

Abstract
Fatty acid biosynthesis is crucial for all living cells. In contrast to higher organisms, bacteria use a type II fatty acid synthase (FAS II) composed of a series of individual proteins, making FAS II enzymes excellent targets for antibiotics discovery. The β-hydroxyacyl-ACP dehydratase (FabZ) catalyzes an essential step in the FAS II pathway. Here, we report the structure of Campylobacter jejuni FabZ (CjFabZ), showing a hexamer both in crystals and solution, with each protomer adopting the characteristic hot dog fold. Together with biochemical analysis of CjFabZ, we define the first functional FAS II enzyme from this pathogen, and provide a framework for investigation on roles of FAS II in C. jejuni virulence.
Keywords: β-Hydroxyacyl-ACP dehydratase, Fatty acid biosynthesis, Crystal structure, Campylobacter jejuni
Introduction
Campylobacter jejuni is an important food-borne pathogen and the leading cause of bacterial gastroenteritis in humans [1]. Moreover, severe post-infectious complications include Guillain-Barré syndrome, a significant cause of acute neuromuscular paralysis [2], and irritable bowel syndrome. Despite being a major diarrheal pathogen and the availability of multiple genome sequences, the details of the molecular pathogenesis of C. jejuni remain elusive. This is in part due to the fact that C. jejuni largely lacks homologs of virulence factors found in other pathogens [3]. Because the incidence of human C. jejuni infection is noticeably increasing, with the rapid emergence of antibiotic-resistant strains, there is growing interest to identify novel virulence factors and pathogenesis mechanisms associated with this pathogen as a key step toward controlling the disease.
Fatty acids and their derivatives are key molecules that contribute to the structural and biochemical nature of cell membranes. Thus, fatty acid biosynthesis is essential for all living cells. For de novo fatty acid biosynthesis, bacteria, plants, and apicomplexan parasites utilize the type II fatty acid synthase (FAS II) pathway consisting of a series of individual enzymes, whereas mammals and fungi employ the FAS I pathway involving large multifunctional enzymes [4]. Due to important structural differences in enzymes, the indispensable bacterial FAS II pathway has been an attractive target for antibacterial drug discovery [4]. The FAS II pathway includes initiation followed by the elongation cycle. Each elongation cycle involves four sequential reactions, which bring about the extension of the two-carbon unit. The dehydration of β-hydroxyacyl-ACP to trans-2-acyl-ACP is catalyzed by the enzymes FabZ or FabA. While FabA is only found in unsaturated fatty acid-producing Gram-negative bacteria, FabZ is the ubiquitous dehydratase of FAS II [5].
Although the enzymatic characterization of the FAS II system has been accomplished for E. coli and a few pathogens, the structural and functional attributes of the C. jejuni FAS II enzymes have not been studied. Following our recent study of a C. jejuni virulence protein Cj0977 [6], which suggested a possible link between virulence and membrane lipid biosynthesis, we have launched structure-function studies on enzymes catalyzing membrane lipid biosynthesis in C. jejuni. Of note, C. jejuni possesses a compact genome, which reveals scarce organization of genes into operons or clusters [7]. Indeed, the gene organizations of the C. jejuni FAS II enzymes differ from those of the E. coli FAS II enzymes, raising possibilities of structural and functional dissimilarities between the E. coli system and pathogenic bacteria.
Here we report the identification, crystal structure, and enzymatic properties of C. jejuni FabZ (CjFabZ), defining the first FAS II enzyme in C. jejuni.
Materials and methods
Cloning, expression, and purification of CjFabZ
The cj0273 gene of NCTC 11168 (predicted to encode FabZ) was amplified from C. jejuni strain 81–176 genomic DNA by using Taq DNA polymerase (Promega) and primers pER_F1 (5′-CCGGAATTCATGATAGATGTAATGCAAATTCAA-3′) and pXh_R146 (5′-CCGCTCGAGTTATTTATCCACTATCAT-3′). For the cloning purpose, EcoRI and XhoI sties (underlined) were introduced to the forward and reverse primers, respectively. PCR fragments were cloned into the pGEX-6p-1 vector (GE Healthcare), and the ligation mix was used to transform E. coli DH5α strain. The cloned gene sequences were confirmed by DNA sequencing.
For expression of CjFabZ, the recombinant plasmid pGEX::CjFabZ was transformed into BL21(DE3). The cells were grown at 37°C in 1 L of Luria-Bertani or SelenoMet (Molecular Dimensions Ltd.) media with 100μg/ml ampicillin. Once OD600nm reached ~0.8, isopropyl-β-D-1-thiogalactopyranoside was added to a final concentration of 0.2 mM, and the culture was incubated at room temperature for 4 hrs. Cells were harvested at 4,000g for 20 minutes, and resuspended in 80 ml of PBS containing 2 % Triton X-100 (Sigma) and Complete™ EDTA-free protease inhibitor (Roche Diagnostics).
After cell lysis using sonication, the total cell lysate was clarified by centrifugation at 40,000g for 20 min. The GST-CjFabZ fusion protein was first isolated by affinity chromatography using Glutathione-Sepharose 4B beads (GE Healthcare). Following the binding step for 2 hrs at 4°C and extensive washing with PBS in the presence of 0.2% Triton X-100 and 1 mM DTT, the CjFabZ portion was cleaved by using 80 units PreScission protease (GE Healthcare) in 3 ml of cleavage buffer (50mM Tris-HCl pH 7.0, 150mM NaCl, 1mM EDTA, 0.2% Triton X-100, and 1 mM DTT) overnight at 4°C, and collected by centrifugation at 1000g for 5 min. The highly pure fractions were combined, concentrated, and further purified using a high resolution gel filtration column (Superdex 75 10/300 GL) equilibrated with buffer-GF (50mM HEPES pH 7.0, 200 mM NaCl, 0.5 mM EDTA, and 5% Glycerol). The highly pure peak fractions were then concentrated to ~4 mg/ml.
Crystallization and structure determination
CjFabZ crystals were grown in a 1:1 mixture of protein (4 mg/ml in buffer-GF) and reservoir solution containing 14% polyethylene glycol monomethyl ether (PEG MME) 550, 0.2 M NaCl, and 0.1 M BICINE, pH 9.0 using the hanging drop vapor diffusion method at 17°C. Rectangular shaped crystals grew to typical dimensions of 150μm × 50μm ×25μm. For data collection, crystals were treated with cryo-solutions containing 20% PEG MME 550, 0.2 M NaCl, 0.1M BICINE, pH 9.0, and glycerol (5, 10, and 20% in three steps) for 10 min, and cooled in liquid N2. Diffraction data were collected using single crystals at 100K at BL19BM (Advanced Photon Source). The data sets were indexed and integrated using HKL3000 and scaled with SCALEPACK to 2.6 Å [8]. CjFabZ crystals belonged to space group P212121 with unit cell dimensions of a=88.589Å, b=93.403Å, c=126.631Å.
The CjFabZ structure was solved by molecular replacement with the structure of H. pylori FabZ (PDBID: 2GLM) as the search model using the program PHASER, refined by using programs CNS1.1 [9] and Refmac5 [10]. Manual adjustment of models was performed using COOT [11]. Solvent molecules were placed at positions where spherical electron density peaks were found above 1.5σ in the |2Fo-Fc| map and above 3.5σ in the |Fo-Fc| map, and where stereo-chemically reasonable hydrogen bonds were allowed. Non-crystallographic symmetry restraint and TLS refinement were applied to the final model using REFMAC5 (Table 1). Structural analysis of the final model using the Protein Data Bank (PDB) validation suite indicated almost all residues are in the most favored regions of the Ramachandran plot. The coordinates and structure factors of the CjFabZ structure have been deposited at PDB ID code 3D6X.
Table 1.
Crystallographic data and refinement statistics.
| Data Collection | |
| Wavelength (Å) | 0.97915 |
| Space group | P212121 |
| Cell parameters (Å) | a = 88.589, b =93.403, c = 126.631 |
| Resolution (Å) | 50-2.6 (2.69-2.60) |
| Reflections (Total / Unique) | 87,762 / 29,418 |
| Completeness (%) a | 88.2 (60.5) |
| Rmerge (%) a,b | 7.2 (26.4) |
| <I/σ (I)>a | 11.7 |
| Structure Refinement | |
| Resolution (Å) | 50-2.6 |
| No. reflections (working/test) | 27,907 / 1,490 |
| |F|/σ(|F|) | >0 |
| R (%) a,c/Rfree d (%) | 21.6 / 25.4 |
| Total number of atoms e | 6636 |
| Protein molecule | 6 |
| Average B factor, Å2 | 56.8 |
| rmsd from ideality | |
| Bonds (Å) | 0.016 |
| Angles (°) | 1.582 |
Overall data, high- resolution shell in parentheses.
Rmerge = Σ|Ii − 〈I〉|/Σ |Ii|, where Ii is the intensity of an observation, 〈I〉 the mean value for that reflection, and the summations are over all equivalents.
R-factor = Σh||Fo(h) | − |Fc(h)||/ΣhFo(h), where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
Rfree was calculated with 5% of the data excluded from the refinement.
includes 15 water molecules.
CjFabZ enzyme assay
The dehydratase activity of CjFabZ was measured by a continuous spectrophotometric assay based on the difference in absorbance at 260 nm due to the conversion of crotonoyl-CoA to β-hydroxybutyryl-CoA and vice versa. The standard assay condition included 8.8 μg CjFabZ, 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, and substrates (various concentrations) in a total volume of 500 μL. All experiments were performed using Cary50 spectrophotometer (Varian) at room temperature. The concentration of crotonoyl-CoA was determined using εM of 22.6× 103 M−1 cm−1 at 260nm [12]. One unit of FabZ activity is defined as the amount of enzyme that produces 1 nmol of product per minute under standard conditions.
Results
Identification and purification of CjFabZ
The C. jejuni fabZ gene predicted to encode FabZ was identified by sequence homology to E. coli FabZ. While many genes involved in fatty acid biosynthesis are clustered as an operon plsX-fabH-fabD-fabG-acpP-fabF in the E. coli paradigm (Fig. 1A), the corresponding genes are scattered in C. jejuni (ε-proteobacteria) genome, suggesting possible divergence in mechanisms of fatty acid and lipid metabolic pathways among different clades of Gram-negative bacteria. Interestingly, the E. coli fabZ gene is not part of the fatty acid biosynthesis gene operon, but is located in a gene cluster involved in Lipid A biosynthesis (Fig. 1B). By contrast, the C. jejuni fabZ gene locus does not involve many Lipid A genes, yet it is located adjacent to the lpxA gene (Fig. 1B). Therefore, the conservation of the gene locus including the fabZ-lpxA pair appears to be remarkably strong among Gram-negative bacteria.
Figure 1. Bacterial fatty acid biosynthesis pathway.

(A) Top, Schematics of the initiation step and the elongation cycle. FabA is only found in unsaturated fatty acid producing Gram-negative bacteria. For example, the C. jejuni genome does not contain FabA/B, but it harbors two genes encoding proteins annotated as homologs of E. coli FabG and FabH, as indicated by stars. Bottom, E. coli (K12 strain) fatty acid biosynthesis gene locus. Many genes in this pathway are clustered except FabZ/A and FabK/I. By contrast, the corresponding genes in C. jejuni are not clustered, but spread over the genome. (B) Gene locus of FabZ. Top, γ-proteobacteria (E. coli and H. influenzae). Bottom, ε-clade (C. jejuni and H. pylori). The coding strand is indicated by the direction of arrows. The gene size and the position are indicated by the nucleotide number below arrows, together with the gene annotation.
CjFabZ corresponds to a protein of 146 amino acids with a theoretical molecular mass of 17 kDa. Clearly, CjFabZ is very similar to its homologs, with the highest sequence identity (51%) to Helicobacter pylori FabZ. To promote protein solubility, CjFabZ was expressed in BL21(DE3) as a GST-CjFabZ fusion protein. Despite a limited solubility of GST-CjFabZ (~15% of total expression), once the soluble fraction was separated from the total lysate, the protein remained soluble and stable during purification and crystallization. The chromatogram of high-resolution gel filtration displayed a single peak with the elution volume of 9.5 ml, indicating that the purified CjFabZ adopts a hexameric structure in solution (Fig. 2A).
Figure 2. Structure of CjFabZ.

(A) Analytical gel filtration chromatograph. The purified CjFabZ protein eluted as a hexamer, with an estimated molecular mass of 100 kDa. (B) Ribbon diagram of the CjFabZ monomer structure. The secondary structures are labeled. N and C denote the N and C termini of CjFabZ, respectively. The central helix (α3) is highlighted in red. (C) Ribbon diagram of the CjFabZ hexamer in the asymmetric unit. The view is from the 3-fold axis of the hexamer. The dimer A/B is colored in red/yellow, the dimer C/D in cyan/blue, and the dimer E/F in magenta/orange. (D) Structure-based sequence alignment of FabZ and FabA. Secondary structures are indicated above and below the primary sequences for FabZ and FabA, respectively. Identical residues are highlighted with gold and silver boxes, for FabZ and FabA, respectively. In FabZ, residues involved in the formation of the hexamer (trimer of dimers) are indicated with red boxes (*). The conserved catalytic residues, His and Glu/Asp, are indicated with blue boxes (+).
Structure of CjFabZ
Molecular replacement showed that the asymmetric unit of the P212121 cell of the crystal contains six subunits forming one hexamer. The final structure is well ordered, and electron densities for all residues were clearly interpretable, except C-terminal residues and the connecting loop between α3 and β3 (three to five residues depending on chains). This loop was also disordered in the FabZ crystal structures of Pseudomonas aeruginosa and Plasmodium falciparum in the absence of ligands, suggesting a very flexible region of the structure [13, 14].
The monomer structure of CjFabZ adopts a typical mixed β + α ‘hot dog’ fold (Fig. 2B), according to the naming of the E. coli β-hydroxydecanoyl thiol ester dehydratase (FabA) [15]. Each subunit consists of six-stranded highly curved anti-parallel β sheet β1/β2/β4/β5/β6/β3 that wraps around a central α-helix α3 (Fig. 2B). Two short α-helices are formed at the N terminus and between β2 and α3.
The structure of the CjFabZ dimer is similar to other known FabZ structures [13,14,16]. The main driving force for CjFabZ dimerization is H-bonds between residues from two β3 strands, resulting in a highly curved 12-stranded anti-parallel sheet. The dimer is further stabilized by hydrophobic interactions in the last two turns of the central helix α3. The dimer interface buries a surface area of ~1180 Å2 per monomer (about 15% of the total surface area). The substrate-binding pocket is formed in the subunit- subunit interface, and thus each dimer harbors two active sites. Analysis of other known FabZ structures allows us to define the active sites of CjFabZ. The binding tunnel is lined by residues His48, Phe49, Ile54, Tyr55, Pro56, Gly57, Val58, Ile60, Phe97, Arg98, Asn99, Pro100 and Val101 from one subunit and Leu11, His 13, Glu62, Ala65, Gln66, Gly69, Phe73, Lys85, Val86, Val87, Tyr88, Phe89, Thr90 and Ala141 from the second subunit (Fig. 2D & Fig. 3). These residues, mostly located at the loop between α2 and α3, the central helix α3, and β3, are well conserved. The proposed catalytic residues, the strictly conserved His48 and Glu62* (* indicates residue from the other subunit of the dimer), reside in the middle of the tunnel (Fig. 3). While the crystal structure of an ACP-FabZ complex remains elusive, the docking site for the ACP moiety onto FabZ can be inferred based on previous studies [17,18]. A wide groove is formed by the curved portion of β-sheets including β3, β6, β5, and β4 from one subunit (Tyr88, Thr90, Gly91, Arg117, Met120, Lys140 and Met 142) and the C-terminal end of β3 of the other subunit (Lys96* and Arg98*) (Fig. 3). Although these residues do not display remarkable identities across homologs, the groove has a common surface structure with the hydrophobic hollow surrounded by conserved basic residues (Arg117, Lys96* and Arg98*). This observation suggests that the interaction mode between bacterial FAS II enzymes and ACP is similar, yet each system contains determinants for its specificity.
Figure 3. Active site of CjFabZ.
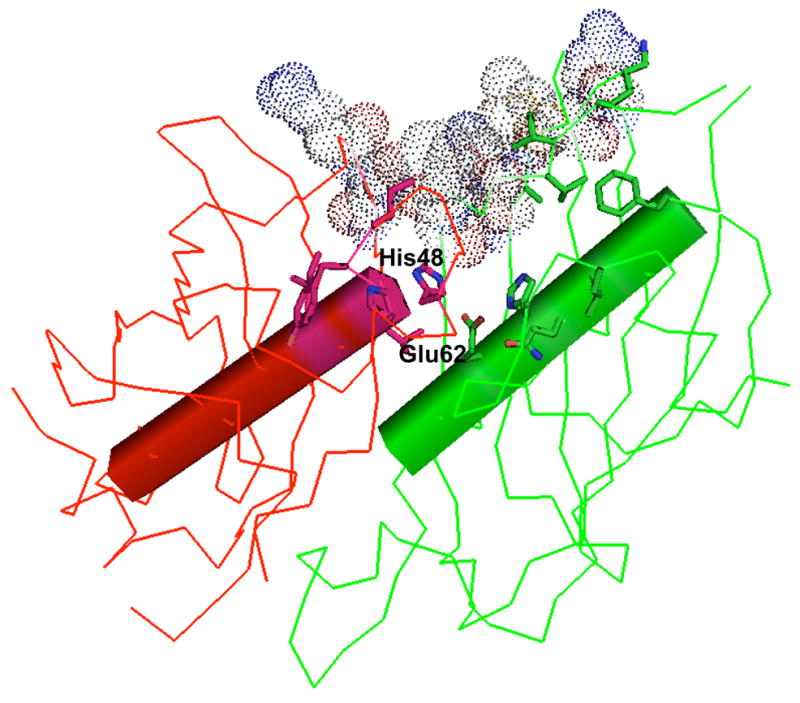
The active sites of CjFaZ were defined by analysis of other known FabZ structures. For clarity, only one active site is depicted. Key amino acid residues forming the substrate-binding tunnel are shown in stick model, and catalytic residues are labeled. The proposed ACP-interaction site is depicted in CPK model.
Consistent with the oligomeric state of CjFabZ in solution, the crystal structure reveals that individual subunits assemble into hexamers in the crystalline lattice. The six molecules in the asymmetric units are superimposed well with r.m.s.d. ranging 0.24 to 0.48 Å between protomers, and structural flexibilities among different protomers are observed mostly in the loop between α3 and β3 and at the N-terminus. The interface of hexamer (trimer of dimers) buries ~940 Å2 per monomer (about 12% of the total surface area), and is very similar to other FabZ structures, reflecting highly conserved amino acid residues involved in the dimer-dimer interface (Fig. 2C & 2D). Therefore, the functional quaternary structure of CjFabZ is defined as a hexamer.
Biochemical properties of CjFabZ
The dehydratase activity of CjFabZ was assessed by spectrophotometric assays. While FabZ catalyzes the reversible dehydration of a (3R)-β-hydroxyacyl-ACP to a trans-2-acyl-ACP in vivo (Fig. 1A), substrate analogs β-hydroxybutyryl-CoA and crotonoyl-CoA have been utilized for the in vitro study. The reverse reaction of the enzyme, from crotonoyl-CoA to β-hydroxybutyryl-CoA, was measured by monitoring the decrease in absorbance at 260 nm. A Km value of 69.7 μM was obtained by our assay with a Vmax value of 181.8 nmol min−1 mg−1 (Fig. 4 and Table 2). The catalytic efficiency of CjFabZ (kcat/Km) for crotonoyl-CoA was calculated as 738.9 M−1 s−1. The forward reaction of the enzyme, from β-hydroxybutyryl-CoA to crotonoyl-CoA, was measured by monitoring the increase in absorbance at 260 nm, resulting in a Km value of 257 μM and a Vmax value of 15.6 nmol min−1 mg−1 (Fig. 4 and Table 2). The catalytic efficiency of CjFabZ (kcat/Km) for β-hydroxybutyryl-CoA was calculated as 17.1 M−1 s−1. The enzymatic characterization has been reported for a few FabZ homologs using substrate analog crotonoyl-CoA [14,19]. The Km values of the FabZ homologs studied to date narrowly ranges from 69.7 (CjFabZ) to 86 (PfFabZ) μM with the kcat/Km values ranging from 157 (HpFabZ) to 738.9 (CjFabZ) M−1s−1. On the other hand, the enzymatic parameters for β-hydroxybutyryl-CoA were reported only for PfFabZ (Km=199 μM; and kcat/Km =80.4 M−1s−1). Therefore, CjFabZ displays similar enzymatic properties to its homologs. Indeed, the equilibrium of the FabZ reaction appears to lie towards the hydration reaction in vitro, whereas FabI, the subsequent enzyme of FabZ in the elongation cycle, pulls the reaction toward the forward direction in vivo.
Figure 4. Enzyme activity of CjFabZ.

Kinetic analysis of CjFabZ with crotonoyl-CoA (A), and β-hydroxybutyryl-CoA (B). Based on non-linear regression analysis, KM values of 69.7 and 257 μM were obtained with crotonoyl-CoA and β-hydroxybutyryl-CoA, respectively.
Table 2.
Activity of CjFabZ in the dehydratase assays
| Substrate | VMax(nmol min−1 mg−1) | kcat (s−1) | KM (μM) | kcat//KM (M−1 s−1 |
|---|---|---|---|---|
| Crotonoyl-CoA | 181.8 ± 16.6 | 0.0515 ± 0.0047 | 69.7 ± 11.2 | 738.9 |
| β-Hydroxybutyryl-CoA | 15.6 ± 1.3 | 0.0044 ± 0.0004 | 257.1 ± 37.0 | 17.1 |
Discussion
While E. coli serves as the paradigm for the FAS II pathway, the observation of dissimilar gene structures and organization among proteobacteria raised issues about the functional assignment of many uncharacterized homologs from diverse genome sequences. Previous work clearly demonstrated that bacterial fatty acid biosynthetic pathways could not be deduced solely by sequence comparison [5,20]. Our analyses revealed that the gene structures encoding C. jejuni FAS II are quite different from E. coli FAS II, including the lack of FAS II gene cluster and the presence of two copies of genes encoding FabG and FabH (Fig. 1), which await functional annotation as do other C. jejuni FASII enzymes.
Thus far, the crystal structures of FabZ have been solved from P. aeruginosa, P. falciparum, and H. pylori [13,14,16,17]. In this study, we demonstrated that CjFabZ is the dehydratase of C. jejuni possessing a comparable enzyme activity to its homologs in vitro. Although FabZ structures display considerable similarity, each structure has unique properties, especially in the loop between α3 and β3 and at the N-terminus. Besides being a potential antibiotics target, FabZ represents an important molecule due to the characteristic hot dog fold found in dehyratases and thioesterase [21]. Oligomerization of proteins denotes an essential strategy for producing protein structural and functional complexity, with potential advantages of genetic economy, structural stability, and increased potential for regulation [22]. The hot dog fold proteins represent an excellent system to investigate specific selection benefits of the quaternary structures observed, as we have collected a significant body of structural and functional data with the hot dog fold. Despite the common fold and enzymatic activities of FabA and FabZ, the latter adopts a hexamer structure, whereas the former adopts a dimer structure. Our structure-based sequence alignment of FabZ/FabA shows specific sequence conservation in the regions involved in the higher order quaternary structure in FabZ (Fig. 2D). Interestingly, hot dog fold thioesterases have been shown to adopt various quaternary structures, whereas FabZ dehydratases appear to adopt one hexameric quaternary assembly, yet distinct from recently reported hexameric thioesterases [23]. It would be interesting to discover the driving force governing the specific quaternary structure of FabZ, and know whether or not different hexameric (or tetrameric) assemblies are associated with an active form of FabZ.
In conclusion, the crystal structure and functional identification of CjFabZ delineate the first functional dehydratase of C. jejuni. Finally, this study provides an impetus for investigation on roles of the C. jejuni FAS II system in virulence, and functional activities associated with CjFabZ dehydratase in vivo.
Acknowledgments
We thank the staff of beamline 19BM of the Structural Biology Center at APS for help during data collection, Dr. Pat Guerry for C. jejuni 81–117 genomic DNA of exceptional quality and Dr. Joe Eichberg for helpful discussions. This work was supported in part by NIH Grant RO1 AI068943 (to H.J.Y.) and by the R. Welch Foundation grant E-1616 (to H.J.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Young KT, Davis LM, DiRita VJ. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol. 2007;5:665–679. doi: 10.1038/nrmicro1718. [DOI] [PubMed] [Google Scholar]
- 2.van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barre syndrome. Lancet Neurol. 2008;7:939–950. doi: 10.1016/S1474-4422(08)70215-1. [DOI] [PubMed] [Google Scholar]
- 3.Guerry P. Campylobacter flagella: not just for motility. Trends Microbiol. 2007;15:456–461. doi: 10.1016/j.tim.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Zhang YM, White SW, Rock CO. Inhibiting bacterial fatty acid synthesis. J Biol Chem. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Cronan JE. Functional replacement of the FabA and FabB proteins of Escherichia coli fatty acid synthesis by Enterococcus faecalis FabZ and FabF homologues. J Biol Chem. 2004;279:34489–34495. doi: 10.1074/jbc.M403874200. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama T, Paek S, Ewing CP, Guerry P, Yeo HJ. Structure of a sigma28-regulated nonflagellar virulence protein from Campylobacter jejuni. J Mol Biol. 2008;384:364–376. doi: 10.1016/j.jmb.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parkhill J, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 8.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode, Methods in Enzymology. Macromol Cryst A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 9.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 10.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. 1999;D55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 11.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 12.Stadtman ER. Preparation and assay of acyl coenzyme A and other thiol esters; use of hydroxylamine. Methods in Enzymology. 1957;3:931–941. [Google Scholar]
- 13.Kimber MS, et al. The structure of (3R)-hydroxyacyl-acyl carrier protein dehydratase (FabZ) from Pseudomonas aeruginosa. J Biol Chem. 2004;279:52593–52602. doi: 10.1074/jbc.M408105200. [DOI] [PubMed] [Google Scholar]
- 14.Kostrewa D, Winkler FK, Folkers G, Scapozza L, Perozzo R. The crystal structure of PfFabZ, the unique beta-hydroxyacyl-ACP dehydratase involved in fatty acid biosynthesis of Plasmodium falciparum. Protein Sci. 2005;14:1570–1580. doi: 10.1110/ps.051373005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: two catalytic activities in one active site. Structure. 1996;4:253–264. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- 16.Swarnamukhi PL, Sharma SK, Bajaj P, Surolia N, Surolia A, Suguna K. Crystal structure of dimeric FabZ of Plasmodium falciparum reveals conformational switching to active hexamers by peptide flips. FEBS Lett. 2006;580:2653–2660. doi: 10.1016/j.febslet.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YM, Rao MS, Heath RJ, Price AC, Olson AJ, Rock CO, White SW. Identification and analysis of the acyl carrier protein (ACP) docking site on beta-ketoacyl-ACP synthase III. J Biol Chem. 2001;276:8231–8238. doi: 10.1074/jbc.M008042200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang YM, Marrakchi H, White SW, Rock CO. The application of computational methods to explore the diversity and structure of bacterial fatty acid synthase. J Lipid Res. 2003;44:1–10. doi: 10.1194/jlr.r200016-jlr200. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Liu W, Hu T, Du L, Luo C, Chen K, Shen X, Jiang H. Structural basis for catalytic and inhibitory mechanisms of beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ) J Biol Chem. 2008;283:5370–5379. doi: 10.1074/jbc.M705566200. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Cronan JE. Only one of the two annotated Lactococcus lactis fabG genes encodes a functional beta-ketoacyl-acyl carrier protein reductase. Biochemistry. 2004;43:11782–11789. doi: 10.1021/bi0487600. [DOI] [PubMed] [Google Scholar]
- 21.Dillon SC, Bateman A. The Hotdog fold: wrapping up a superfamily of thioesterases and dehydratases. BMC Bioinformatics. 2004;5:109. doi: 10.1186/1471-2105-5-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Willis MA, Zhuang Z, Song F, Howard A, Dunaway-Mariano D, Herzberg O. Structure of YciA from Haemophilus influenzae (HI0827), a hexameric broad specificity acyl-coenzyme A thioesterase. Biochemistry. 2008;47:2797–2805. doi: 10.1021/bi702336d. [DOI] [PubMed] [Google Scholar]