail to apically constrict in sax-7(eq1); hmr-1(RNAi) embryos
The gastrulation defects seen in sax-7(eq1); hmr-1(RNAi) embryos could have multiple causes. First, it is possible that endodermal fate is not properly specified; failure of Ea and Ep ingression is often due to misspecification (Maduro and Rothman, 2002). With correct endoderm specification, the Ea/Ep cell division cycle is about 25 minutes longer than that of MSxx (Fig. 3A) (Knight and Wood, 1998; Schierenberg et al., 1980; Sulston et al., 1983). The expected cell cycle delay of 20–25 minutes is observed in sax-7(eq1); hmr-1(RNAi) embryos (Supplemental Table S1). In addition, birefringent gut granules, markers of endoderm specification, were observed in all sax-7(eq1); hmr-1(RNAi) embryos examined (20/20), as was end-3::gfp expression (46/46; Supplemental Fig. 2), which is a marker for differentiation of Ea and Ep (Maduro et al., 2005). Thus, Ea/Ep are properly specified as endodermal precursors.
Second, when Ea/Ep ingress, the apical surfaces of Ea/Ep decrease in area as MSxx and P4 move over them (Lee and Goldstein, 2003). Reduction in the apical surfaces of Ea/Ep occurred in wildtype, sax-7(eq1) alone, and hmr-1(RNAi) alone embryos (Fig. 3B, C), with the apical membrane length of Ea shrinking from about 12 μm to 5 μm and the apical membrane length of Ep shrinking from about 8 μm to 4 μm over the same time period. Ea/Ep did not apically constrict in sax-7(eq1); hmr-1(RNAi) embryos, consistent with their failure to ingress (Fig. 3C). In addition, the exposed apical surface of Ep is larger in double mutants than in wild-type embryos (double mutants significantly different from other genotypes for both Ea and Ep, p < 0.02, n = 5 embryos of each genotype examined). Thus, loss of SAX-7 and the CCC together leads to failure to reduce the apical surfaces of Ea and Ep.
SAX-7 and the CCC cooperatively regulate blastomere compaction
Although it is well established that the CCC regulates blastomere compaction in metazoans (De Vries et al., 2004), it is not known whether L1CAMs function similarly. We therefore assessed whether SAX-7 and the CCC cooperate in blastomere compaction. We observed that all cells in sax-7(eq1); hmr-1(RNAi) embryos appear to be more rounded than in wildtype or in sax-7(eq1) alone. We devised a metric for the “flatness” of the apical surface of Ea/Ep (see Methods and Materials), and determined this value for Ea/Ep in various genetic backgrounds. In wild-type and sax-7(eq1) embryos the apical surface flattens over time as cells ingress (Fig. 3B, D). In hmr-1(RNAi) embryos the apical surface of Ea/Ep remains somewhat rounded, even as ingression occurs, although this difference from wildtype was not quite statistically significant. The apical surface is least flat in sax-7(eq1); hmr-1(RNAi) embryos and does not flatten over time (for Ea, the final value was significantly different from wildtype, p < 0.02; for Ep, the double mutants were significantly different from wildtype and sax-7, p < 0.01. n = 5 embryos each). Thus HMR-1 and SAX-7 appear to cooperatively regulate apical flattening of Ea and Ep in intact embryos.
The eggshell and vitelline envelope are likely important as a passive mechanical influence on blastomere packing in the embryo and may mask effects on blastomere compaction. A similar effect on blastomere compaction has been reported in mouse embryos (De Vries et al., 2004). We therefore removed the eggshell and vitelline envelope and observed cells in isolation. By isolating the cell P1 and letting it divide, one can observe apical constriction without the influence of other cells. Experiments with isolated blastomeres have previously shown that Ea/Ep ingression is primarily due to apical constriction and not dependent on signals from distant cells (Lee and Goldstein, 2003).
We observed Ea/Ep in wildtype, sax-7(eq1), hmr-1(RNAi), and sax-7(eq1); hmr-1(RNAi) blastomeres (Fig. 4A; n = 5 for each genotype). Surprisingly, sax-7(eq1); hmr-1(RNAi) cells adhere to one another, and after being forcibly separated, can re-adhere strongly within a few minutes of being placed into contact with blastomeres of the same genotype (data not shown). Thus, despite loss of HMR-1 and SAX-7, cells still retain some adhesiveness, presumably due to other cell-cell adhesion molecules. However, blastomeres isolated from double mutants are less compacted, since their surface area of contact with neighbors is obviously reduced (Fig. 4A, B). sax-7 mutant blastomeres do not show reduced contact compared to wildtype (Fig. 4B). Loss of hmr-1 alone led to a greater reduction in contact, but not quite at the level required for statistical significance at the 95% confidence level compared to wildtype in posthoc tests. The double mutant combination led to further reduction in contact significantly different from wildtype and sax-7 (p < 0.0001, n = 5), but this difference was not sufficient to lead to a statistically significant difference from hmr-1 alone at the 95% confidence level given the number of cases examined (Fig. 4B). Similar results were obtained using circularity measurements: while sax-7 cells are not different from wildtype, hmr-1(RNAi) cells have a significantly more rounded shape (Fig. 4C; p < 0.01, n = 5 ) which is not significantly different from sax-7;hmr-1 cells. Isolated mutant cells are correctly specified, as indicated by the timing of blastomere divisions relative to wild-type isolates, the relative size of daughter blastomeres, and the consistent presence of gut granules in terminal isolates in all cases (data not shown).
Autonomous apical constriction of Ea/Ep is observed following P4 birth in wild-type embryos (compare P4 and MSx(x) position at 20 minutes vs. 48 minutes in Fig. 4A). As previously observed (Lee and Goldstein, 2003), configurations of isolated blastomeres in culture is variable, as is the degree of Ea/Ep movement (partially due to movements along the Z-axis). In blastomeres isolated from hmr-1(RNAi) or sax-7(eq1) embryos, Ea/Ep constrict similarly to wild-type embryos. However, this apical constriction is not seen in sax-7(eq1); hmr-1(RNAi) embryos (Fig. 4A). Instead, movements appear to be random, with P4 moving in one direction and MSxx moving in the other direction, (toward the asterisk in Fig. 4A) and MSxx moving in the other direction. Blastomeres from double mutants are much less compact, forming loose clusters by 60 minutes, instead of the more rigid linear structure typically observed in other conditions (Fig. 4A).
We next measured the change in angle of P4 and MSxx position with respect to the Ea/Ep axis, which has been used previously to assess apical constriction in isolated blastomeres (Lee and Goldstein, 2003). The initial direction of movement of P4 was considered to be an indicator of the apical side of the blastomeres. P4 and MSxx moved towards each other over the apical surface of Ea/Ep in wild-type, hmr-1(RNAi) and sax-7(eq1) isolates (Fig. 4D). Movements were variable between isolates, partially due to movements along the Z-axis. Wildtype and sax-7(eq1) isolates showed similar overall movements, while hmr-1(RNAi) blastomeres showed somewhat reduced movements. In contrast, P4 and MSxx move in opposite directions in sax-7(eq1); hmr-1(RNAi) embryos (Fig. 4D; double mutant angular distribution significantly different for MSxx, p < 0.05, n = 5 of each genotype). Thus, we conclude that in addition to decreased compaction, directed apical constriction does not occur in sax-7(eq1); hmr-1(RNAi) blastomeres.
PAR-2::GFP and PAR-6::GFP localization indicates Ea/Ep are properly polarized in sax-7(eq1); hmr-1(RNAi) embryos
In blastomeres the pattern of cell-cell contacts establishes apical-basal PAR asymmetry (Nance and Priess, 2002). Thus, cell-cell adhesion can influence cell polarity. Non-myosin II (NMY-2) accumulation at the apical surface depends on PAR proteins: PAR depletion leads to delayed ingression of Ea/Ep (Nance et al., 2003). We therefore examined the apical-basal polarity of Ea and Ep in sax-7(eq1); hmr-1(RNAi) embryos with the cell polarity markers PAR-2::GFP and PAR-6::GFP. In wildtype embryos PAR-2::GFP is predominantly basolateral (14/14 embryos) (Fig. 5), as expected (Lee et al., 2006). (Note that in P4, which gives rise to the germ cells, PAR-2 is cortically localized because the promoter used in the expression construct is highly active in P4.) Similar localization of PAR-2::GFP was observed in hmr-1(RNAi) (12/12), sax-7(eq1) (14/14), and sax-7(eq1); hmr-1(RNAi) (12/12) embryos. PAR-6::GFP, is apically enriched in Ea/Ep in wildtype embryos (26/26), but not as obviously in other cells. Similar localization of PAR-6::GFP was observed in hmr-1(RNAi) (21/21), sax-7(eq1) (18/18), and sax-7(eq1); hmr-1(RNAi) (20/20) embryos (Supplemental Fig. S3). In conclusion, Ea/Ep appear properly polarized in sax-7(eq1); hmr-1(RNAi) embryos and loss of overt polarity cannot explain Ea/Ep ingression defects.
Fig. 5.
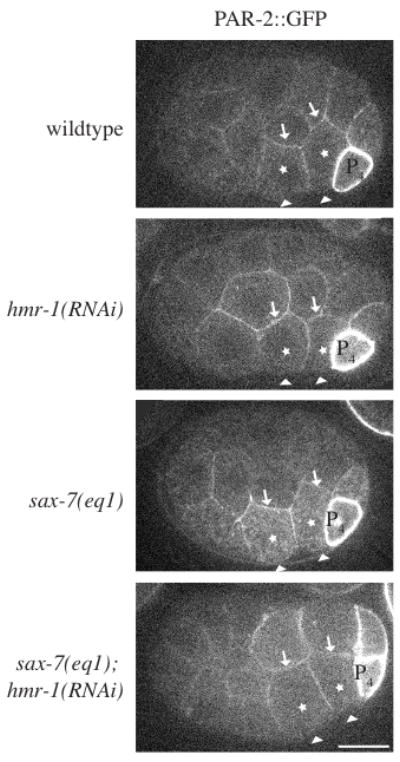
PAR localization indicates Ea/Ep are correctly polarized in sax-7(eq1); hmr-1(RNAi) embryos. PAR-2::GFP was imaged in Ea/Ep shortly following P4 birth in wild-type and mutant embryos. Although GFP distribution was monitored throughout all focal planes, single focal planes are shown. The arrowheads point to the apical surface of Ea/Ep (note that vitelline envelope appears white in sax-7(eq1), but is not the apical surface). Arrows point to the basal surface. The promoter used in the PAR-2 expression construct is highly active in P4 leading to cortical localization. In some embryos PAR-2::GFP is brighter in D than in other cells. This occurs more frequently in sax-7(eq1); hmr-1(RNAi), but has been observed in all genotypes.
hmr-1(RNAi); sax-7(eq1) embryos fail to accumulate NMY-2/myosin II on the apical surfaces of Ea/Ep and fail to exclude NMY-2 from the Ea/Ep border
Previously, NMY-2/myosin II has been shown to accumulate on the apical surfaces of Ea/Ep (Nance and Priess, 2002) along with an accumulation of phosphorylated light chains, which is thought to drive the contraction of the apical surface of ingressing cells (Lee et al., 2006). Thus, accumulation of NMY-2 is essential for apical constriction. We observed that NMY-2::GFP accumulates comparably in wild type, hmr-1(RNAi), and sax-7(eq1) embryos, but not in sax-7(eq1); hmr-1(RNAi) (Fig. 6A, Movies 3–6). We measured NMY-2::GFP levels at the apical surfaces of Ea/Ep and found that NMY-2::GFP fails to accumulate in sax-7(eq1); hmr-1(RNAi) embryos (Fig. 6B), but that in sax-7(eq1) or hmr-1(RNAi) embryos, NMY-2::GFP concentrates at levels similar to wildtype. Thus, loss of both hmr-1 and sax-7 function leads to failure of NMY-2::GFP concentration at the apical surface, but either SAX-7 or HMR-1 is sufficient to achieve an NMY-2 concentration that drives ingression of Ea/Ep. The failure to accumulate NMY-2 when HMR-1 and SAX-7 are absent presumably prevents apical constriction and ingression of Ea/Ep.
We also observed that in wild-type embryos, the NMY-2::GFP localized between Ea/Ep decreases dramatically as it accumulates on their apical surface (Fig. 6C). However, in sax-7(eq1); hmr-1(RNAi) embryos, NMY-2::GFP levels at the Ea/Ep border actually increase over time (Fig. 6C). The level of NMY-2::GFP on the Ea/Ep border in sax-7(eq1) or hmr-1(RNAi) embryos is intermediate to the levels in wild-type and double loss of function embryos. The striking differences observed at the Ea/Ep border in various genotypes (Movies 3–6), suggests that HMR-1 and SAX-7 play complementary roles at the Ea/Ep border to direct NMY-2 away from their shared border to the apical surface.
Later gastrulation defects are also observed following loss of CCC and SAX-7 function
Additional cells ingress later in gastrulation to internalize the mesoderm and endoderm. At this time a ventral gastrulation cleft is visible (Nance and Priess, 2002; Sulston et al., 1983), but by 40 minutes after the entry of the last cells into the gastrulation cleft, it is normally closed over by neuroblasts (Fig. 7). In sax-7(eq1); hmr-1(RNAi) embryos, the ventral gastrulation cleft is larger, fails to close, and cells subsequently spill out (Fig. 7, Table 2)). Interestingly, all hmr-1(RNAi) embryos have a larger gastrulation cleft than seen in wild-type embryos, but it closes 92% of the time, while sax-7(eq1) embryos have a normally sized gastrulation cleft. As with hmr-1(RNAi), removal of other core members of the CCC in the sax-7(eq1) background leads to late gastrulation defects or other milder defects during ventral enclosure, when the hypodermis wraps around and encloses the embryo (time 130 in Fig. 7), or later during elongation (Table 2).
Fig. 7.

Later gastrulation defects are also observed following loss of CCC and SAX-7 function. Nomarski images of the ventral surface of wild-type and mutant embryos. Open gastrulation clefts are outlined with dashed lines. White stars indicate extruded cells.
Table 2.
Other morphogenetic defects following CCC and SAX-7/L1CAM loss of function*
| Embryo | % Gastrulation cleft closure failure | % Rupture during ventral enclosure | % Hmr phenotype | % Hmp-like dorsal balloon | % Embryos Survive | n |
|---|---|---|---|---|---|---|
| Wildtype | 0 | 0 | 0 | 0 | 100 | 19 |
| hmr-1(RNAi) | 8 | 17 | 75 | 0 | 0 | 36 |
| sax-7(eq1) | 0 | 0 | 0 | 0 | 100 | 21 |
| sax-7(eq1); hmr-1(RNAi) | 100 | 0 | 0 | 0 | 0 | 39 |
| sax-7(RNAi); hmr-1(RNAi) | 92 | 8 | 0 | 0 | 0 | 24 |
| hmp-1(RNAi) | 0 | 0 | 25 | 75 | 0 | 40 |
| hmp-2(RNAi) | 3 | 8 | 75 | 14 | 0 | 40 |
| sax-7(eq1); hmp-1(RNAi) | 43 | 57 | 0 | 0 | 0 | 46 |
| sax-7(eq1); hmp-2(RNAi) | 33 | 67 | 0 | 0 | 0 | 51 |
| sax-7(eq1); jac-1(RNAi) | 0 | 0 | 0 | 0 | 100 | 47 |
Developmental events are shown from earliest to latest: the most severe developmental defects are on the left, starting with late gastrulation.
Knockdown of candidate SAX-7 interactors does not identify synergistic functional interactions with HMR-1 during gastrulation
The CCC links cells together via connections to the actin cytoskeleton (Gates and Peifer, 2005). How SAX-7 functions in adhesion in the early embryo is unknown, though it could also link to the actin cytoskeleton. The intracellular domain of SAX-7 contains a FERM domain binding motif, an ankyrin binding domain and a PDZ binding domain (Pocock et al., 2007). Candidate linker molecules were tested by injection RNAi or by using mutant alleles along with hmr-1(RNAi) and scoring for gastrulation defects.
L1 and NrCAM appear to function through ankyrins in mice (Scotland et al., 1998; Zhou et al., 1998). Similarly, SAX-7 recruits the C. elegans ankyrin, UNC-44, to sites of cell-cell contact in post-gastrulation embryos (Chen et al., 2001). We therefore assessed synergy between hmr-1 and unc-44. However, hmr-1(RNAi); unc-44(e362) embryos did not have gastrulation defects (Supplemental Table S2). We also knocked down expression of the C. elegans spectrins: sma-1/βH-spectrin, spc-1/α-spectrin, and unc-70/β-spectrin, but their loss along with hmr-1(RNAi) does not lead to increased gastrulation defects, even though knockdown for each displays the later morphogenetic phenotypes previously reported, e.g., (Norman and Moerman, 2002), indicating that knockdown did occur (Supplemental Table S2). Thus, SAX-7 does not appear to have a stringent requirement for the spectrin-ankyrin cytoskeleton during gastrulation.
L1CAMs also bind ezrin, a member of the Ezrin/Radixin/Moesin membrane-cytoskeletal linker family (Dickson et al., 2002). erm-1 is the only ERM family member required for C. elegans development and depletion of erm-1 perturbs gut tubulogenesis (Gobel et al., 2004; Van Furden et al., 2004). Although we observed similar gut defects in later development, we did not observe an effect of hmr-1(RNAi); erm-1(RNAi) on gastrulation, so there does not appear to be a stringent requirement for ERM-1 in conjunction with HMR-1 during gastrulation.
Finally, neuroglian, the Drosophila homologue of SAX-7, has been shown to bind to Echinoid (Islam et al., 2003). Echinoid has also been shown to function with the CCC (Wei et al., 2005) in the Drosophila wing disc epithelium. Although we did not have access to probes to easily assess extent of knockdown of the Echinoid homologue, icgm-1, and igcm-1 knockdown does not have any obvious phenotypes (our unpublished observations), we nevertheless performed knockdown of igcm-1, and hmr-1 together; this treatment does not result in gastrulation defects (Supplemental Table S2).
In summary, these experiments suggest that if SAX-7 links to the actin cytoskeleton in early embryogenesis, it likely does so via an unknown pathway, and how it facilitates apical constriction and blastomere compaction in the early embryo remains unclear.
Loss of HMR-1 and SAX-7 leads to delayed contact between C and ABar and failure to orient the ABar division
In addition to redundant roles in gastrulation, HMR-1 and SAX-7 have synergistic roles in ABar spindle alignment. Correct ABar spindle alignment depends on a Wnt signal transmitted by contact with the C blastomere (Walston et al., 2004). Shortly after its birth, C extends to contact ABar; the two cells rapidly develop a large area of close contact. This reorients the ABar spindle as ABar begins to divide 5–6 minutes after C birth. We observed that knockdown of members of the CCC leads to defects in alignment of the ABar spindle, resulting in abnormal cell division orientation (Supplemental Fig. S4A, Table S3).
In embryos where we could clearly observe the surface of C, we measured the time C first contacts ABar. Significantly, when C-ABar contact is delayed or fails altogether, the division orientation of ABar is more likely to be abnormal (because it begins to divide before being oriented) (Supplemental Fig. S4B). Contact is delayed in hmr-1(RNAi) embryos and even more so in hmr-1(RNAi); sax-7(eq1) embryos. Thus, SAX-7 loss of function enhances defects in ABar division orientation. Most previously reported mutants that affect ABar division orientation are in the Wnt signaling pathway (mom-1/Porcupine, mom-2/Wnt, mom-3/mig-14/wntless, mom-5/Fz, dsh-2/disheveled, mig-5/disheveled) (Rocheleau et al., 1999; Thorpe et al., 1997; Walston et al., 2004). However, the defects we report here appear to depend on cell-cell contact at the appropriate time, (as observed previously for ABar defects in other mutants (Walston et al., 2004) and for EMS division orientation (Goldstein et al., 2006)). When C contacts ABar early enough the division orientation is correct in mutants, implying a functional Wnt signaling pathway is present. Thus, the ABar defect presumably arises because C is prevented from extending to touch ABar sufficiently early. Another possibility is that loss of HMR-1 leads to a mild disorganization of the early embryo, causing delayed contact between C and ABar. However, cells appear to be correctly positioned in pre-gastrulation double mutant embryos, making this possibility unlikely.
Interestingly, we also observed a cell division orientation defect in isolates. In wild-type embryos, the division orientation of blastomeres starts with the future germline, the P lineage, always posterior. A switch in the division orientation occurs for the P2 division, resulting in the smaller cell P3 being born anterior to the larger cell C (Supplemental Fig. S5). In this way the germline maintains contact with the future intestine, which later nourishes the germ cells (Schierenberg, 1987). The cells P1 and P2 are the most posterior cells in isolated blastomeres as well. However, in half (4/8) of the sax-7(eq1); hmr-1(RNAi) P1 isolates, the P2 blastomere divided incorrectly so that the P3 blastomere was positioned posterior to C (Supplemental Fig. S5). This defect is not likely a result of altered cell fate because the timing of cell divisions of the daughter cells P3 and C, and the division orientation of P3, are comparable to wildtype (data not shown). Instead, this division defect may be due to impaired blastomere compaction that reduces the surface area of contact between blastomeres, preventing the correct polarization signal from reaching P2.
DISCUSSION
HMR-1/cadherin and SAX-7/L1CAM cooperatively regulate multiple processes in the early embryo
This is the first study to demonstrate shared developmental roles for two major cell-cell adhesion systems: L1CAMs and cadherins. We show that a cadherin, HMR-1, and an L1CAM, SAX-7, play redundant roles in several key processes in the early C. elegans embryo. Given the importance of cell adhesion in metazoans, it is surprising how little attention has been paid to functional redundancy of cell-cell adhesion molecules in early embryos. To our knowledge this issue has not been explored previously in the case of cadherins and L1CAMs. N-cadherin and L1CAM function together in axonal regeneration in chick explants in vitro, but the mechanisms underlying this interaction are unknown (Blackmore and Letourneau, 2006). In vertebrate tumor cells, L1CAM and cadherins appear to interact very indirectly, via transcriptional feedback. Increased levels of L1CAM in tumors are associated with decreased levels of E-cadherin (Boo et al., 2007), and nuclear β-catenin can activate L1CAM expression, transforming cells (Gavert et al., 2005). L1CAM expression disrupts adherens junctions and increases motility in MCF breast carcinoma cells (Shtutman et al., 2006). Our study is the first to examine an L1CAM and a cadherin together in vivo in early embryos. Moreover, since adhesion in the early C. elegans embryo does not require new transcription (Costa et al., 1998; Raich et al., 1999), transcriptional mechanisms cannot account for the synergy we observe, and hence the functional redundancy must involve more direct mechanisms.
HMR-1/cadherin and SAX-7/L1CAM cooperatively regulate blastomere compaction
The more rounded apices of Ea and Ep in intact hmr-1; sax-7 loss of function embryos suggests that HMR-1 and SAX-7 influence blastomere compaction in the early C. elegans embryo. Further analysis of isolated blastomeres confirms this view. Loss of both adhesion systems together severely decreases the surface area of contact between Ea and Ep. Such loss of contact is consistent with an overall decrease in blastomere adhesion, or an increase in cortical tension, or both. Cadherins engage in complex reciprocal interactions with Rho family GTPases (Braga and Yap, 2005; Nelson, 2008); it is possible that L1CAMs can also affect these actin modulators. Further experiments involving assessment of Rho GTPase activity, as well as cell- and temporally specific alteration of Rho GTPase function in backgrounds compromised for HMR-1 and SAX-7 will be required to resolve these issues.
The other straightforward explanation for the combined effects of HMR-1 and SAX-7 involves cell-cell adhesion. Surprisingly, adhesion is not totally abrogated when both proteins are removed, even when the constraining influence of the vitelline envelope is removed. This suggests that other adhesion molecules cooperate with HMR-1 and SAX-7 in the early embryo, in contrast to results from early mammalian embryos, in which E-cadherin alone appears crucial (De Vries et al., 2004). Such molecules could presumably be identified through their enhancement of cell-cell adhesion defects in hmr-1 or sax-7 loss-of-function backgrounds.
One issue in our experiments involves the use of RNAi knockdown to remove hmr-1 function. We chose this methodology for two reasons, First, hmr-1 nulls are zygotic lethal, but there is appreciable maternal hmr-1 mRNA loaded into zygotes (Costa et al., 1998). Knockdown of hmr-1 via injection RNAi remove both maternal and zygotic hmr-1 mRNA. Second, although complete abrogation of expression of maternal gene products can be accomplished using germline mosaic embryos, generating sufficient embryos simultaneously mutant for sax-7 to perform the dynamic analyses described here make this method technically challenging. In our hands, as in previous reports, injection RNAi leads to phenotypes indistinguishable from hmr-1 germline mosaic embryos (Raich et al., 1999), indicating that knockdown is essentially complete. Moreover, our staining experiments show no detectable HMR-1 protein in the early embryo. It is nonetheless possible that undetectable levels of HMR-1 persist. Even if HMR-1 is present at low levels, however, we have still uncovered a novel cooperative role for SAX-7 in blastomere adhesion.
Several other cell-cell contact events in the early embryo appear to redundantly require HMR-1 and SAX-7. These proteins also function redundantly in late gastrulae, where their simultaneous loss results in failure of the ventral neuroblasts to seal the gastrulation cleft. Although the molecular events that underlie this process are unclear, it involves numerous cell surface interactions, including signaling events associated with Eph/ephrin and Slit/Robo signaling (reviewed in (Chisholm and Hardin, 2005; Ghenea et al., 2005)). Our results suggest that redundant homotypic interactions mediated by cadherins and L1CAMs also aid cleft closure.
Loss of HMR-1 and SAX-7 affects cell division orientation
Our experiments implicate HMR-1 and SAX-7 in the timing of C contact with ABar, an event that is normally accompanied by a rapid increase in surface area of contact between an anterior extension of C and the posterior surface of ABar (Walston et al., 2004). A stratighfoward interpretation of our results is that these cells must establish a firm adhesion for C to transmit its Wnt-based signal to ABar, thereby orienting the ABar spindle (Rocheleau et al., 1999; Thorpe et al., 1997) (previously observed for ABar in other mutants (Walston et al., 2004)). The cell division orientation of the ABar blastomere is directed by a Wnt signal from the C blastomere. The division orientation of the EMS blastomere also depends on a Wnt signal (Schlesinger et al., 1999), yet we did not observe any abnormalities in division of EMS in intact embryos or isolated blastomeres (data not shown), so Wnt signals are apparently intact in sax-7(eq1); hmr-1(RNAi) embryos. One explanation for this difference may be the time course of establishment of the contact between P2/EMS and C/ABar. In the former case, the inducing and induced cells lie next to one another continuously after their birth, due to the geometry of cell divisions during second cleavage. In the latter case, C must extend a small extension anteriorly to contact ABar. Loss of HMR-1 and SAX-7 perturbs this event, thus preventing transmission of the orienting Wnt signal. We show here that when HMR-1 is knocked down, C sometimes fails to contact ABar in time to orient its division. SAX-7 is not required for C to contact ABar when HMR-1 is present, but its loss along with members of the CCC increases ABar polarity defects. Such defects are likely related to cooperative adhesion provided by SAX-7, although we cannot completely rule out subtle secondary effects that perturb either the ability of C to produce a cortical extension or the substratum of underlying blastomeres along which C extends anteriorly.
An additional defect in division orientation was observed in isolated blastomeres: the division orientation of the P2 blastomere depends on the redundant function of HMR-1 and SAX-7. In sax-7(eq1); hmr-1(RNAi) blastomeres, the P2 division orientation is randomized, resulting in C being more anterior to P3 half the time. This phenotype has been reported once previously: Berkowitz et al. cite a personal communication with E. Schierenberg that indicates mes-1 mutant blastomeres do not display the polarity reversal observed in the P2 division in wild-type embryos (Berkowitz and Strome, 2000). Thus, it is possible that the reduced adhesive surface between P2 and EMS prevents sufficient MES-1 signal from reaching P2, resulting in random division orientation. Intact mes-1 mutant embryos display symmetric P2 and P3 divisions, resulting in similarly sized daughter cells (Berkowitz and Strome, 2000). We do not observe this phenotype, so we believe that the MES-1 signal is still produced, but may fail to be transmitted between isolated sax-7(eq1); hmr-1(RNAi) blastomeres due to further reduction of the surface area of contact when the vitelline envelope is removed. Further studies will be required to clarify these issues.
HMR-1 and SAX-7 in gastrulation
Our evidence demonstrates that either HMR-1 or SAX-7 is sufficient for gastrulation. Phenotypes for removal of HMR-1 by RNAi, in germline mosaics, or in various mutants include a small percentage of gastrulation cleft-sealing defects, but no early gastrulation defects (our results and (Costa et al., 1998; Raich et al., 1999). SAX-7 has not previously been shown to play a role in gastrulation, but only in later cell positioning events (Sasakura et al., 2005; Wang et al., 2005). Only when these two adhesion systems were removed simultaneously did the redundant requirement become apparent. While hmr-1 loss of function affects the ability of the apical surfaces of Ea and Ep to flatten as they ingress, it does not disrupt ingression. Simultaneous loss of hmr-1 and sax-7 function further compromises this flattening and completely blocks Ea/Ep ingression movements. This failure is accompanied by loss of NMY-2/non-muscle myosin II accumulation at the apical surfaces of Ea and Ep. In isolated blastomeres we observed similar effects on ingression movements and the shape of blastomeres.
C. elegans gastrulation is initiated by the ingression of the intestinal precursors Ea/Ep. For Ea and Ep to ingress properly, they must by properly specified (Nance et al., 2005). Moreover, they must constrict their apical surfaces, an event that is thought to require accumulation and activation of myosin at the apical surfaces of Ea and Ep. Several preexisting events are required for correct apical myosin accumulation and activation. Apical-basal polarized distribution of PAR proteins is essential for the accumulation of myosin on the ventral surfaces of Ea/Ep (Nance et al., 2003). Once myosin is accumulated, Wnt signaling is required for activation of actomyosin contractility, which leads to apical constriction of Ea/Ep and draws neighboring cells over their surfaces (Lee et al., 2006). We show here that apical myosin accumulation requires the presence of HMR-1/cadherin or SAX-7/L1CAM. However, this requirement does not appear to act upstream of PAR-dependent polarization, since both PAR-2 and PAR-6 accumulate in hmr-1; sax-7 loss of function embryos.
HMR-1 and SAX-7 play redundant roles in apical myosin accumulation and complementary roles in preventing myosin accumulation at the Ea/Ep border
There are several ways in which cadherins and L1CAMs might redundantly regulate gastrulation movements in C. elegans. As discussed above, general effects on cortical stiffness could be involved. Since apical constriction of Ea/Ep and/or the lateral movements of cells adjacent to them that occur during gastrulation (Nance et al., 2005) could be affected by general changes in cortical stiffness, we cannot rule out this possibility. Given available evidence, however, we propose that increased adhesion and subsequent signaling conferred by HMR-1 and SAX-7 at the Ea/Ep border is vital to ingression primarily through effects on NMY-2 accumulation. In other situations in which myosin accumulation is blocked at the apical surface of Ea/Ep, e.g., in par mutants (Nance et al., 2003; Nance and Priess, 2002), or in cases when myosin is not activated, either through pharmacological treatments (Lee and Goldstein, 2003) or as a result of blocking Wnt signaling (Lee et al.), ingression of Ea and Ep fails in a manner very similar to what we have observed. Failure of NMY-2 to accumulate at the apical surface in sax-7(eq1); hmr-1(RNAi) embryos thus provides the simplest explanation for failure of Ea and Ep to ingress.
In other organisms the CCC and various myosins appear to functionally interact and affect one another’s recruitment. For example, myosin VI and Armadillo/β-catenin are mutually dependent in Drosophila border cells (Geisbrecht and Montell, 2002). Similarly, mammalian myosin VI is recruited to cadherin-based adhesions in an E-cadherin dependent fashion, and is need for formation of a functional junction (Maddugoda et al., 2007). In epithelial cells, following E-cadherin ligation, Rho kinase recruits and activates myosin II, which in turn recruits more cadherin at cell-cell contacts (Shewan et al., 2005). We are not aware of other studies showing a link between an L1CAM and myosin, but we propose that together HMR-1 and SAX-7 are likewise involved in signaling that leads to myosin recruitment in the early C. elegans embryo.
The mechanisms by which HMR-1 and SAX-7 cooperate to regulate myosin accumulation are unclear. Using a candidate gene approach we attempted to identify SAX-7 effectors that might play a role in Ea/Ep ingression, but were unable to identify any obvious candidates. Moreover, HMR-1 and SAX-7 are largely absent from the apical surface of Ea/Ep and so do not appear to directly localize myosin to the apical surface. (Although Nance et al reported that SAX-7 is not restricted to basolateral surfaces of cells, we do not observe SAX-7 staining at the apical surface (Nance et al., 2003).) HMR-1 and SAX-7 could function at the lateral surfaces of Ea/Ep to stabilize structures that initiate a myosin scaffold that spans the apex. Consistent with this possibility, in intact embryos the apical surface of Ea/Ep is significantly more rounded in double mutants than in either loss of function mutant alone.
Alternatively, apical redistribution of NMY-2 in Ea/Ep could occur via exclusion of myosin from the shared Ea/Ep border and its redistribution to the apical surface. Such a mechanism would be similar to the role proposed for E-cadherin in MDCK cells forming de novo contacts in vitro (Yamada and Nelson, 2007). Lengthening of de novo contacts in MDCK cells requires E-cadherin, which excludes myosin II from the center of the contact and drives it towards cortical actin bundles at the edges of the growing contact. Similarly, we observed that in wild-type embryos the level of NMY-2 at the shared border drops dramatically over time, while increasing at the apex. The loss of either HMR-1 or SAX-7 results in steady levels of NMY-2 at the Ea/Ep border, but some increase in apical NMY-2 concentration over time; in this case, apical constriction still occurs. In contrast, loss of HMR-1 and SAX-7 together results in an increase in NMY-2 concentration at the shared border over time, and virtually no increase in apical NMY-2 concentration. In this case, apical constriction fails, presumably because there is insufficient NMY-2 to construct an actomyosin contractile network. In MDCK cells these events depend on Rac and Rho signaling (Yamada and Nelson, 2007); these molecules may also be used to ferry NMY-2 away from the cell-cell border to the free surface in Ea/Ep in C. elegans embryos as well. NMY-2 is presumably regulated via phosphorylation of its associated regulatory light chain (Lee et al., 2006), which could in turn result from signaling from these small GTPases. We attempted to detect phospho-myosin light chain via traditional means and via tyramide enhancement but were unable to reproducibly detect signal, as has been reported by others (Gally et al., 2009). Accumulation of myosin light chains depends on accumulation of the associated heavy chains, whose rod domains are required for filament assembly in metazoans, including C. elegans (Landsverk and Epstein, 2005). Since the primary defects we identified involves NMY-2 mislocalization, it is unlikely that light chain regulation is a major mode of synergistic regulation by SAX-7 and HMR-1. Future experiments will be needed to refine out understanding of how these two cell surface proteins coordinately regulate gastrulation.
Supplementary Material
Acknowledgments
We are grateful to Lihsia Chen for SAX-7 antibodies, the outcrossed sax-7(eq1) strain, LH81, and sharing unpublished data. We thank Yuji Kohara for providing numerous cDNAs, Jim Priess and Jeremy Nance for providing gfp reporter strains, and Morris Maduro for providing pMM446. Some strains were provided by the C. elegans Genetics Center, which is funded by the NIH National Center for Research Resources. We thank current and former members of the Hardin lab, especially Ryan King, Allison Lynch, Joshua Roehrich, Timothy Walston, and Jindong Wang for helpful interactions. This work was supported by an American Cancer Society Postdoctoral Fellowship awarded to TMG, a NIH Postdoctoral Fellowship awarded to EAC and NIH grant GM058038 awarded to JH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Theresa M. Grana, Email: tgrana@umw.edu.
Elisabeth A. Cox, Email: coxe@geneseo.edu.
Allison M. Lynch, Email: amlynch2@wisc.edu.
References
- Axang C, Rauthan M, Hall DH, Pilon M. The twisted pharynx phenotype in C. elegans. BMC Dev Biol. 2007;7:61. doi: 10.1186/1471-213X-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb SG, Barnett J, Doedens AL, Cobb N, Liu Q, Sorkin BC, Yelick PC, Raymond PA, Marrs JA. Zebrafish E-cadherin: expression during early embryogenesis and regulation during brain development. Dev Dyn. 2001;221:231–7. doi: 10.1002/dvdy.1132. [DOI] [PubMed] [Google Scholar]
- Berkowitz LA, Strome S. MES-1, a protein required for unequal divisions of the germline in early C. elegans embryos, resembles receptor tyrosine kinases and is localized to the boundary between the germline and gut cells. Development. 2000;127:4419–31. doi: 10.1242/dev.127.20.4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore M, Letourneau PC. L1, beta1 integrin, and cadherins mediate axonal regeneration in the embryonic spinal cord. J Neurobiol. 2006;66:1564–83. doi: 10.1002/neu.20311. [DOI] [PubMed] [Google Scholar]
- Boo YJ, Park JM, Kim J, Chae YS, Min BW, Um JW, Moon HY. L1 expression as a marker for poor prognosis, tumor progression, and short survival in patients with colorectal cancer. Ann Surg Oncol. 2007;14:1703–11. doi: 10.1245/s10434-006-9281-8. [DOI] [PubMed] [Google Scholar]
- Braga VM, Yap AS. The challenges of abundance: epithelial junctions and small GTPase signalling. Curr Opin Cell Biol. 2005;17:466–74. doi: 10.1016/j.ceb.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Ong B, Bennett V. LAD-1, the Caenorhabditis elegans L1CAM homologue, participates in embryonic and gonadal morphogenesis and is a substrate for fibroblast growth factor receptor pathway-dependent phosphotyrosine-based signaling. J Cell Biol. 2001;154:841–55. doi: 10.1083/jcb.200009004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm AD. Gastrulation: Wnts signal constriction. Curr Biol. 2006;16:R874–6. doi: 10.1016/j.cub.2006.09.028. [DOI] [PubMed] [Google Scholar]
- Chisholm AD, Hardin J. Epidermal morphogenesis. WormBook. 2005:1–22. doi: 10.1895/wormbook.1.35.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Raich W, Agbunag C, Leung B, Hardin J, Priess JR. A putative catenin-cadherin system mediates morphogenesis of the Caenorhabditis elegans embryo. J Cell Biol. 1998;141:297–308. doi: 10.1083/jcb.141.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox EA, Tuskey C, Hardin J. Cell adhesion receptors in C. elegans. J Cell Sci. 2004;117:1867–70. doi: 10.1242/jcs.01177. [DOI] [PubMed] [Google Scholar]
- De Vries WN, Evsikov AV, Haac BE, Fancher KS, Holbrook AE, Kemler R, Solter D, Knowles BB. Maternal beta-catenin and E-cadherin in mouse development. Development. 2004;131:4435–45. doi: 10.1242/dev.01316. [DOI] [PubMed] [Google Scholar]
- Dickson TC, Mintz CD, Benson DL, Salton SR. Functional binding interaction identified between the axonal CAM L1 and members of the ERM family. J Cell Biol. 2002;157:1105–12. doi: 10.1083/jcb.200111076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar LG. Blastomere culture and analysis. Methods Cell Biol. 1995;48:303–21. doi: 10.1016/s0091-679x(08)61393-x. [DOI] [PubMed] [Google Scholar]
- Fang X, Ji H, Kim SW, Park JI, Vaught TG, Anastasiadis PZ, Ciesiolka M, McCrea PD. Vertebrate development requires ARVCF and p120 catenins and their interplay with RhoA and Rac. J Cell Biol. 2004;165:87–98. doi: 10.1083/jcb.200307109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–30. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- Gally C, Wissler F, Zahreddine H, Quintin S, Landmann F, Labouesse M. Myosin II regulation during C. elegans embryonic elongation: LET-502/ROCK, MRCK-1 and PAK-1, three kinases with different roles. Development. 2009;136:3109–19. doi: 10.1242/dev.039412. [DOI] [PubMed] [Google Scholar]
- Gates J, Peifer M. Can 1000 reviews be wrong? Actin, alpha-Catenin, and adherens junctions. Cell. 2005;123:769–72. doi: 10.1016/j.cell.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Gavert N, Conacci-Sorrell M, Gast D, Schneider A, Altevogt P, Brabletz T, Ben-Ze’ev A. L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol. 2005;168:633–42. doi: 10.1083/jcb.200408051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbrecht ER, Montell DJ. Myosin VI is required for E-cadherin-mediated border cell migration. Nat Cell Biol. 2002;4:616–20. doi: 10.1038/ncb830. [DOI] [PubMed] [Google Scholar]
- Ghenea S, Boudreau JR, Lague NP, Chin-Sang ID. The VAB-1 Eph receptor tyrosine kinase and SAX-3/Robo neuronal receptors function together during C. elegans embryonic morphogenesis. Development. 2005;132:3679–90. doi: 10.1242/dev.01947. [DOI] [PubMed] [Google Scholar]
- Gobel V, Barrett PL, Hall DH, Fleming JT. Lumen morphogenesis in C. elegans requires the membrane-cytoskeleton linker erm-1. Dev Cell. 2004;6:865–73. doi: 10.1016/j.devcel.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Goldstein B, Takeshita H, Mizumoto K, Sawa H. Wnt signals can function as positional cues in establishing cell polarity. Dev Cell. 2006;10:391–6. doi: 10.1016/j.devcel.2005.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P, Echeverri C, Oegema K, Coulson A, Jones SJ, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, Hannak E, Kirkham M, Pichler S, Flohrs K, Goessen A, Leidel S, Alleaume AM, Martin C, Ozlu N, Bork P, Hyman AA. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000;408:331–6. doi: 10.1038/35042526. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–34. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–37. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- Hardin J. Local shifts in position and polarized motility drive cell rearrangement during sea urchin gastrulation. Dev Biol. 1989;136:430–45. doi: 10.1016/0012-1606(89)90268-6. [DOI] [PubMed] [Google Scholar]
- Hardin J, Lockwood C. Skin tight: cell adhesion in the epidermis of Caenorhabditis elegans. Curr Opin Cell Biol. 2004;16:486–92. doi: 10.1016/j.ceb.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Heasman J, Ginsberg D, Geiger B, Goldstone K, Pratt T, Yoshida-Noro C, Wylie C. A functional test for maternally inherited cadherin in Xenopus shows its importance in cell adhesion at the blastula stage. Development. 1994;120:49–57. doi: 10.1242/dev.120.1.49. [DOI] [PubMed] [Google Scholar]
- Islam R, Wei SY, Chiu WH, Hortsch M, Hsu JC. Neuroglian activates Echinoid to antagonize the Drosophila EGF receptor signaling pathway. Development. 2003;130:2051–9. doi: 10.1242/dev.00415. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–7. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kane DA, Hammerschmidt M, Mullins MC, Maischein HM, Brand M, van Eeden FJ, Furutani-Seiki M, Granato M, Haffter P, Heisenberg CP, Jiang YJ, Kelsh RN, Odenthal J, Warga RM, Nusslein-Volhard C. The zebrafish epiboly mutants. Development. 1996;123:47–55. doi: 10.1242/dev.123.1.47. [DOI] [PubMed] [Google Scholar]
- Kane DA, McFarland KN, Warga RM. Mutations in half baked/E-cadherin block cell behaviors that are necessary for teleost epiboly. Development. 2005;132:1105–16. doi: 10.1242/dev.01668. [DOI] [PubMed] [Google Scholar]
- Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645–9. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- Knight JK, Wood WB. Gastrulation initiation in Caenorhabditis elegans requires the function of gad-1, which encodes a protein with WD repeats. Dev Biol. 1998;198:253–65. [PubMed] [Google Scholar]
- Landsverk ML, Epstein HF. Genetic analysis of myosin II assembly and organization in model organisms. Cell Mol Life Sci. 2005;62:2270–82. doi: 10.1007/s00018-005-5176-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue L, Ohsugi M, Hirchenhain J, Kemler R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A. 1994;91:8263–7. doi: 10.1073/pnas.91.17.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer JS, Bazzicalupo P, Wood WB. Segregation of developmental potential in early embryos of Caenorhabditis elegans. Cell. 1980;19:569–77. doi: 10.1016/s0092-8674(80)80033-x. [DOI] [PubMed] [Google Scholar]
- Lee JY, Goldstein B. Mechanisms of cell positioning during C. elegans gastrulation. Development. 2003;130:307–20. doi: 10.1242/dev.00211. [DOI] [PubMed] [Google Scholar]
- Lee JY, Marston DJ, Walston T, Hardin J, Halberstadt A, Goldstein B. Wnt/Frizzled signaling controls C. elegans gastrulation by activating actomyosin contractility. Curr Biol. 2006;16:1986–97. doi: 10.1016/j.cub.2006.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddugoda MP, Crampton MS, Shewan AM, Yap AS. Myosin VI and vinculin cooperate during the morphogenesis of cadherin cell cell contacts in mammalian epithelial cells. J Cell Biol. 2007;178:529–40. doi: 10.1083/jcb.200612042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro MF. Endomesoderm specification in Caenorhabditis elegans and other nematodes. Bioessays. 2006;28:1010–22. doi: 10.1002/bies.20480. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Hill RJ, Heid PJ, Newman-Smith ED, Zhu J, Priess JR, Rothman JH. Genetic redundancy in endoderm specification within the genus Caenorhabditis. Dev Biol. 2005;284:509–22. doi: 10.1016/j.ydbio.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Rothman JH. Making worm guts: the gene regulatory network of the Caenorhabditis elegans endoderm. Dev Biol. 2002;246:68–85. doi: 10.1006/dbio.2002.0655. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Miller JR, McClay DR. Characterization of the role of cadherin in regulating cell adhesion during sea urchin development. Dev Biol. 1997;192:323–39. doi: 10.1006/dbio.1997.8740. [DOI] [PubMed] [Google Scholar]
- Morton DG, Shakes DC, Nugent S, Dichoso D, Wang W, Golden A, Kemphues KJ. The Caenorhabditis elegans par-5 gene encodes a 14–3–3 protein required for cellular asymmetry in the early embryo. Dev Biol. 2002;241:47–58. doi: 10.1006/dbio.2001.0489. [DOI] [PubMed] [Google Scholar]
- Myster SH, Cavallo R, Anderson CT, Fox DT, Peifer M. Drosophila p120catenin plays a supporting role in cell adhesion but is not an essential adherens junction component. J Cell Biol. 2003;160:433–49. doi: 10.1083/jcb.200211083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance J, Lee JY, Goldstein B. Gastrulation in C. elegans. WormBook. 2005:1–13. doi: 10.1895/wormbook.1.23.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance J, Munro EM, Priess JR. C. elegans PAR-3 and PAR-6 are required for apicobasal asymmetries associated with cell adhesion and gastrulation. Development. 2003;130:5339–50. doi: 10.1242/dev.00735. [DOI] [PubMed] [Google Scholar]
- Nance J, Priess JR. Cell polarity and gastrulation in C. elegans. Development. 2002;129:387–97. doi: 10.1242/dev.129.2.387. [DOI] [PubMed] [Google Scholar]
- Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36:149–55. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman KR, Moerman DG. Alpha spectrin is essential for morphogenesis and body wall muscle formation in Caenorhabditis elegans. J Cell Biol. 2002;157:665–77. doi: 10.1083/jcb.200111051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa M, Kemler R. The membrane-proximal region of the E-cadherin cytoplasmic domain prevents dimerization and negatively regulates adhesion activity. J Cell Biol. 1998;142:1605–13. doi: 10.1083/jcb.142.6.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M, Yap AS. Traffic control: p120-catenin acts as a gatekeeper to control the fate of classical cadherins in mammalian cells. J Cell Biol. 2003;163:437–40. doi: 10.1083/jcb.200310090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt J, Cox EA, Broadbent ID, Flett A, Hardin J. The Caenorhabditis elegans p120 catenin homologue, JAC-1, modulates cadherin-catenin function during epidermal morphogenesis. J Cell Biol. 2003;162:15–22. doi: 10.1083/jcb.200212136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock R, Benard CY, Shapiro L, Hobert O. Functional dissection of the C. elegans cell adhesion molecule SAX-7, a homologue of human L1. Mol Cell Neurosci. 2007 doi: 10.1016/j.mcn.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Raich WB, Agbunag C, Hardin J. Rapid epithelial-sheet sealing in the Caenorhabditis elegans embryo requires cadherin-dependent filopodial priming. Curr Biol. 1999;9:1139–46. doi: 10.1016/S0960-9822(00)80015-9. [DOI] [PubMed] [Google Scholar]
- Reynolds AB. p120-catenin: Past and present. Biochim Biophys Acta. 2007;1773:2–7. doi: 10.1016/j.bbamcr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmacher D, Brinkmann V, Birchmeier C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci U S A. 1995;92:855–9. doi: 10.1073/pnas.92.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau CE, Yasuda J, Shin TH, Lin R, Sawa H, Okano H, Priess JR, Davis RJ, Mello CC. WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell. 1999;97:717–26. doi: 10.1016/s0092-8674(00)80784-9. [DOI] [PubMed] [Google Scholar]
- Sasakura H, Inada H, Kuhara A, Fusaoka E, Takemoto D, Takeuchi K, Mori I. Maintenance of neuronal positions in organized ganglia by SAX-7, a Caenorhabditis elegans homologue of L1. Embo J. 2005;24:1477–88. doi: 10.1038/sj.emboj.7600621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierenberg E. Reversal of cellular polarity and early cell-cell interaction in the embryos of Caenorhabditis elegans. Dev Biol. 1987;122:452–63. doi: 10.1016/0012-1606(87)90309-5. [DOI] [PubMed] [Google Scholar]
- Schierenberg E, Miwa J, von Ehrenstein G. Cell lineages and developmental defects of temperature-sensitive embryonic arrest mutants in Caenorhabditis elegans. Dev Biol. 1980;76:141–59. doi: 10.1016/0012-1606(80)90368-1. [DOI] [PubMed] [Google Scholar]
- Schlesinger A, Shelton CA, Maloof JN, Meneghini M, Bowerman B. Wnt pathway components orient a mitotic spindle in the early Caenorhabditis elegans embryo without requiring gene transcription in the responding cell. Genes Dev. 1999;13:2028–38. doi: 10.1101/gad.13.15.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotland P, Zhou D, Benveniste H, Bennett V. Nervous system defects of AnkyrinB (-/-) mice suggest functional overlap between the cell adhesion molecule L1 and 440-kD AnkyrinB in premyelinated axons. J Cell Biol. 1998;143:1305–15. doi: 10.1083/jcb.143.5.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield M, Loveless T, Hardin J, Pettitt J. C. elegans Enabled exhibits novel interactions with N-WASP, Abl, and cell-cell junctions. Curr Biol. 2007;17:1791–6. doi: 10.1016/j.cub.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewan AM, Maddugoda M, Kraemer A, Stehbens SJ, Verma S, Kovacs EM, Yap AS. Myosin 2 is a key Rho kinase target necessary for the local concentration of E-cadherin at cell-cell contacts. Mol Biol Cell. 2005;16:4531–42. doi: 10.1091/mbc.E05-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtutman M, Levina E, Ohouo P, Baig M, Roninson IB. Cell adhesion molecule L1 disrupts E-cadherin-containing adherens junctions and increases scattering and motility of MCF7 breast carcinoma cells. Cancer Res. 2006;66:11370–80. doi: 10.1158/0008-5472.CAN-06-2106. [DOI] [PubMed] [Google Scholar]
- Simske JS, Koppen M, Sims P, Hodgkin J, Yonkof A, Hardin J. The cell junction protein VAB-9 regulates adhesion and epidermal morphology in C. elegans. Nat Cell Biol. 2003;5:619–25. doi: 10.1038/ncb1002. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Tepass U, Gruszynski-DeFeo E, Haag TA, Omatyar L, Torok T, Hartenstein V. shotgun encodes Drosophila E-cadherin and is preferentially required during cell rearrangement in the neurectoderm and other morphogenetically active epithelia. Genes Dev. 1996;10:672–85. doi: 10.1101/gad.10.6.672. [DOI] [PubMed] [Google Scholar]
- Thorpe CJ, Schlesinger A, Carter JC, Bowerman B. Wnt signaling polarizes an early C. elegans blastomere to distinguish endoderm from mesoderm. Cell. 1997;90:695–705. doi: 10.1016/s0092-8674(00)80530-9. [DOI] [PubMed] [Google Scholar]
- Uemura T, Oda H, Kraut R, Hayashi S, Kotaoka Y, Takeichi M. Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the Drosophila embryo. Genes Dev. 1996;10:659–71. doi: 10.1101/gad.10.6.659. [DOI] [PubMed] [Google Scholar]
- Van Furden D, Johnson K, Segbert C, Bossinger O. The C. elegans ezrin-radixin-moesin protein ERM-1 is necessary for apical junction remodelling and tubulogenesis in the intestine. Dev Biol. 2004;272:262–76. doi: 10.1016/j.ydbio.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Wacker S, Grimm K, Joos T, Winklbauer R. Development and control of tissue separation at gastrulation in Xenopus. Dev Biol. 2000;224:428–39. doi: 10.1006/dbio.2000.9794. [DOI] [PubMed] [Google Scholar]
- Walston T, Tuskey C, Edgar L, Hawkins N, Ellis G, Bowerman B, Wood W, Hardin J. Multiple Wnt signaling pathways converge to orient the mitotic spindle in early C. elegans embryos. Dev Cell. 2004;7:831–41. doi: 10.1016/j.devcel.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Wang X, Kweon J, Larson S, Chen L. A role for the C. elegans L1CAM homologue lad-1/sax-7 in maintaining tissue attachment. Dev Biol. 2005;284:273–91. doi: 10.1016/j.ydbio.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Wei SY, Escudero LM, Yu F, Chang LH, Chen LY, Ho YH, Lin CM, Chou CS, Chia W, Modolell J, Hsu JC. Echinoid is a component of adherens junctions that cooperates with DE-Cadherin to mediate cell adhesion. Dev Cell. 2005;8:493–504. doi: 10.1016/j.devcel.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Yamada S, Nelson WJ. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J Cell Biol. 2007;178:517–27. doi: 10.1083/jcb.200701058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen JA, Kirch SA, Bargmann CI. Genes required for axon pathfinding and extension in the C. elegans nerve ring. Development. 1999;126:3679–92. doi: 10.1242/dev.126.16.3679. [DOI] [PubMed] [Google Scholar]
- Zar J. Biotsatisical Analysis. Pearson/Prentice-Hall; Upper Saddle River, NJ: 2010. [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Opperman K, Wang X, Chen L. unc-44 Ankyrin and stn-2 gamma-syntrophin regulate sax-7 L1CAM function in maintaining neuronal positioning in Caenorhabditis elegans. Genetics. 2008;180:1429–43. doi: 10.1534/genetics.108.091272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.