

Abstract
In regenerative medicine, hydrogels are employed to fill defects and support the infiltration of cells that can ultimately regenerate tissue. Gene delivery within hydrogels targeting infiltrating cells has the potential to promote tissue formation, but the delivery efficiency of nonviral vectors within hydrogels is low hindering their applicability in tissue regeneration. To improve their functionality, we have conducted a mechanistic study to investigate the contribution of cell migration and matrix degradation on gene delivery. In this report, lipoplexes were entrapped within hydrogels based on poly(ethylene glycol) (PEG) crosslinked with peptides containing matrix metalloproteinase degradable sequences. The mesh size of these hydrogels is substantially less than the size of the entrapped lipoplexes, which can function to retain vectors. Cell migration and transfection were simultaneously measured within hydrogels with varying density of cell adhesion sites (Arg-Gly-Asp peptides) and solids content. Increasing RGD density increased expression levels up to 100-fold, while greater solids content sustained expression levels for 16 days. Increasing RGD density and decreasing solids content increased cell migration, which indicates expression levels increase with increased cell migration. Initially exposing cells to vector resulted in transient expression that declined after 2 days, verifying the requirement of migration to sustain expression. Transfected cells were predominantly located within the population of migrating cells for hydrogels that supported cell migration. Although the small mesh size retained at least 70% of the lipoplexes in the absence of cells after 32 days, the presence of cells decreased retention to 10% after 16 days. These results indicate that vectors retained within hydrogels contact migrating cells, and that persistent cell migration can maintain elevated expression levels. Thus matrix degradation and cell migration are fundamental design parameters for maximizing gene delivery from hydrogels.
Keywords: gene delivery, hydrogel, migration, three-dimensional culture, non-viral vectors
Introduction
Tissue engineering strategies aim to construct functional tissue replacements in regenerative medicine through the manipulation of the local environment to direct cellular processes. The major strategy toward promoting regeneration is to present a combination of signals to recruit and direct progenitor cell differentiation and migration, facilitating morphogenesis [1, 2]. Tissue engineering scaffolds function to support cellular adhesion and cell infiltration, and the infiltrating cells are responsible for regenerating the lost or damaged tissue. Hydrogels are implemented as a scaffold in a variety of tissue engineering applications, as their physical properties closely resemble native tissue [3] and can be readily manipulated upon crosslinking conditions [4, 5]. Additionally, hydrogels are employed as drug delivery vehicles, and gene delivery in particular offers a versatile approach to direct tissue formation by inducing the expression of tissue inductive factors. Taken together, gene delivery within hydrogels to induce the expression of tissue inductive factors may enhance the capabilities of infiltrating cells to regenerate the lost tissue [6–8].
A number of hydrogels that have been employed for regenerative medicine have been investigated for their potential use in gene delivery, however, many result in limited transfection profiles [8]. Hydrogels tested for non-viral gene delivery include both natural hydrogels such as fibrin [7, 9] and collagen [6], synthetic hydrogels such as poly(ethylene glycol) (PEG) [10–12], and composites such as chitosan-g-PEG-folate [13] and PEG-hyaluronic acid [14]. Hydrogels in tissue regeneration support cell adhesion and migration, enabling infiltration of cells targeted for gene delivery. Transfected cells can then function as bioreactors for the transient expression of tissue inductive proteins within the injury site. In previous work, many hydrogel designs have focused on obtaining a sustained release of the vectors [6, 11, 12, 15, 16], which may contribute to the low levels of gene transfer within hydrogels, as infiltrating cells would encounter low concentrations of vector if released too rapidly into the surrounding microenvironment. The limited levels and duration of expression has hindered the broad applicability of this strategy. These limitations to gene delivery motivate the need to identify the fundamental hydrogel design parameters that maximize gene transfer from hydrogels.
This mechanistic study investigates the hypothesis that cell migration and matrix degradation must be balanced to maximize gene transfer within the hydrogel. PEG-based hydrogels, formed by crosslinking the PEG with peptides containing matrix metalloproteinase (MMP)-degradable sequences, were employed to investigate the hydrogel design parameters that impact transfection. The density of adhesion sites incorporated into the matrix and hydrogel crosslink density cooperate to control cell migration in hydrogels [17–19]. The adhesion site density alters cell-matrix interactions and was controlled through the RGD concentration. Cell migration through hydrogels can occur by localized proteolytic degradation of the peptide crosslinkers by enzymes secreted from cells, or non-proteolytically with amoeboid locomotion through the mesh [10, 19, 20]. A greater crosslink density tends to slow migration, and crosslink density can be controlled by the solids content. The reported mesh size of these PEG hydrogels measured by swelling experiments are on the order of 25 nm [21], substantially smaller than the diameter of lipoplexes [22]. Thus, initial studies investigated the retention of the vector within the gel, which would be necessary to locally transfect the migrating cells. Subsequently, cell migration and transfection were monitored simultaneously to determine the contribution of cell migration on the level and duration of expression. The relationship of cell migration and matrix degradation on transgene expression was further characterized by identifying the distribution of transfected cells within the hydrogel, measuring the release of DNA in the presence of cells, and determining the effect of the initial exposure of cells to DNA. The impact of persistent cell migration on sustaining expression levels was investigated using hydrogels of varying volumes. This report identifies key design parameters for hydrogels that impact gene delivery, which can be employed in developing novel hydrogels, or modifying existing hydrogels to maximize gene transfer for applications in regenerative medicine.
Materials and Methods
Materials
Plasmids encoding for cell-secreted Gaussia Luciferase (pGLuc) and Firefly luciferase/enhanced green fluorescent fusion protein (pEGFP-Luc) each containing the CMV promoter were produced with a Qiagen Maxiprep kit (Valencia, CA). The pGLuc reporter plasmid was used for quantifying transgene expression, while the pEGFP-Luc plasmid was implemented for imaging the localization of transfected cells. Gaussia luciferase assay kit was purchased from New England BioLabs (Ipswich, MA). TransFast transfection reagent was purchased from Promega (Madison, WI). Four-arm poly(ethylene glycol) vinyl sulfone was synthesized using previously reported techniques [5]. The adhesion peptide sequence GCGYGRGDSPG and enzymatically degradable peptide crosslinker sequence GKCD-GPQG-IWGQ-DCKG were custom syntheses purchased from Celtek Bioscience LLC (Nashville, TN). Human thrombin and fibrinogen were obtained as a generous gift from Baxter Healthcare (Deerfield, IL). α-32P-dATP was purchased from PerkinElmer (Waltham, MA). NIH/3T3 cells were purchased from ATCC (Manassas, VA) and HT-1080 cell line was generously provided by Dr. J. Jones (Northwestern University, Chicago, IL). Four-arm PEG-acryl (20 kDa) was obtained from SunBio (Orinda, CA) and Igracure 2959 (I2959) photoinitiator was a generous gift from Ciba (Tarrytown, NY). All other reagents were purchased from Fisher Scientific (Waltham, MA) unless otherwise noted.
Hydrogel formation, characterization, and co-encapsulation of DNA and cells
Hydrogels were crosslinked using Michael-Type addition chemistry, in which the 4-arm PEG-VS was crosslinked by the enzymatically degradable, bifunctional peptide through the free thiols of its cysteine residues. Prior to addition of the crosslinking peptide and subsequent gelation, RGD adhesion peptides containing cysteine residues were conjugated to 4-arm PEG in 0.3 M triethanolamine (TEOA), pH 8.0 for 15 minutes. Rheology measurements were conducted with a Paar Physica MCR Rheometer (Anton Paar, Graz, Austria) with parallel plate geometry at 25 °C under a humidified environment. Amplitude sweeps were performed initially to ensure subsequent measurements were conducted in the linear viscoelastic regime. Mechanical spectra were obtained from frequency sweeps at a constant strain of 0.05. Lipoplexes were formed by adding TransFast to plasmid in a 1.5:1.0 μL:μg ratio and vortexing gently. Lipoplex z-average diameter was measured with a Zetasizer Nano ZS (Malvern, Worcestershire, United Kingdom). Cells and DNA lipoplexes were co-encapsulated within the hydrogels upon gelation by adding the enzymatically degradable crosslinking peptide dissolved in 0.3 M TEOA, pH 10.0 to the PEG precursor solution in a 1:1 molar ratio of thiol:VS. The reaction was carried out for 30 minutes to ensure complete gelation. Hydrogels were washed repeatedly with 200 μL cDMEM within the first 4 hours to remove buffer and unreacted monomers followed by a media change every day thereafter. Transfection studies were performed with either HT-1080 or NIH/3T3 cell lines, both of which secrete MMPs [23], cultured in EMEM supplemented with 10% FBS and 1% Pen/strep or DMEM supplemented with 10% FCS, respectively. Cells were split using trypsin and cultured at 37°C and 5% CO2 environment.
DNA retention studies
The retention of plasmid within the hydrogel was determined for varying solids content (7.5% and 10% PEG) and RGD concentrations (0, 2 and 5 mM). Plasmid was radiolabeled with α-32P dATP using a nick translation kit (Amersham Pharmacia Biotech, Piscataway, NJ) following the manufacturer’s protocol with minor modifications [24]. Lipoplexes were formed at a ratio of 1.5 μL Transfast to 1.0 μg DNA. Lipoplexes were added to precursor solutions to yield a final concentration of 3.0 μg DNA/gel, and hydrogels were crosslinked upon addition of the enzymatically degradable crosslinker. Hydrogels were washed with PBS repeatedly within the first several hours followed by periodic washing for 30 days; all washes were collected and added to 5 mL of Biosafe II scintillation cocktail (Research Products International Corp., Mount Prospect, IL) for measurement with a scintillation counter. At the end of the study, hydrogels were completely degraded by 1 mg/mL collagenase I solution and added to scintillation cocktail to measure the amount of DNA remaining in the hydrogel. The cumulative retention was calculated as the amount of DNA remaining in the gel after each time point, divided by the total amount of DNA released by the end of the study plus the DNA remaining in the hydrogel at the end of the study.
Cell migration and expression level measurements
Cell migration and the extent of transgene expression were measured simultaneously utilizing live cell measurement methods to monitor the same hydrogels over the 16 day culture period. The method of measuring cell migration is based on that reported by Raeber et al [21]. HT-1080 cell-dense fibrin clots were formed by adding 25 mg/mL fibrinogen solution to a cell pellet to yield a final concentration of 4.1 × 107 cells/mL and pipetting 1 μL of this solution to 1 μL thrombin. Fibrin clots were transferred to the PEG-RGD precursor solution and co-encapsulated with DNA lipoplexes within hydrogels containing 7.5% or 10% PEG at a final concentration of 4 μg per hydrogel. For migration measurements, the hydrogels were imaged with phase microscopy at 5× magnification using an inverted microscope and multiple images were stitched together. The area of the fibrin clot was measured the day of encapsulation and the migrating cell front was measured subsequently at multiple time points in ImageJ. Radial cell migration was calculated by subtracting the radius of the initial fibrin clot from the radius of the migrated cell front at each time point. To measure the extent of transgene expression, media was collected once per day throughout the 16 day cultured and stored at −80°C. Following one freeze-thaw cycle, the cell-secreted luciferase activity was measured using the Gaussia Luciferase assay kit and luminometer (Turner Design, Sunnyvale, CA), with values reported in relative light units (RLUs) per hydrogel.
Imaging transfected cell localization
The location of transfected cells within the hydrogel was identified by co-encapsulating fibrin clots containing HT-1080 cells and pEGFP-Luc lipoplexes. The hydrogels were formed with 10% PEG containing 0, 2, or 5 mM RGD. Phase images of cells and fluorescence images of EGFP expressing cells (green) were captured at days 2 and 5 at 5× magnification. Images were overlapped in Adobe Photoshop to identify the transfected cells.
Transfection in two- and three-dimensions
Transgene expression following an initial exposure of cells to DNA was investigated for two- and three-dimensional culture. An initial exposure of lipoplexes to cells was performed by incubation of HT-1080 cells (450,000) with 18 μg DNA lipoplexes encoding for Gaussia Luciferase for 30 minutes at +4 °C. Following incubation, cells were collected by centrifugation and washed twice with fresh cEMEM to remove DNA that was not cell associated. For two-dimensional culture, a cell suspension (37,500 cells/mL) was seeded onto hydrogels (50 μL) formed within a 96 well plate. For three-dimensional studies, the 96 well plate was initially covered with a 40 μL PEG-acryl non-adherent hydrogel base, which was photocrosslinked by exposure to UV light for 90 sec at 365 nm wavelength using 0.05% of 600 mg/mL I259 in N-vinyl-2-pyrrilidone (PVP) [25]. Cells were then suspended at a concentration of 106 cells/mL within the precursor solution. Gels were formed with a final volume of 15 μL gels which were transferred to wells containing PEG bases and washed 1 hour with 200 μL cEMEM following encapsulation. The hydrogels used for these studies were either fibrin hydrogels, prepared to yield a final concentration of 12.5 mg/mL fibrinogen and 25 U/mL thrombin, or PEG hydrogels, prepared with 10% PEG-VS with 5 mM RGD. The extent of transgene expression was determined by collecting media from each well. Media was stored at −80°C until analysis. After one freeze-thaw cycle, the luciferase content in the media was measured and values were recorded in RLUs per well.
DNA distribution and cell-association quantifications
The distribution of DNA within the hydrogel, culture media, and cells was investigated using radiolabeled DNA. Fibrin clots containing HT-1080 cells were encapsulated within 7.5% or 10% PEG and 5 mM RGD as described above using radiolabeled lipoplexes. Culture media was collected at each time point to determine the quantity released into the media. To measure the quantity of DNA entrapped within the hydrogel or associated with cells, hydrogels were washed with 200 μL PBS followed by the addition of CellScrub Buffer (Gene Therapy Systems, Inc., San Diego, CA, 150 μL) to collagenase type I solution (150 μL of 2 mg/mL ) dissolved in 1X HBSS with CaCl2. CellScrub was added to remove extracellular lipoplexes and prevent cell association of liberated DNA during hydrogel degradation in collagenase. After 30 minutes of incubation, cells were separated from solution by centrifugation and aspiration. The radioactivity within the aspirated solution was measured to quantify DNA within the hydrogel. Cells were resuspended in 200 μL PBS and added to 5 mL Biosafe II scintillation cocktail for subsequent counting with a scintillation counter, for which the counts were employed to determine the quantity of cell associated DNA. The counts were correlated to DNA concentration using a standard curve and results are reported as the percentage remaining in the gel, released, and cell associated.
Statistics
Statistical analyses were performed using JMP software (SAS Institute, Inc. Cary, NC). Experiments were performed at minimum in triplicate with replication. Multiple comparisons tests were performed using a Tukey multivariable comparison method with a 95% confidence level. Comparisons between pairs at each time point were conducted using a one-way analysis of variance (ANOVA) with a 95% confidence level. Mean values with standard deviation are reported in all figures.
Results
DNA retention within PEG-based hydrogels
Hydrogels for gene delivery in regenerative medicine target cells infiltrating the scaffold. We thus initially investigated retention of the vector within the matrix, which would function to maintain elevated concentrations in the hydrogel into which cells are infiltrating. More than 70% of the entrapped lipoplexes were retained within the hydrogel throughout the 32-day incubation period for both 7.5% and 10% PEG hydrogel compositions (Fig. 1). The 10% PEG hydrogels retained more than 80% of the encapsulated DNA, which may result from the increased crosslink density relative to the 7.5% PEG hydrogels that retained statistically less DNA (70%, p < 0.05). The mean lipoplex diameter measured was 486 nm, which is consistent with the diameter of lipoplexes commonly used for gene delivery [22, 26]. The diameter of the lipoplexes is larger than the mesh size of the hydrogel (~25 nm) [21], which likely limits release from the hydrogel. During the initial 24 hours, approximately 8% of DNA was lost, likely resulting from a combination of hydrogel washing and matrix swelling. The hydrogels swell to their maximum size within 4 hours, and DNA release afterwards likely occurs by diffusion of DNA that is present near the surface of the hydrogel.
Figure 1.
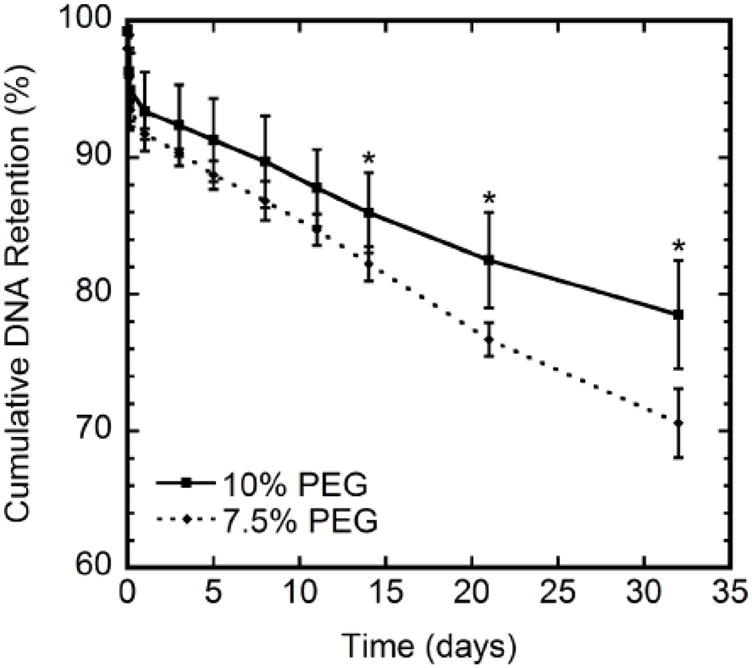
Lipoplex retention for 7.5% and 10% PEG hydrogels containing 5 mM RGD. Significant differences in release at each time point based on a t-test are denoted by an asterisk (p < 0.05).
Cellular migration rates and expression level profiles
A fibrin clot assay was subsequently used to simultaneously measure cell migration and transgene expression resulting from the entrapped lipoplexes throughout the 16-day culture. Consistent with previous reports [19], the distances traveled by two cell types: a more readily transfected HT-1080 cells and the more difficult to transfect NIH/3T3 cells increased with increasing RGD concentration and decreasing PEG content (Fig. 2). Minimal cell movement was measured for hydrogels without RGD, and the small displacements reflect a localized matrix degradation and reorganization. Maximal cell migration was achieved for hydrogels containing 5 mM RGD. Importantly, cells migrated to the outer edge of the gel within 12 days in 7.5% PEG hydrogels. For 10% PEG gels, 16 days of culture were necessary for cells to migrate to the hydrogel boundary. Intermediate levels of RGD incorporation (2 mM) resulted in displacements that were intermediate between the displacements obtained for the 0 and 5 mM RGD concentrations for both cell types. Cell viability was not significantly affected following encapsulation or at subsequent time points (data not shown), consistent with previous reports [10].
Figure 2.

Cell migration. HT-1080 (A, B) or NIH/3T3 (C, D) cells were encapsulated within PEG hydrogels containing 0, 2, or 5 mM RGD. Cells were entrapped within a fibrin clot that was encapsulated within 7.5% (A, C) or 10% (B, D) PEG hydrogels. Hydrogels contained 4 μg DNA encoding for Gaussia Luciferase. Significant difference between conditions at each time point is indicated by different letters (A, B, or C) based on ANOVA with p < 0.05. In these panels and subsequent figures, groups are statistically different if they are not marked by a common letter. For example, on day 6 of panel B, 2 and 5 mM RGD have the letter A in common, and are thus not statistically different (p > 0.05). Similarly, the 0 and 2 mM RGD conditions have the letter B in common and are not significantly different (p > 0.05). However, data for 0 and 5 mM RGD are marked with letters B and A, respectively, and are significantly different (p < 0.05). Insets compare migration of each cell type within hydrogels containing 5 mM RGD. Note that cells migrated to the outer edge of the hydrogel by day 12 for both HT-1080 and NIH/3T3 cells within 7.5% PEG containing 5 mM RGD. The day 16 data point for these conditions were removed for clarity.
Transfection levels were measured concurrently with migration and also exhibited a dependence on the RGD density and PEG content, both of which impact migration. Hydrogels lacking RGD had the lowest levels of expression (Fig. 3). Hydrogels containing 5 mM RGD, which had the fastest migration, exhibited significantly greater levels of transgene expression than 0 or 2 mM RGD (p < 0.05). For these hydrogels, expression levels were maximal at the 2 day time point, reaching levels that were an order of magnitude greater than 0 mM RGD hydrogels, which is the condition that did not support cell migration. For the 2 mM condition, the mean expression levels were intermediate between the 0 and 5 mM conditions, and were reduced for the NIH/3T3 cells relative to the HT-1080 cells. Interestingly, the duration of expression was a function of the hydrogel solids content. For 7.5% PEG, the expression levels decreased from the maximum that was achieved at day 2 in most cases. However, expression persisted within 10% PEG hydrogels for longer times relative to the 7.5% PEG hydrogel. For HT-1080 cells, expression remained steady or increased in 10% hydrogels throughout the study. For NIH/3T3 cells, expression increased throughout culture at the greatest RGD content, yet persisted for approximately 10 days at the lower RGD concentrations.
Figure 3.

Gene expression profiles. HT-1080 (A, B) or NIH/3T3 (C, D) cells were encapsulated within PEG hydrogels containing 0, 2, or 5 mM RGD. Cells were entrapped within a fibrin clot that was encapsulated within 7.5% PEG (A, C) or 10% (B, D) PEG hydrogels. Hydrogels contained 4 μg DNA encoding for Gaussia Luciferase. Significant difference between conditions at each time point is indicated by different letters (A, B, or C) based on ANOVA with p < 0.05. Expression levels of cells within 7.5% PEG containing 5 mM RGD decreased from day 2 to day 16 for both cell types, while levels remained elevated for 10% PEG containing 5 mM RGD. Insets compare expression levels of each cell type within hydrogels containing 5 mM RGD.
Transfected cell localization
The localization of transfected cells within the hydrogels was investigated for a relationship between migration and transfection. Following 2 days of culture, cells were primarily within the fibrin clot (Fig. 4A-C), with some limited cell migration into the PEG hydrogel for the 2 and 5 mM RGD concentrations (Fig. 4B, C). Transfected cells were observed in the fibrin clot of PEG hydrogels lacking RGD (Fig. 4A), and transfected cells were observed both within the fibrin clot and within the PEG hydrogel for the 2 and 5 mM RGD conditions. At day 5, cells were restricted to the fibrin clot for the hydrogels without RGD, some of which were transfected (Fig. 4D). For the 2 and 5 mM RGD conditions, cell migration from the fibrin clot increased, and transfection was observed for migrating cells within the PEG hydrogel (Fig. 4E, F). Minimal transfection was observed within the fibrin clot for 2 mM RGD, while no transfected cells were observed within the fibrin clot at this later time point for 5 mM RGD. Thus, the majority of transfected cells after 2 days are outside of the initial fibrin clot, which likely results from cells migrating from the clot.
Figure 4.

Localization of transfected cells within hydrogels. Fibrin clots (FC) containing HT-1080 cells encapsulated within 10% PEG (P) containing 0, 2, or 5 mM RGD and 4 μg DNA were imaged with fluorescence microscopy at days 2 (A-C) and 5 (D-G), respectively. Migrating cells (MC) are observed at both time points in hydrogels containing RGD. White arrows denote transfected cells. The inset of (F) denoted by the white box is shown in (G). Scale bars represent 500 μm. Note the smaller scale bars for images taken at day 5 with 2 and 5 mM RGD (E, F), respectively, indicating a larger area is shown to capture cell migration. The fibrin clot dimensions may appear different as images were captured from different orientations.
DNA distribution and cell-association
The impact of cell migration on the quantity and distribution of DNA initially entrapped within the hydrogel was investigated. The percentage of DNA retained within the hydrogel was 60% to 67% at day 1, which decreased significantly to 15% and 12% by days 9 and 16 (p < 0.05), respectively (Fig. 5A), which contrasts with the 86% to 89% that was retained through day 9 in the initial retention study. The quantity of DNA that was released from the hydrogel or associated with cells was subsequently measured. The percentage of DNA released into the culture media was approximately 30% to 37% at day 1 and 83% by day 9 (Fig. 5B). The amount of DNA associated with cells was a relatively small percentage of the total DNA within the system (Fig. 5C). The amount of DNA retained or released was not statistically different between PEG contents (p < 0.05); however, the amount of DNA associated with cells declined with the 7.5% PEG hydrogels and remained steady for the 10% PEG hydrogels (p < 0.05).
Figure 5.

DNA distribution throughout the hydrogel culture. For hydrogels with encapsulated HT-1080 cells, percentage of DNA that was remaining within the hydrogel matrix (A), released in the cell culture media (B), and cell-associated (C) was measured at days 1, 2, 9, 16. Hydrogels were composed of 7.5% and 10% PEG containing 5 mM RGD. Statistical significance based on ANOVA between PEG contents at each time point as well as statistical significance between time points at each PEG content are denoted by different letters (A, B, C, or D) (p < 0.05). For example, in panel C on day 1, the 7.5 and 10 % PEG are both marked with the letter A and are not significantly different (p > 0.05), whereas, 7.5 % PEG on day 1 and day 9 have no letters in common and are significantly different (p < 0.05).
DNA internalization of two- and three-dimensional cell cultures
The mechanism regulating the level and duration of expression within the hydrogel was investigated by controlling the initial exposure of cells to DNA. After mixing cells and lipoplexes, cells were washed to remove excess DNA, and cells were then either seeded onto two-dimensional culture surfaces or within hydrogels without DNA (Fig. 6). Cells cultured within or on PEG hydrogels had expression that persisted at near maximum levels for approximately 2 days (Fig 6B). A similar trend was observed for fibrin hydrogels, indicating that this response is not dependent on a specific hydrogel. After day 2, expression levels declined significantly (p < 0.05), typically by more than an order of magnitude from the maximal values. This decline in the duration of expression with only an initial exposure was also observed for culture on tissue culture polystyrene. Taken together, these results indicate that the initial exposure provides transient expression and suggests that the sustained expression may result from cell migration increasing the exposure of cells to the vectors.
Figure 6.

Gene expression with initial vector exposure. Extent of transgene expression measured of cells cultured in two- and three-dimensions using tissue culture polystyrene (PS) surfaces, fibrin or 10% PEG containing 5 mM RGD. HT-1080 cells exposed to lipoplexes were cultured on hydrogel surfaces (2D) (A) or encapsulated within hydrogels (3D) (B). Expression levels on days 1, 2, 4, and 7 are reported. The asterisks refer to statistical significance relative to maximal expression (day 2) for the same condition based on a Tukey multivariable analysis (p < 0.05). Transfection levels at day 2 of cells cultured on PS were statistically greater than cells cultured on either fibrin or PEG (p < 0.05).
Persistence of cell migration
The relationship between cell migration and the duration of transgene expression was also investigated by varying the volume of hydrogel composed of 10% PEG containing 5 mM RGD, which influenced the time required for cells to migrate to the edge of the hydrogel. The time required for the migrating cell front to reach the hydrogel edge decreased as the hydrogel volume decreased (Fig. 7A). Interestingly, expression levels declined at varying times for the three hydrogel volumes (Fig. 7B), and the decline in expression was similar to the time required for cells to migrate to the hydrogel edge. These results suggest that persistent cell migration is required for sustained levels of transgene expression.
Figure 7.

Cell migration and gene expression profiles in hydrogels of varying volume. HT-1080 cells were encapsulated within multiple volumes (5, 7.5 and 15 μL) of 10% PEG hydrogels containing 5 mM RGD. Cell migration (A) and extent of transgene expression (B) were simultaneously measured. For (A), statistical significance in migration between conditions at each time point is denoted by different letters (A or B) based on ANOVA with p < 0.05. Migration reached maximum values on days 6, 9 and 12 for 5, 7.5 and 15 μL hydrogels, respectively. For (B), significant differences in expression levels relative to maximal expression (day 2) for the same condition based on ANOVA with p < 0.05 are denoted by the letter A.
Discussion
Hydrogels capable of localized gene delivery could potentially control the local environment for numerous applications in tissue engineering; however, currently, limited levels or duration of expression hinders their use in regenerative medicine. Hydrogels have increasingly been investigated for use in a number of tissue engineering applications because of their mechanical properties [3] and ease of tailorability [4, 8, 19]. Delivery of therapeutic factors including DNA from hydrogels can be challenging due to the mass-transfer limitations in the high water content environment. Many past hydrogel designs target sustained vector release primarily by diffusion [6, 11, 12, 15, 16], which may hinder gene transfer within hydrogels by limiting local vector concentration near infiltrating cells if the release is too rapid.
In this report, we employ a previously developed synthetic hydrogel system to study the transfection mechanisms of cells as they infiltrate hydrogels in order to identify the fundamental hydrogel design parameters that maximize gene delivery within hydrogels. Through a series of mechanistic studies, we have tested the hypothesis that gene delivery requires a balance between cell migration into the matrix and hydrogel matrix degradation to limit vector loss from the cellular microenvironment and transfect these migrating cells. Our results indicated a relationship between the cell migration in the hydrogel and the extent and duration of transgene expression, with transfected cells observed throughout the hydrogel for gels that supported cell migration.
Cell migration within the hydrogel facilitates access of the cells to the entrapped vector to increase gene transfer. MMP-degradable PEG hydrogels have been widely used to study three-dimensional cellular growth and served as the model biomaterial as they support cell migration and their mesh size would allow for vector retention [10, 20–22]. These hydrogels have a reported mesh size on the order of 25 nm [21], sufficient to promote nutrient exchange yet much smaller than the measured hydrodynamic radius of DNA complexes. In the absence of cells, more than 70% of the DNA complexes were retained by the PEG hydrogel over a prolonged time period (Fig. 1), indicating the importance of degradation of the crosslinks via cell migration to free the entrapped vector from the matrix to facilitate cellular uptake. Previous reports have suggested that continued or repeated internalization of the vector as a factor that impacts duration of expression [9, 27, 28]. In the report herein, initially exposing cells to DNA prior to hydrogel culture was insufficient to maintain expression levels past day 2 regardless of hydrogel type (Fig. 6). This transient expression is typically attributed to clearance of the vector from the cell, or dilution of the plasmid with cell division [29]. To maintain expression levels, cells must repeatedly come in contact with DNA by migrating through the matrix. Varying hydrogel properties implicated in cell migration [19], namely the density of adhesion sites and solids content, led to similar migration trends for HT-1080 cells and NIH/3T3 cells (Fig. 2). Interestingly, increased cell migration by increasing the density of adhesion sites corresponded with an increased extent of transgene expression of the entrapped DNA, while the duration of expression was prolonged with increasing solids content (Fig. 3). Hydrogels containing no RGD did not support cell migration. These non-migratory cells had relatively low expression levels compared to hydrogels containing RGD, which is attributed to inaccessibility of cells to the vector. For hydrogels that supported cell migration, transfected cells are observed throughout the cell populations and in greater numbers relative to the non-migratory conditions, which supports the need for vector retention within the hydrogel (Fig. 4). In addition to migration, the varied density of RGD (2 or 5 mM) may impact other cellular processes, such as adhesion strength and cell morphology, that may impact transfection. Taken together, the results suggest that cell migration enables their access to the entrapped vectors to increase gene transfer.
Matrix degradation was occurring in the presence of migrating cells, as evidenced by increased release of the vector relative to cell-free gels, and this degradation also supported cell migration. During migration, cells can remodel their matrix by secreting MMPs, which has been previously reported to support migration through these hydrogels [10, 20]. To meet the objective of sustained, localized gene delivery for regenerative medicine applications, the challenge will be to retain the vector within the hydrogel despite hydrogel degradation to maintain expression levels. However, in the presence of cells, vector release was greatly increased in this study and was independent of solids content (Fig. 5B). Despite 85% of the encapsulated DNA being released from the hydrogel, the number of plasmid copies per cell based on amount of cell-associated DNA measured and the number of cells in the matrix was approximately 9 × 104, indicating cells were still able to capture a significant amount of DNA. Although release was statistically the same between solids content, transgene expression declined for 7.5% PEG while expression levels were maintained for 10% PEG (Fig. 3); which was consistent to the amount of cell-associated DNA measured (Fig. 5C). Interestingly, expression levels declined significantly once cells reached their maximum displacement (i.e., the edge of the hydrogel) (Figs. 2A,C and 3A,C and 7). Increasing the distance and time over which cells are able to migrate by increasing hydrogel volumes extended the duration of elevated transfection (Fig. 7). Taken together, these results indicate that retention of the vectors within the hydrogel can lead to contact of the DNA with migrating cells, and that persistent cell migration can maintain elevated expression levels within hydrogels by allowing cells to repeatedly contact sufficient quantities of DNA.
Cell migration and matrix degradation contributed to the duration of expression; however, other factors, such as intracellular trafficking, vector clearance or hydrogel mechanics, which are impacted by the solids content [21], may also contribute [29–31]. Substrate mechanics influence cell morphology, migration, proliferation, and the cytoskeletal architecture [32], all of which could impact intracellular trafficking of DNA. Transfection levels were previously reported to increase exponentially with increasing matrix rigidity for cells cultured in two-dimensions on hydrogels [31]. The hydrogels used herein had initial storage moduli ranging from 341 to 597 Pa and loss moduli ranging from 49.5 to 67.7 Pa, consistent with previous reports [10, 21]. Future studies that isolate the impact of mechanics, cell migration, and vector retention would further elucidate the mechanisms of gene transfer within hydrogels given the complex interactions within the system.
Conclusions
This report identifies key hydrogel design parameters that enhance transfection levels of cells infiltrating hydrogels. Solids content and density of adhesion sites, both of which regulate cell migration, modulates both the extent and duration of transgene expression within the hydrogel. The PEG hydrogels retained the vectors due to the mesh size being smaller than the lipoplex diameter, which maintained elevated concentrations locally. However, the presence of cells that degraded the crosslinks as a component of cell migration decreased the quantity retained. The expression level and number of transfected cells increased for hydrogels with a greater density of RGD peptides, which also led to increased cell migration throughout the hydrogel. Interestingly, increased solids content prolonged the duration of gene expression. This extended expression could not be replicated with only an initial vector exposure, suggesting that cell migration is required for persistent expression. The identification of hydrogel design parameters that maximize gene transfer can enhance their utility for numerous applications in regenerative medicine.
Acknowledgments
The authors thank Dr. Ariella Shikanov for synthesis of 4-arm PEG-vinyl sulfone, Michael S. Weiss of Northwestern University for valuable scientific discussions, and Christine E. Wang of Northwestern University for technical assistance. Financial support for this research was provided by PL1EB008542, R21EB006520, RO1EB005678.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 2.Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 3.Peretti GM, Xu J-W, Bonassar LJ, Kirchhoff CH, Yaremchuk MJ, Randolph MA. Review of injectable cartilage engineering using fibrin gel in mice and swine models. Tissue Eng. 2006;12:1151–1168. doi: 10.1089/ten.2006.12.1151. [DOI] [PubMed] [Google Scholar]
- 4.Andreopoulos FM, Beckman EJ, Russell AJ. Light-induced tailoring of PEG-hydrogel properties. Biomaterials. 1998;19:1343–1352. doi: 10.1016/s0142-9612(97)00219-6. [DOI] [PubMed] [Google Scholar]
- 5.Lutolf MP, Hubbell JA. Synthesis and physiochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition. Biomacromolecules. 2003;4:713–722. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 6.Fang J, Zhu Y-Y, Smiley E, Bonadio J, Rouleau JP, Goldstein SA, et al. Stimulation of new bone formation by direct transfer of osteogenic plasmid genes. Proc Natl Acad Sci USA. 1996;93:5753–5758. doi: 10.1073/pnas.93.12.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trentin D, Hall H, Wechsler S, Hubbell JA. Peptide-matrix-mediated gene transfer of an oxygen-insensitive hypoxia-inducible factor-1alpha variant for local induction of angiogenesis. Proc Natl Acad Sci USA. 2006;103:2506–2511. doi: 10.1073/pnas.0505964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Laporte L, Shea LD. Matrices and scaffolds for DNA delivery in tissue engineering. Adv Drug Deliv Rev. 2007;59:292–307. doi: 10.1016/j.addr.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.des Rieux A, Shikanov A, Shea LD. Fibrin hydrogels for non-viral vector delivery in vitro. J Control Release. 2009;136:148–154. doi: 10.1016/j.jconrel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei Y, Segura T. DNA delivery from matrix metalloproteinase degradable poly(ethylene glycol) hydrogels to mouse mouse cloned mesenchymal stem cells. Biomaterials. 2009;30:254–265. doi: 10.1016/j.biomaterials.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quick DJ, Anseth KS. DNA delivery from photocrosslinked PEG hydrogels: Encapsulation efficiency, release profiles, and DNA quality. J Control Release. 2004;96:341–351. doi: 10.1016/j.jconrel.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 12.Quick DJ, Anseth KS. Gene delivery in tissue engineering: A photopolymer platform to coencapsulate cells and plasmid DNA. Pharm Res. 2003;20:1730–1737. doi: 10.1023/b:pham.0000003368.66471.6a. [DOI] [PubMed] [Google Scholar]
- 13.Chan P, Kurisawa M, Chung JE, Yang Y-Y. Synthesis and characterization of chitosan-g-poly(ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery. Biomaterials. 2007;28:540–549. doi: 10.1016/j.biomaterials.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 14.Wieland JA, Houchin-Ray T, Shea LD. Non-viral vector delivery from PEG-hyaluronic acid hydrogels. J Control Release. 2007;120:233–241. doi: 10.1016/j.jconrel.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chun KW, Lee JB, Kim SH, Park TG. Controlled release of plasmid DNA from photo-cross-linked pluronic hydrogels. Biomaterials. 2005;26:3319–3326. doi: 10.1016/j.biomaterials.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 16.Megeed Z, Haider M, Li D, O’Malley BW, Cappello J, Ghandehari H. In vitro and in vivo evaluation of recombinant silk-elastinlike hydrogels for cancer gene therapy. J Control Release. 2004;94:433–445. doi: 10.1016/j.jconrel.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 17.Segura T, Anderson BC, Chung PH, Webber RE, Shull KR, Shea LD. Crosslinked hyaluronic acid hydrogels: A strategy to functionalize and pattern. Biomaterials. 2005;26:359–371. doi: 10.1016/j.biomaterials.2004.02.067. [DOI] [PubMed] [Google Scholar]
- 18.Drury JL, Mooney DJ. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 19.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc Natl Acad Sci USA. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raeber GP, Lutolf MP, Hubbell JA. Mechanisms of 3-D migration and matrix remodeling of fibroblasts within artificial ECMs. Acta Biomater. 2007;3:615–629. doi: 10.1016/j.actbio.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Raeber GP, Lutolf MP, Hubbell JA. Molecularly engineered PEG hydrogels: A novel model system for proteolytically mediated cell migration. Biophys J. 2005;89:1374–1388. doi: 10.1529/biophysj.104.050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bengali Z, Pannier AK, Segura T, Anderson BC, Jang J-H, Mustoe TA, et al. Gene delivery through cell culture substrate adsorbed DNA complexes. Biotechnol Bioeng. 2005;90:290–302. doi: 10.1002/bit.20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoon S-O, Park S-J, Yoon SY, Yun C-H, Chung A-S. Sustained production of H2O2 activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-κB pathway. J Biol Chem. 2002;277:30271–30282. doi: 10.1074/jbc.M202647200. [DOI] [PubMed] [Google Scholar]
- 24.A Amersham International plc. Labeling of DNA with 32P by nick translation. Tech Bull. 1980:80. [Google Scholar]
- 25.Bryant SJ, Nuttleman CR, Anseth KS. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J Biomater Sci Polym Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 26.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shea LD, Smiley E, Bonadio J, Mooney DJ. DNA delivery from polymer matrices for tissue engineering. Nat Biotechnol. 1999;17:551–554. doi: 10.1038/9853. [DOI] [PubMed] [Google Scholar]
- 28.Fukunaka Y, Iwanaga K, Morimoto K, Kakemi M, Tabata Y. Controlled release of plasmid DNA from cationized gelatin hydrogels based on hydrogel degradation. J Control Release. 2002;80:333–343. doi: 10.1016/s0168-3659(02)00026-3. [DOI] [PubMed] [Google Scholar]
- 29.Pannier AK, Shea LD. Controlled release systems for DNA delivery. Mol Ther. 2004;10:19–26. doi: 10.1016/j.ymthe.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 30.Bengali Z, Rea JC, Gibly RF, Shea LD. Efficacy of immobilized polyplexes and lipoplexes for substrate-mediated gene delivery. Biotechnology and Bioengineering. 2008;102:1679–1691. doi: 10.1002/bit.22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong HJ, Liu J, Riddle K, Matsumoto T, Leach K, Mooney DJ. Non-viral gene delivery regulated by stiffness of cell adhesion substrates. Nature Mater. 2005;4:460–464. doi: 10.1038/nmat1392. [DOI] [PubMed] [Google Scholar]
- 32.Peyton SR, Raub CB, Keschrumrus VP, Putnam AJ. The use of poly(ethylene glycol) hydrogels to investigate the impact of ECM chemistry and mechanics on smooth muscle cells. Biomaterials. 2006;27:4881–4893. doi: 10.1016/j.biomaterials.2006.05.012. [DOI] [PubMed] [Google Scholar]