

Abstract
Little is known about the molecular mechanisms by which STAT proteins promote tumorigenesis. Drosophila is an ideal system for investigating this issue, as there is a single STAT (Stat92E), and its hyperactivation causes overgrowths resembling human tumors. Here we report the first identification of a dominant-active Stat92E protein, Stat92EΔNΔC, which lacks both N- and C-termini. Mis-expression of Stat92EΔNΔC in vivo causes melanotic tumors, while in vitro it transactivates a Stat92E-luciferase reporter in the absence of stimulation. These gain-of-function phenotypes require phosphorylation of Y711 and dimer formation with full-length Stat92E. Furthermore, a single point mutation, an R442P substitution in the DNA-binding domain, abolishes Stat92E function. Recombinant Stat92ER442P translocates to the nucleus following activation but fails to function in all assays tested. Interestingly, R442 is conserved in most STATs in higher organisms, suggesting conservation of function. Modeling of Stat92E indicates that R442 may contact the minor groove of DNA via invariant TC bases in the consensus binding element bound by all STAT proteins. We conclude that the N- and C- termini function unexpectedly in negatively regulating Stat92E activity, possibly by decreasing dimer dephosphorylation or increasing stability of DNA interaction, and that Stat92ER442 has a nuclear function by altering dimer:DNA binding.
Keywords: STAT, JAK, Unpaired, Drosophila, constitutively active, in vitro reporter, in vivo reporter, structure function, signal transduction
Introduction
The Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway is evolutionarily conserved and is critical for numerous biological processes, including immunity and proliferation (reviewed in (Arbouzova and Zeidler, 2006; Levy and Darnell, 2002)). STATs are a family of latent cytoslic transcription factors that are activated by tyrosine phosphorylation, which allows the formation of an activated dimer through reciprocal phosphorylated tyrosine-Src Homology 2 (SH2) interactions between two STAT monomers. Studies in cultured cells have led to a model in which JAK non-receptor tyrosine kinases, constitutively associated with transmembrane receptors, are activated following ligand binding (Fig. 1A). JAK activation leads to the subsequent tyrosine phosphorylation of receptor sites to which unphosphorylated STAT dimers dock. STAT dimers are activated by JAK-dependent tyrosine phosophorylation and are then able to bind to consensus sequences in target genes and influence their transcription (Becker et al., 1998; Braunstein et al., 2003; Chen et al., 2003; Chen et al., 1998; Kretzschmar et al., 2004; Mao et al., 2005; Neculai et al., 2005; Novak et al., 1998; Schroder et al., 2004; Stancato et al., 1996). The activity of phosphorylated STAT dimers is transient, and these dimers are dephosphorylated in the nucleus and are exported to the cytoplasm (Mertens et al., 2006; Reich and Liu, 2006; Zhong et al., 2005).
Figure 1. Model of the Drosophila JAK/STAT pathway and Stat92E transgenes.
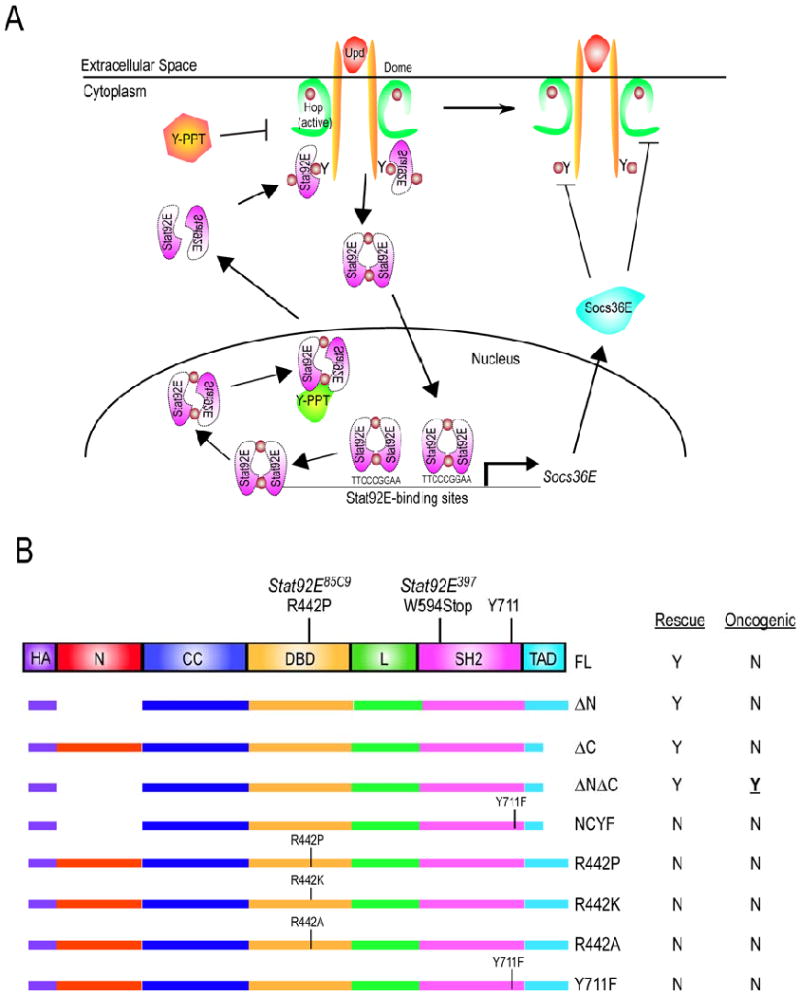
(A) Model of the Drosophila JAK/STAT pathway. Hop is constitutively associated with Dome and is activated by Upd binding to dimerized for of Dome (Brown et al., 2003). Activated Hop subsequently phosphorylates one or more tyrosine residues in the Dome cytoplasmic domain. Inactive, anti-parallel Stat92E dimers dock at the phospho-tyrosine residues on the receptor. Stat92E dimers are then phosphorylated by the activated Hop proteins, assume an activated dimer conformation and translocate to the nucleus where they influence the transcription of target genes. Subsequently, activated Stat92E dimers no longer bind to Stat92E binding sites on DNA; they are dephosphorylated by an unidentified nuclear tyrosine phophatase (green Y-PPT); they are then exported as unphosphorylated inactive dimers to the cytoplasm. The basal phosphorylation of Dome, Hop and/or Stat92E balanced by the actions of cytoplasmic tyrosine phosphatases (orange Y-PPT). (B) Stat92E domains include an N-terminal (N) (red), a coiled-coil (CC) (royal blue), a DNA binding (DBD) (yellow), a linker (L) (green), an SH2 (pink), and a C-terminal trans-activation (TAD) (turquoise) domain. The critical tyrosine in Stat92E is located at residue 711. Deletions and substitutions were made to a UASP-3HA-Stat92EFL transgene containing three N-terminal HA tags (purple). Constructs include deletion of residues 1-133 (ΔN); 725-761 (ΔC); 1-133 and 725-761 simultaneously (ΔNΔC). Substitution constructs include a mutation of the critical Y711 to F in ΔNΔC (ΔNΔCY711F); of R442 to P (R442P), R442 to K (R442K) and R442 to A (R442A); of the critical Y711 to F (Y711F). Constructs were tested in two Stat92E mutant backgrounds: Stat92E85C9, which results from an R442P substitution, and Stat92E397, which results from a premature stop at W594. “Rescue” refers to the abilty of a Stat92E variant to rescue Stat92E loss-of-function phenotypes in vivo. “Oncogenic” refers to the abilty of a Stat92E variant to cause overgrowths or melanotic tumors in vivo. Y = yes and N = no.
Mammals have seven STAT proteins (STAT1-4, 5a, 5b, and 6) that share a similar a conserved domain structure, including N-terminus, coiled-coil, DNA-binding, linker, SH2, and C-terminus (Fig. 1B and (Becker et al., 1998; Chen et al., 1998; Vinkemeier et al., 1998)). The N-terminal domain (residues ∼1-130) is required for formation of tetramers as well as of non-phosphorylated dimers, for tyrosine dephosphorylation, for transcriptional activation and for protein-protein interactions (Chang et al., 2003; Chen et al., 2003; Murphy et al., 2000; Ota et al., 2004; Shuai et al., 1996; Vinkemeier et al., 1998; Xu et al., 1996). A helical coiled-coil domain beginning around residue 130 mediates interaction between several proteins including c-Jun (Zhang et al., 1999). The DNA-binding domain of STATs (DBD) (residues ∼320 to 490) has limited contact with both the major and minor grooves of DNA (Chen et al., 1998). The linker domain (residues ∼490 to 580) modulates the rate of STAT:DNA interactions, ultimately controlling transcriptional activation of STAT target genes (Yang et al., 2002). An SH2 domain (residues ∼580-680) is required for the formation of an activated STAT dimer by mediating reciprocal interactions with a phosphorylated conserved tyrosine residue at position ∼700 that exists in all STATs (Chen et al., 1998; Levy and Darnell, 2002). The phosphorylation of this tyrosine residue is required for STAT function, which is abolished by its mutation to Phe in all species tested (Levy and Darnell, 2002). Lastly, the carboxy-terminal transactivation domain (TAD) varies in length from 38 to 200 amino acids interacts and is required for transcriptional co-activation of mammalian STATs (Horvath, 2000).
Gain-of-function mutations in the Drosophila JAK hopscotch (hop) were the first to link the JAK/STAT pathway to cancer. These hop alleles result in hyperactive kinases that cause an over-proliferation of blood cells, leading to fly “leukemia” and lethality (Binari and Perrimon, 1994; Hanratty and Dearolf, 1993; Harrison et al., 1995; Luo et al., 1995). Similarly, sustained activation of the JAK/STAT pathway is a causal event in human leukemia and myeloproliferative disorders (Baxter et al., 2005; James et al., 2005; Jones et al., 2009; Kilpivaara et al., 2009; Lacronique et al., 1997; Levine et al., 2005; Olcaydu et al., 2009). Persistent activation of Stat3 is associated with a dozen types of human cancer, including all classes of carcinoma (Darnell, 2005). Moreover, a dominant-active form of Stat3, called Stat3-C, generated by conversion of two residues in the C-terminal loops of the SH2 domain to Cys, is oncogenic and causes tumors in nude mice (Bromberg et al., 1999). The constitutive transcriptional abilities of Stat3-C require tyrosine phosphorylation of Y705 and likely arise because the cysteine mutations increase occupancy of the STAT dimer on DNA and/or prevent its dephosphorylation, which results in the accumulation of activated STAT dimers (Li and Shaw, 2006; Liddle et al., 2006). Inhibition of Stat3 arrests the growth of primary human cancer cells, which makes Stat3 an attractive target for cancer therapy (Blaskovich et al., 2003; Chiarle et al., 2005; Song et al., 2005; Sun et al., 2005).
Redundancy in components of the mammalian JAK/STAT pathway exists at each level of this signaling cascade. In contrast, the Drosophila pathway is a complete yet simplified version of its mammalian counterpart (Arbouzova and Zeidler, 2006). Three related IL-6-like cytokines, Unpaired (Upd), Upd2 and Upd3, activate a gp-130-like receptor Domeless (Dome), which leads to the activation of the sole JAK Hop and the sole STAT Stat92E (Fig. 1A and (Agaisse et al., 2003; Binari and Perrimon, 1994; Brown et al., 2001; Chen et al., 2002; Gilbert et al., 2005; Harrison et al., 1998; Hombria et al., 2005; Hou et al., 1996; Sefton et al., 2000; Yan et al., 1996)). Activated Stat92E regulates expression of target genes, such as Socs36E, which encodes a negative regulator of the pathway (Callus and Mathey-Prevot, 2002; Issigonis et al., 2009; Karsten et al., 2002; Rawlings et al., 2004). This pathway plays important roles in many aspects of Drosophila development, including embryonic and eye development and larval hematopoiesis (reviewed in (Arbouzova and Zeidler, 2006)). Sustained JAK/STAT signaling in the fly eye during development leads to an enlarged eye that is 2-3 times larger than wild type (Bach et al., 2003; Juni et al., 1996; Tsai and Sun, 2004). Furthermore, loss of tumor suppressor genes tsg-101 and vps25 lead to excessive activation of JAK/STAT pathway signaling and over-growth of the eye, and this phenotype is suppressed by removal of one copy of Stat92E (Herz et al., 2006; Moberg et al., 2005; Thompson et al., 2005; Vaccari and Bilder, 2005). In contrast, global loss of Stat92E during eye development leads to an ablated eye and lethality prior to adulthood (Ekas et al., 2006; Hou et al., 1996; Tsai et al., 2007; Yan et al., 1996).
Stat92E is a 761 amino acid protein that shares a similar domain structure to other STATs and that is most similar to human STAT5 with 37% identity (Hou et al., 1996; Yan et al., 1996; Zeidler et al., 2000). Despite the sequence similarity between mammalian STATs and Stat92E, the functional requirements of the different domains in Stat92E are largely unknown, and an in-depth structural analysis of this protein has not been undertaken. To address this issue, we designed in vivo rescue assays and in vitro readouts to investigate the role of the N- and C-terminal domains in Stat92E. We also examined the requirement of Arg442, which is conserved in the majority of mammalian STATs, and is mutated to Pro in the strong hypomorphic allele Stat92E85C9 (Silver and Montell, 2001; Wang and Levy, 2006a). Surprisingly, we found that neither the first 133 nor the last 36 amino acids are required for Stat92E function. Furthermore, removal of both of these domains simultaneously resulted in a constitutively active form of Stat92E, the oncogenic activity of which depends on phosphorylation of Y711. We also demonstrated that Arg442 is required for Stat92E function, presumably because it forms a key STAT:DNA contact point.
Materials and Methods
Genetics
These stocks are described in FlyBase: Stat92E85C9; Stat92E397; Stat92E06346; Stat92Ej6C8; outstretched (os); UAS-upd; UAS-hop; tub-Gal4; Df(3R)H-B79; UAS-GFP; ey-Gal4 (Hauck et al., 1999); P[AyGAL4]25 P[UAS-GFP.S65T]T2; hs-flp MKRS/TM6B; ey-Gal4, UAS-flp (Stowers and Schwarz, 1999); M(3)96C; Ribosomal protein S3 (RpS3)1; and FRT82B ry506. We also used 10×STAT-GFP (Bach et al., 2007). actin-Gal425/CyO was a gift of Norbert Perrimon. Crosses were maintained at 25°C.
We used the Mosaic Analysis with a Repressible Cell Marker (MARCM) technique to generate positively-marked Stat92E85C9 clones that over-expressed Stat92EΔNΔC (Lee and Luo, 1999).
For the MARCM analyses, we crossed w; UAST-3HA-Stat92EΔNΔC/UAST-3HA-Stat92EΔNΔC; FRT82B Stat92E/TM6C,Tb,Sb flies to ey-flp, UAS-GFP; tub-Gal4/CyO; FRT82B tub-Gal80/TM6B,Tb or hs-flp, UAS-GFP 6xMyc, tub-Gal4; +/+; FRT82B tub-Gal80/TM6B,Tb.
In animals possessing the ey-Gal4, UAS-flp (EGUF (Stowers and Schwarz, 1999)) chromosome, the ey promoter drives expression of the yeast Gal4 transactivator from the earliest stages of eye development (Stowers and Schwarz, 1999). Gal4 stimulates the expression of genes under the control of Upstream Activating Sequences (UAS) via the UAS/Gal4 technique (Brand and Perrimon, 1993). In this case, Gal4 induces flp, and FLP induces mitotic recombinantion between homologous Flippase Recognition Target (FRT) sites by means of the FLP/FRT technique (Xu and Rubin, 1993). Minutes are mutations in ribosomal genes that cause slow growth and recessive lethality in cells possessing the wild type chromosome, providing a growth advantage to the homozygous (e.g., Stat92E) tissue (Lambertsson, 1998; Morata and Ripoll, 1975). Minute clones were made using FRT82B M(3)96C, arm-lacZ or FRT82B RpS31, ubi-GFP stocks. FRT82B ry506 was used as the control + chromosome. When EGUF flies also carry a UAS-3HA-Stat92E transgene, ey-Gal4 drives expression of both UAS-flp and UAS-3HA-Stat92E in the eye disc. This results in the generation of homozygous Stat92E mutant tissue and the expression of 3HA-Stat92E proteins specifically in eye disc cells.
Clones mis-expressing hop and Stat92EΔNΔC were generated by crossing UAS-hop or UAS-Stat92EΔNΔC flies with P[AyGAL4]25 P[UAS-GFP.S65T]T2; hs-flp MKRS/TM6B flies, in which FLP is under the control of the heat-shock promoter (Ito et al., 1997).
Transgene generation
Constructs
UASp-3HA-Stat92EFL (Ekas et al., 2006)
UASp-3HA-Stat92EΔN
UASp-3HA-Stat92EΔC
UASp-3HA-Stat92EΔNΔC
UAST-3HA-Stat92EΔNΔCY711F
UAST-3HA-Stat92EY711F
UASp-3HA-Stat92ER442P
UAST-3HA-Stat92ER442K
UASp-3HA-Stat92ER442A
Ac5c-hop-myc-his
socs1-luc
Primers
Stat92EΔN
5′DELTA1-133
5′ TTTTTGGATCCATGAACAACACGCCCATGGTTACCGGG 3′
3′DELTA1-133
5′ GTTGGTGGCGCCAGTTCTTGAGCTCG 3′
Stat92EΔC
5′delta725-761
5′ CAGATCCGTGTGTGGACCCTGTCCTTAC 3′
3′delta725-761
5′ TTTTGCGGCCGCTCCGTTTCTACAAACGTGAACATGCAATG 3′
Stat92EΔNΔCY711F
5′ Primer
5′ CTCGTCCTAGATCCTGTGACCGGTTTTGTGAAGAGCACATTGCATGTTCAC 3′
3′ Primer
5′ GTGAACATGCAATGTGCTCTTCACAAAACCGGTCACAGGATCTAGGACGAG 3′
Stat92EY711F
5′ B YF NEW
.5′ GTTCTAGATCCTGTGACCGGTTTTGTGAAGAGCACATTGCATGTTCAC 3′
3′ B YF NEW
5′ GGGCTATGCGGCCGCAAAGTTCTCAAAGTTTGTAATCGTATC 3′
Stat92ER442P
5′R442PB
5′ GCCCTTCTTTTCTGCCGGCTTGATCTTCTTCAG 3′
3′R422PA
5′ CTGAAGAAGATCAAGCCGGCAGAAAAGAAGGGC 3′
Stat92ER442K
5′R442K B
5′ CTGAAGAAGATCAAGAAGGCAGAAAAGAAGGGC 3′
3′R442K A
5′ GCCCTTCTTTTCTGCCTTCTTGATCTTCTTCAG 3′
Stat92ER442A
5′ R442A B
5′ CTGAAGAAGATCAAGGCGGCAGAAAAGAAGGGC 3′
3′ R442A A
5′ GCCCTTCTTTTCTGCCGCCTTGATCTTCTTCAG 3′
Ac5c-hop-myc-his
5′ Oligo (Oligo 37)
5′ GAGTATCTGCAATCTGGTTCCTTCGAC 3′
3′ Oligo (Oligo 38) (to put an XbaI site at 3′ end of hop cDNA and subclone in frame with Myc and His tags)
5′ GGAAATCTAGAAACTCGGCATCCGTCGGCTGATTCGGCGGCGAC 3′
socs1-luc
5′ Oligo
5′-TTTTTAGATCTGACTGTTTACCGCTTGCGGGTCGCATTTC-3′ (BglII site)
3′ Oligo
5′-TTTTTGGATCCCCTTAACAACTGGCTTGAACTTATGTTTA-3′ (BamHI site).
Transgene generation
A full-length Stat92E cDNA containing three N-terminal HA tags was ligated into UASp to generate the UASp-3HA-Stat92EFL transgene as described in (Ekas et al., 2006). We have subsequently discovered that 3HA-Stat92EFL transgene has a silent point mutation that results in an Ala substitution of Ser at position 8. This mutation does not effect the function of the 3HA-Stat92EFL protein since it can rescue Stat92E loss-of-function phenotypes ((Ayala-Camargo et al., 2007; Ekas et al., 2006), this study and data not shown). The constructs in this study were engineered by polymerase chain reaction (PCR) using the primers above and the C5HA3-3HA-Stat92EFL plasmid as a template. C5HA3 is a pBluescript KS-based vector C5HA3 (Nybakken et al., 2005). The 5′ end of the C5HA3 polylinker includes an ATG immediately upstream of sequence encoding three HA epitopes, followed by an in-frame BamHI site. At the 3′ end, a NotI site immediately precedes and is in frame with a stop codon. All Stat92E constructs in this study were designed to have BamHI (5′) and NotI (3′) ends. After digestion with BamHI and NotI, the insert was ligated into C5HA3 that had also been digested with BamHI and NotI. For the 3HA-Stat92EΔN, 3HA-Stat92EΔC and 3HA-Stat92ER442P plasmids, the insert was excised from C5HA3 by digestion with BssHII. The 3′ recessed termini were filled in with Klenow and were ligated into UASp (Rorth, 1996) that had been cut with BamHI, filled in Klenow and treated with Shrimp Alkaline Phosphatase (SAP) (Roche). For 3HA-Stat92EΔNΔC, a ∼400 bp fragment deleting the first 133 amino acids was generated by PCR using the primers for the 3HA-Stat92EΔN construct. This fragment was digested with BamHI and ClaI and ligated into the C5HA3-3HA-Stat92EΔC vector that had been digested with BamHI and ClaI. It was subsequently cloned into UASp as described above For 3HA-Stat92EΔNΔCY711F, 3HA-Stat92ER442K, 3HA-Stat92ER442A, and 3HA-Stat92EY711F, oligos containing an AvrII restriction site were made to the 5′ and 3′ ends of the Stat92E transgenes and PCR was then used to remove the entire transgenic sequence (including the 3 HA tags). Inserts were cut with AvrII and ligated into UAST (Brand and Perrimon, 1993) that had been cut with XbaI and treated with SAP. Throughout the text, these constructs are frequently referred to without the “UAS” preface. Transgenic lines were by injection into w1118 Drosophila embryos (CBRC Transgenic Fly Core, Charlestown, MA). Multiple insertions of each UAS-3HA-Stat92E transgene were tested in each assay, and similar results were obtained for each construct.
To make the Ac5c-hop-myc-his construct, we subcloned the entire ∼5 kb ApaI-XbaI fragment of the hop cDNA from Bluescript-hop (Binari and Perrimon, 1994) into pcDNA3.1(-)/myc-His B (Invitrogen). The resulting plasmid was called pcDNA3.1-hop-myc-His-ApaI-XbaI. The parental pcDNA3.1(-)/myc-His B plasmid contains C-terminal Myc and His epitodes tags followed by a stop codon and preceded by unique restriction enzyme sites. To remove the stop coding from the pcDNA3.1-hop-myc-His-ApaI-XbaI plasmid, we amplified by PCR a 600 bp fragment of the C-terminal end of hop and mutated the stop codon, inserted an XbaI site and engineered it to be in frame with the Myc and His tags. This fragment was digested with PflMI (a unique site in hop) and XbaI and the resulting 355 bp band was purified and ligated into the pcDNA3.1-hop-myc-His-ApaI-XbaI plasmid vector at the PflMI and XbaI sites. The resulting plasmid pcDNA3.1-hop-myc-his was cut with NotI and PmeI, which removes 500 bp of the 5′UTR of the hop cDNA but leaves the start site, the epitopes tags, stop codon intact. The NotI-PmeI fragment was treated with SAP and subcloned into the Actin5c (Ac5c) plasmid that had been digested with NotI and HpaI and treated with SAP. The construct was verified by sequencing and by Western blotting (data not shown). In the text describing in the in vitro transfection assays, “Hop-Myc-His” is also referred to as “Hop”.
To generate the socs1-luc reporter we used PCR to amplify an 862 bp fragment of genomic Socs36E intron 1 DNA from isogenized FRT42 flies (Janody et al., 2004) with BglII (5′) and BamHI (3′) ends. After digestion with BglII and BamHI, the insert was ligated into a pGL3 Basic Vector (Promega) at the BglII site. socs1-luc was excised from pGL3 at SmaI and XbaI sites and ligated in pCaSpeR 4 at SpeI and XbaI sites.
in situ hybridization and antibody staining
in situ hybridization and antibody stainings were performed as described in (Bach et al., 2003; Flaherty et al., 2009). We used mouse anti-Discs large (Dlg) (1:50) (Developmental Hybridoma Studies Bank (DHSB)); rabbit anti-β-galactosidase (1:100) (Cappel); rat anti-HA (1:1000) (clone 3F10, Roche); 4′,6-diamidino-2-phenylindole (DAPI) (1:1000) (Invitrogen); rabbit anti-Stat92E (1:1000) (Flaherty et al., in press) and fluorescent secondary antibodies at 1:250 (Jackson Laboratories). Fluorescent images were taken of eye discs (at 25×) using a Zeiss LSM 510 confocal microscope and of cells (at 40×) using a Nikon Eclipse TE2000-E microscope and digital camera. Bright field pictures were taken at 5× using a Leica MZ 8 microscope with an Optronics digital camera or at 20× using a Zeiss Axioplan microscope with a Nikon Digital Sight DL-UL camera.
Western blot analysis
Extracts from S2 cells were prepared by lysis in a buffer containing 1×PBS, 150 mM NaCl, 1% Triton, 1× Complete protease inhibitors (Roche), 2 mM sodium orthovanadate, and 0.5 mM EDTA. Immunoprecipitation was performed as described in (Bach et al., 1996) using rat anti-HA (1:1000) (clone 3F10, Roche) and Protein A-Sepharose (Amersham Biosciences). Immunoprecipitated proteins were eluded, subjected to SDS-PAGE and immunoblotted using standard methods. We used PY-20 (1:1000) (BD Biosciences); mouse anti-HA (1:000) (Covance); and rabbit anti-Stat92E (1:1000) (Flaherty et al., in press). Horseradish peroxidase secondary antibodies (Jackson ImmunoResearch) were used (1:10,000). The blots were developed using Amersham ECL Western Blotting Analysis System (GE Healthcare).
Luciferase assay
Luciferase assays were performed as described in (DasGupta et al., 2005). 6×104 S2 cells were seeded in 96-well plates and transfected using Effectene (Qiagen) with 40 ng of Ac5c-Gal4 (Zeidler et al., 2004), 40 ng of UAS-3HA-Stat92E transgene, 40 ng of socs1-luc, 40 ng of Renilla-luciferase and 4 ng of Ac5c-hop-myc-his. Ac5c was added as needed to maintain a constant concentration of DNA. After 48 hours, firefly luciferase (from socs1-luc) and Renilla luciferase activity were monitored in cell lysates using Dual-Glo Luciferase Assay System (Promega) and a Perkin Elmer Wallac EnVision 2103 multilabel plate reader. Experiments were performed four times in quadruplicate. For each data point, firefly luciferase values were divided by appropriate Renilla luciferase value. Relative luciferase units were then calculated by Nexperimental/Nconstant where Nexperimental is the value of firefly luciferase/Renilla luciferase and Nconstant is Ac-Gal4 firefly luciferase/Renilla Luciferase.
Stat92E homology model
The homology model was built as described in (Cardozo et al., 1995) using the ICM-Pro software program (Molsoft, LLC, La Jolla, CA). Briefly, the crystal structure of phosphorylated STAT1 was used as a template (pdb code 1bf5) for the sequence of Stat92E (Q24151). The similarity between the sequence of the template and the sequence of Stat92E is high enough to virtually guarantee that Stat92E adopts the same overall 3D fold as the template. After threading the Stat92E sequence onto the STAT1 crystal structure using the alignment between the two sequences as a guide, side-chains and loops were energy minimized to obtain the final 3D structural model.
Alignments
Protein sequences were obtained from NCBI. The DNA binding domains were aligned by the Clustral-W method using DS Gene 1.5 software. Optimal STAT binding elements used in this study were originally reported in (Decker et al., 1997; Wang and Levy, 2006b; Yan et al., 1996).
Results
Generation of UAS-3HA-Stat92E transgenes
We previously reported the generation of a full length Stat92E transgene UAS-3HA-Stat92EFL, which encodes the 761 amino acid Stat92E protein tagged at the N-terminus by 3 tandem HA epitopes (Ekas et al., 2006). This transgene fully rescues Stat92E phenotypes in the developing Drosophila eye (Fig. 3D and (Ekas et al., 2006)). To determine the functionally important sequences in Stat92E, we made deletions and substitutions to the 3HA-Stat92EFL construct and then assessed their performance in several assays (Fig. 1B).
Figure 3. Stat92E phenotypes are rescued by Stat92EΔN, Stat92EΔC and Stat92EΔNΔC but not by Stat92EY711F or the Stat92ER442 mutants.
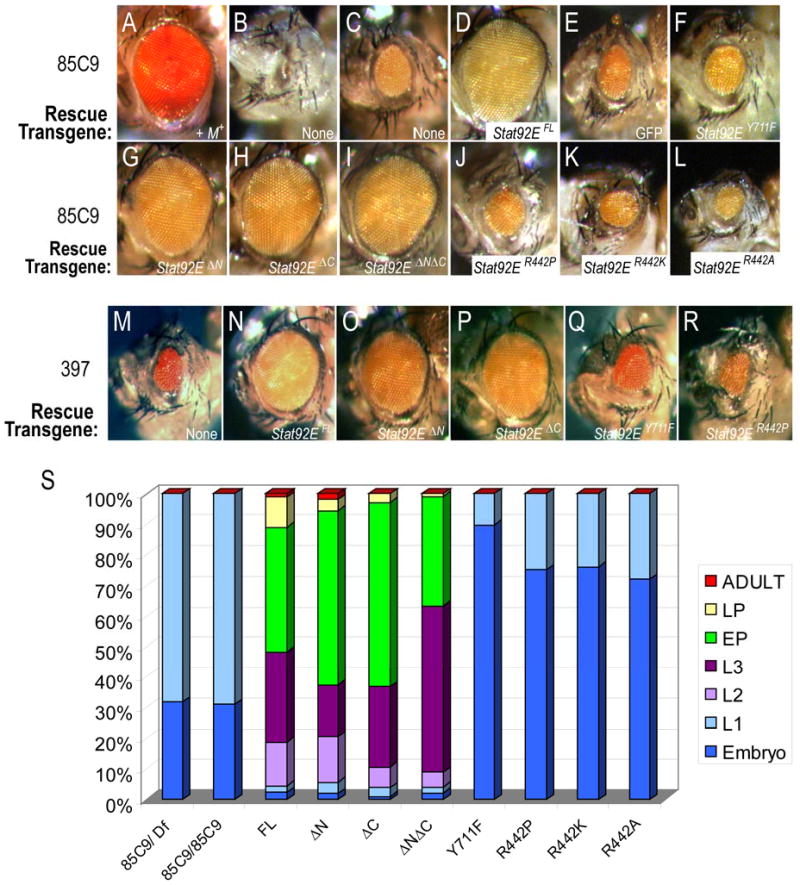
Large patches of Stat92E mutant tissue were generated using an EGUF chromosome and the Minute technique, where ey-GAL4 drives UAS-flp, which then stimulates mitotic recombination, as well as expression of UAS-3HA-Stat92E rescue transgenes. (A) A +M+ adult control eye is the same size as wild type. (B,C) Stat92E85C9 M+ adults exhibit a no-eye (B) or small-eye (C) phenotype. (D-L) A Stat92EFL transgene rescues the Stat92E85C9 phenotype (D), as do Stat92EΔN (G), Stat92EΔC (H), and Stat92EΔNΔC (I). In contrast, UAS-GFP (E), Stat92EY711F (F), Stat92ER442P (J), Stat92ER442K (K), and Stat92ER442A (L) do not. (M-R) Stat92E transgenes behave similarly when tested in EGUF Stat92E397 M+ eye discs. (S) Lethal phase analysis of Stat92E variants. Stat92E transgenes were expressed using tub-Gal4 in 85C9/85C9 zygotic mutants that still have maternal Stat92E mRNAs and the genotype w; tub-Gal4/UAS-3HA-Stat92E; 85C9/85C9. The FL, ΔN, ΔC, and ΔNΔC transgenes significantly delayed the 85C9/85C9 lethal phase to late pupal or infrequently adult stages. In contrast, the Y711F, R442P, R442K and R442A transgenes do not. Rather they increased the embryonic lethality in 85C9/85C9 animals. First (L1), second (L2), and third (L3) larval instar, early (EP) and late (LP) pupal stage. Animals counted: 85C9/Df=129; 85C9/85C9=100; FL=91; ΔN=107; ΔC=103; ΔNΔC=101; Y711F=194; R442P=157; R442K=160; R442A=111.
Stat92E allelic series
First, we determined the Stat92E mutant background in which the in vivo experiments should be performed. We used lethal-phase analysis to assess the strongest Stat92E loss-of-function allele. The Stat92E85C9 and Stat92E397 are ethyl methanesulfonate-induced alleles that are caused by a substitution of Arg at position 442 to a Pro and of a Trp to a stop codon at position 594, respectively (Silver and Montell, 2001). Both Stat92E06346 and Stat92Ej6C8 have P-element insertions 5′ to the start site of the Stat92E gene, which presumably lower transcription of this gene (Hou et al., 1996; Spradling et al., 1999). While P-element mutations are typically hypomorhpic, Stat92E06346 has been reported to be a null (Henriksen et al., 2002; Hou et al., 1996; Mukherjee et al., 2005; Zeidler et al., 1999). However, the level of Stat92E mRNA or protein in this mutant has never been published. We monitored the zygotic lethal phase of these Stat92E alleles placed in trans to a chromosomal deficiency Df(3R)H-B79 that removes the Stat92E gene. 30% of Stat92E85C9/Df, 30% of Stat92E397/Df, 22% of Stat92E06346/Df and 24% of Stat92Ej6C8/Df animals died during embryogenesis. In all four crosses, the embryos that did hatch subsequently died in the first larval instar (Fig. 2A). These data indicate that 85C9 and 397 are stronger alleles than 06346 and j6C8. In support of this conclusion, we found that Stat92E85C9 or Stat92E397 clones in the eye-antennal, wing or leg disc generated significantly stronger phenotypes than Stat92E06346 or Stat92E j6C8 clones ((Ayala-Camargo et al., 2007; Ekas et al., 2006) and data not shown). We propose this Stat92E allelic series, starting with the strongest: 85C9 = 397 > j6C8 >06346.
Figure 2. Stat92E85C9 and Stat92E397 are strong hypomorphic alleles.
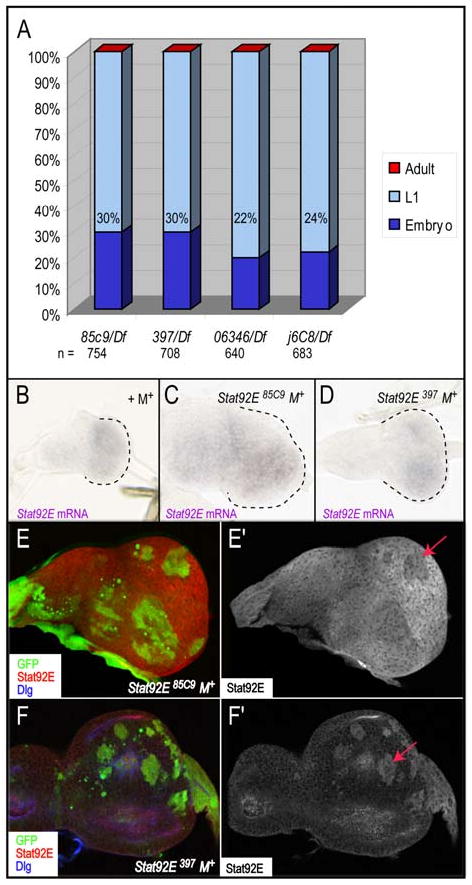
(A) Lethal phase analysis of Stat92E85C9, Stat92E397, Stat92E06346, Stat92E j6C8 alleles in trans to the Df(3R)H-B79 deficiency that removes the Stat92E gene. Both 85C9/Df and 397/Df result in 30% embryonic lethality, while 06346/Df and j6C8/Df result in 22% and 24%, respectively. (B-F) Large Stat92E clones were generated using ey-Gal4, UAS-FLP (EGUF) in a Minute (M+) background. (A-C) Stat92E mRNA is produced in both Stat92E85C9 and Stat92E397 mutant backgrounds. Second instar eye discs in a control EGUF +M+ (A), an EGUF Stat92E85C9 M+ or (B), or an EGUF Stat92E397 M+ background (C). (D,E) Stat92E antibody staining in EGUF Stat92E85C9M+ (H) or EGUF Stat92E397M+ (I) eye discs. Stat92E homozygous mutant tissue is marked by the absence of GFP (green), Stat92E is red and Dlg, which marks cell outlines, is blue. The Stat92E antibody detects Stat92E protein in Stat92E85C9 M+ tissue (D). However, this antibody does not recognize antigen in Stat92E397 M+ tissue because Stat92E397 lacks the region to which the antibody binds (last 15 amino acids of Stat92E) (E). An arrow marks GFP+ Stat92E-/+ heterozygous tissue in D,E. For all figures, eye discs and adult eyes are oriented anterior to the left and dorsal to the top.
Characterization of Stat92E85C9 and Stat92E397
To further characterize the 85C9 and 397 alleles we established assays in the developing eye-antennal disc, which is derived from a primordial of ∼50 progenitor cells and gives rise to the adult eye, antenna and head capsule (Dominguez and Casares, 2005). In wildtype eye discs, activation of the JAK/STAT pathway promotes proliferation of these progenitor cells and formation of the eye field (Bach et al., 2003; Chao et al., 2004; Ekas et al., 2006; Reynolds-Kenneally and Mlodzik, 2005; Tsai and Sun, 2004). An eye-antennal disc composed of almost entirely homozygous Stat92E mutant tissue (hereafter referred to as Stat92E M+) was generated using ey-Gal4, UAS-flippase (flp) (EGUF) and Minute techniques (see Materials and Methods). Stat92E mRNA was ubiquitously expressed in an eye disc from animals carrying a control wild type chromosome in an EGUF Minute background (referred to as +M+) (Fig. 2B). It was also ubiquitously expressed in eye discs in EGUF Stat92E85C9 M+ or Stat92E397 M+ animals (Fig. 2C,D). These data indicate that 85C9 and 397 alleles are not RNA nulls. To determine whether 85C9 is a protein null, we stained Stat92E M+ eye discs with an antibody that recognizes the last 15 amino acids of Stat92E, a region lacking in Stat92E397 (Chen et al., 2002; Flaherty et al., in press; Silver and Montell, 2001). As expected, in Stat92E397 M+ eye discs, the Stat92E antibody staining specifically overlapped with heterozygous tissue, which have one wild type copy of Stat92E (Fig. 2F, arrow). In contrast, Stat92E85C9 M+ eye discs exhibited ubiquitous Stat92E staining in homozygous mutant tissue and reduced expression in GFP-positive heterozygous tissue (Fig. 2E, arrow), demonstrating that this allele is not a protein null.
in vivo function of Stat92E variants
The majority of animals harboring Stat92E85C9 M+ or Stat92E397 M+ clones generated by EGUF do not hatch from the pupal case and those that do eclose exhibit small or ablated eyes (Fig. 3B,C and (Ekas et al., 2006)). We designed a rescue assay that would generate eye disc cells which were homozygous mutant for Stat92E and also mis-expressed a recombinant 3HA-Stat92E protein (see Materials and Methods). Using this assay, we found that the phenotypes and hatching rate in EGUF Stat92E85C9 M+ or Stat92E397 M+ animals were completely rescued by the expression of UAS-3HA-Stat92FL but not by an irrelevant UAS-linked gene (Fig. 3E and (Ekas et al., 2006)). Mis-expression of Stat92EΔN, which lacks the first 133 amino acids, Stat92EΔC, which lacks the last 36 amino acids (residues 726-761), or Stat92EΔNΔC, which lacks both the first 133 and the last 36 amino acids, fully restored the size of the Stat92E85C9 M+ eye to wild type size (Fig. 3G-I). In addition, all of the pupae in these crosses hatched from their pupal cases. By contrast, mis-expression of Stat92EY711F did not increase the size of the Stat92E M+ small eye, confirming that Tyr711 is critical for Stat92E function in vivo as it is in vitro (Fig. 3F and (Yan et al., 1996)). We quantified the eye area of 20 females of each genotype, which confirms these qualitative results (Fig. S1). Importantly, all UAS-3HA-Stat92E variants gave identical results in the Stat92E397 background, indicating that Stat92E85C9 phenotypes are not due to an additional lethal mutation on the 85C9 chromosome (Fig. 3M-R and data not shown). All transgenes were expressed in the eye disc, although Y711F (and non-functional R442 mutants, see below) had lower expression in Stat92E M+ mutant tissue (Fig. S2,A-J). The reduced expression of HA in these discs might result from aberrations in eye field patterning and/or to ectopic expression of signaling molecules like the Wnt family member Wingless, which represses JAK/STAT signaling, and the Notch pathway ligand Serrate (Ayala-Camargo et al., 2007; Ekas et al., 2006; Flaherty et al., 2009; Tsai et al., 2007). Nevertheless, these data demonstrate that Tyr711, but not the N-terminal 133 and/or the C-terminal 36 amino acids, are required for Stat92E function in vivo.
To confirm that the in vivo functionality of these variants is not specific to eye development, we tested the ability of the Stat92E variants to delay the Stat92E85C9 lethal phase. We used the UAS/Gal4 technique to mis-express UAS-Stat92E transgenes in a homozygous Stat92E85C9 background using tubulin-Gal4 (tub-Gal4), which is ubiquitously expressed during all developmental stages (Brand and Perrimon, 1993). It should be noted that these animals still have maternal Stat92E mRNAs. 30% of 85C9/85C9 homozygotes died during embryonic stages, the same value as 85C9/Df (Fig. 2A,3S). Mis-expression of Stat92EFL significantly delayed this lethal phase and the majority of animals died during pupal stages (Fig. 3S). Mis-expression of Stat92EΔN, Stat92EΔC or Stat92EΔNΔC also delayed the lethal phase in a manner similar to Stat92EFL (Fig. 3S). It is unclear why these transgenes did not rescue Stat92E85C9 homozyotes to adulthood because the pupae from these crosses appeared normal upon dissection (data not shown). Another constitutive driver (actin-Gal4) shifted the 85C9/85C9 lethal phase to late larval stages with mis-expressing Stat92EFL (data not shown). These data suggest that these drivers are not strong enough during larval/pupal stages or do not target one or more critical tissues at sufficient levels to insure survival to adult stages. Stat92EY711F could not delay the lethal phase of Stat92E85C9 homozygous animals. Moreover, it had dominant negative effects as more than 95% of these animals died during embryonic stages (Fig. 3S). Importantly, all UAS-3HA-Stat92E variants gave identical results in Stat92E397 zygotic homozygotes (data not shown). These data suggest that the N- and C-terminal domains are not required for Stat92E function during all stages of Drosophila development, while Tyr711 is essential.
in vitro function of Stat92E variants
To investigate whether the 3HA-Stat92E variants can activate gene transcription in vitro, we generated a transcriptional reporter socs1-luc by placing an 880 bp fragment from intron 1 of the Socs36E gene containing four putative Stat92E binding sites upstream of luciferase (Bach et al., 2007; Baeg et al., 2005; Karsten et al., 2002). We transfected S2 cells with Ac5c-hop-myc-his that drives constitutive expression of Hop-Myc-His, with Ac5c-Gal4 that induces UAS transgenes in vitro (Klueg et al., 2002), and with UAS-3HA-Stat92EFL. We used 4 ng of Ac5c-hop-myc-his, which activated the reporter only slightly above the Ac5c-Gal4 control (Fig. 4A, Lanes 1,2). Expression of Stat92EFL alone did not activate the reporter above Hop-Myc-His control (Fig. 4A, Lane 3). By contrast, Stat92EFL expressed together with Hop-Myc-His resulted in a signficant increase in luciferase activity (Fig. 4A, Lane 4).
Figure 4. Transcriptional activation by and expression of Stat92E variants.
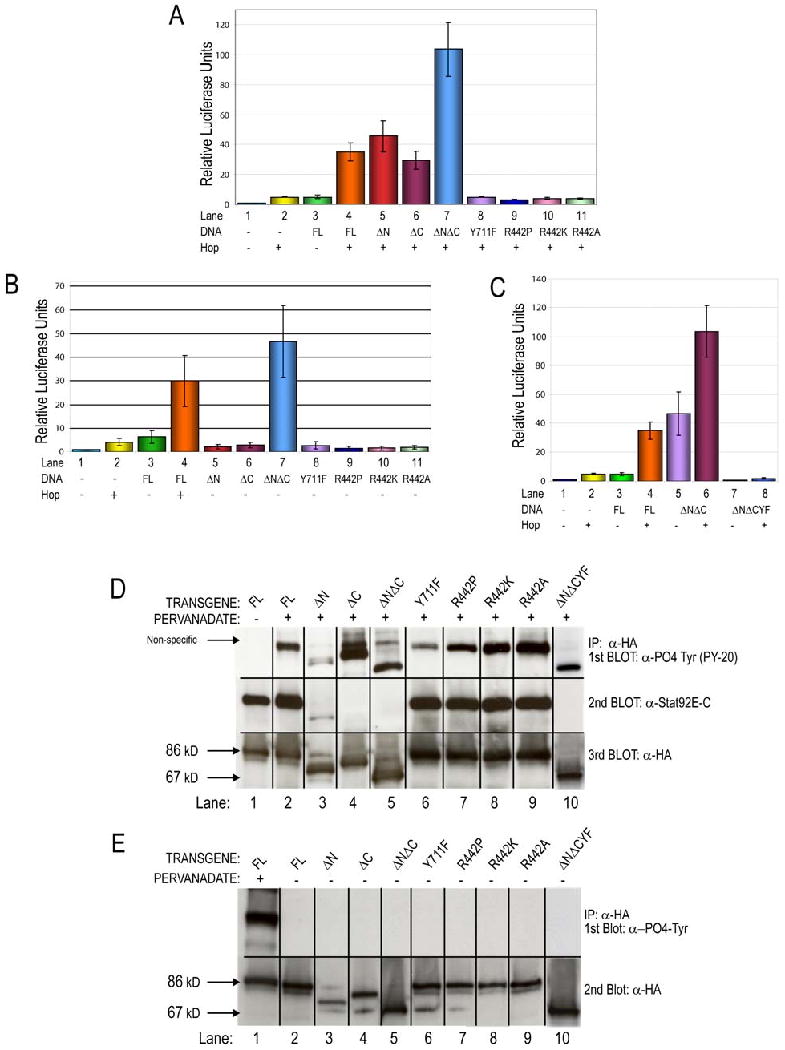
(A,B) S2 cells transiently transfected with the socs1-luc reporter, Ac5c-Gal4, and Ac5c-hop-myc-his as noted. (A) Ac5c-Gal4 alone, Ac5c-hop-myc-his alone and Stat92EFL alone do not significantly activate socs1-luc. In the presence of Hop, Stat92EFL, Stat92EΔN, Stat92EΔC, and Stat92EΔNΔC activate the reporter over baseline levels. In contrast, Stat92EY711F, Stat92ER442P, Stat92ER442K, and Stat92ER442A do not. (B) In the absence of Hop, only Stat92EΔNΔC activates socs1-luc, suggesting that it is a dominant-active mutation (lane 7). (C) The constitutive activity of Stat92EΔNΔC depends on phosphorylation of Y711. Mutation of Tyr711 to Phe (Stat92EΔNΔCY711F, abbreviated Stat92E NCYF) abolishes Stat92EΔNΔC activity either in the presence or absence of Hop (lanes 7 and 8). (D,E) Western blot analysis confirms predicted expression and activation of Stat92E variants in S2 cells which were transiently transfected and stimulated as indicated. (D) In the absence of pervanadate, Stat92EFL is expressed but not tyrosine phosphorylated (lane 1). In contrast, all Stat92E variants are phosphorylated in response to pervanadate treatment (lanes 2-10). Reprobing the blot with C-terminal Stat92E specific or HA specific antibodies reveals that all Stat92E variants are expressed at their predicted Mr. Stat92EΔC, Stat92EΔNΔC and Stat92ENCYF (lanes 4,5 and 10) are not detected by the Stat92E antibody because antigen recognized by this antibody. (E) None of the Stat92E variants are tyrosine phosphorylated in the absence of pervanadate (lanes 2-10). Activation of Stat92EFL by pervanadate (lane 1) serves as a positive control for tyrosine phosphorylation.
We assessed which residues of Stat92E were required for the activation of socs1-luc. Stat92EΔN and Stat92EΔC robustly activated the reporter to levels observed with Stat92EFL and only when expressed with Hop-Myc-His (Fig. 4A,B, Lanes 5,6). In contrast, Stat92EY711F could not activate the reporter socs1-luc when expressed alone or with Hop-Myc-His (Fig. 4A,B, Lanes 8). Western blotting demomstrated that all Stat92E constructs were expressed at similar levels and at the predicted Mr (Fig. 4D). We further showed that all constructs were tyrosine phosphorylated after treatment of the cells with pervanadate, which activates Stat92E in a ligand-independent manner (Sweitzer et al., 1995), but not in untreated cells (Figs. 4D,E). Stat92E contains thirteen tyrosine residues, including Y711, and presumably it is these other tyrosine residues that are phosphorylated after treatment with pervanadate (Hou et al., 1996; Yan et al., 1996).
Stat92EΔNΔC is a constitutive transcriptional co-activator and has dominant-active behavior in vivo
Strikingly, in the presence of Hop-Myc-His, Stat92E lacking both the N and C termini (Stat92EΔNΔC) induced luciferase activity significantly more robustly than Stat92EFL or than the single N- and C-terminal truncations, demonstrating that it has increased transcriptional activation capablities (Fig. 4A, Lane 7). Furthermore, Stat92EΔNΔC alone induced high levels of socs1-luc in the absence of Hop-Myc-His (Fig. 4A,B, Lanes 7). These data strongly suggest that the Stat92EΔNΔC protein is constitutively active. Importantly, ectopic expression of Stat92EΔNΔC with ey-Gal4 resulted in moderate overgrowth of eye tissue (Fig. 5C). Despite low penetrance of this phenotype (∼5%), the Stat92EΔNΔC-induced overgrowth is significant because this gain-of-function phenotype is never observed with ectopic expression of other Stat92E transgenes (Fig. 5A,B and Fig. S1).
Figure 5. Stat92EΔNΔC exhibits constitutive activity.
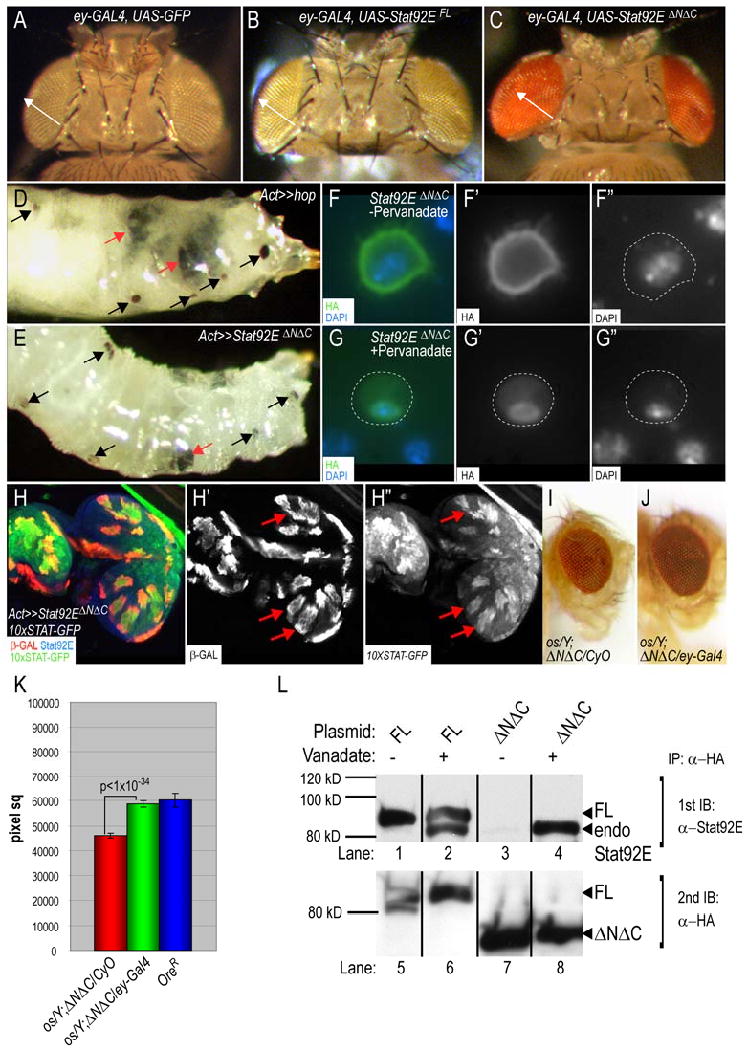
(A-C) Dorsal view of adult heads from animals in which ey-Gal4 (Hauck et al., 1999) drives expression of UAS-GFP (A), of UAS-Stat92EFL (B) or of UAS-Stat92EΔNΔC (C) during larval eye development. Arrow (white) denotes the distance from the medial posterior to the anterior lateral edge of the eye. This standard was used to demonstrate that ectopic expression of Stat92EΔNΔC (C) but not Stat92EFL (B) leads to eye overgrowth. (D,E) Third instar larvae with randomly induced clones expressing Hop (Act≫hop) (D) or Stat92EΔNΔC (Act≫Stat92EΔNΔC) have melanotic tumors (large tumors, red arrows; small tumors, black arrows in D,E). (F,G) Stat92EΔNΔC is not constitutively localized to the nucleus. S2 cells expressing Stat92EΔNΔC were unstimulated (F) or stimulated with pervanadate (G). HA (green), DAPI (blue). In the absence of stimulation, Stat92EΔNΔC is cytoplasmic (F), but moves into the nucleus upon activation (G). (H) A third instar eye disc harboring Stat92EΔNΔC-expressing-clones (Act≫Stat92EΔNΔC) and the 10×STAT-GFP reporter. Clones (anti-β GAL (red)), 10×STAT-GFP (green), anti-Stat92E-C (blue). Ectopic expression of Stat92EΔNΔC induces the expression of the Stat92E transcriptional reporter 10×STAT-GFP in a cell-autonomous manner (red arrows). (I-J) Stat92EΔNΔC acts downstream of upd. An os/Y; UAS-Stat92EΔNΔC/CyO male has small eyes (I). This phenotype was rescued in os/Y; UAS-Stat92EΔNΔC/ey-Gal4 flies, where Stat92EΔNΔC is specifically expressed in the eye disc (J). (K) Quantification of the eye area in os/Y; UAS-3HA-Stat92EΔNΔC/CyO (red bar), os/Y; UAS-3HA-Stat92EiNΔC/CyO (green bar) or wild type +/Y males (blue bar). 80=eyes in each os/Y genotype. n=20 in wild type. The difference between the values of the red and green values is statistically significant (p<10-34, Student's T test). (L) Stat92EFL and Stat92EΔNΔC dimerize with endogenous Stat92E. S2 cells were transiently transfected with the indicated plasmids and were unstimulated or treated with pervanadate. 3HA-Stat92E proteins were immunoprecicipated with an HA antibody and Western blotted first with Stat92E-C and subsequently HA antibodies. Endogenous Stat92E has a predicted Mr of 85 kDa (lanes 2,4), the 3HA-Stat92EFL protein 90 kDa (lane 1) and 3HA-Stat92EΔNΔC 67 kDa (lane 3). Endogenous Stat92E does not co-immunoprecipitate with either variant in the absence of pervanadate (lanes 1 and 3). However, after activation of the pathway, endogenous Stat92E present in anti-HA precipitates (lanes 2 and 4).
To determine whether Stat92EΔNΔC exhibits constitutive activity in other tissues, we examined its ability to induce melanotic tumors. Ectopic expression of UAS-hop in flip-out clones resulted in ligand-independent, autonomous activation of Stat92E and gave rise to melanotic tumors (Fig. 5D and (Ekas et al., 2006)). Similarly, ectopic expression of Stat92EΔNΔC in flip-out clones also caused tumors (Fig. 5E). Melanotic tumors were never seen in clones mis-expressing Stat92EFL (data not shown). Ectopic expression of Stat92EΔNΔC or Hop also induced cell-autonomous expression of an in vivo Stat92E transcriptional reporter (Fig. 5H, data not shown and (Bach et al., 2007)). By contrast, mis-expression of Stat92EFL did not activate this reporter (data not shown). We tested constitutive nuclear localization or constitutive tyrosine phosphorylation as possible mechanisms for the dominant active behavior of Stat92EΔNΔC. Stat92EΔNΔC is cytoplasmic in S2 cells in the absence of stimulation and translocates to the nucleus after activation, ruling out the former model (Fig. 5F,G). Stat92EΔNΔC was not tyrosine phosphorylated in untreated cells (Fig. 4E, Lane 5) but became tyrosine phosphorylated in response to pervanadate (Fig. 4D, Lane 5). These data suggest that the latter model is also not correct. However, Stat92EΔNΔC could contain levels of phosphotyrosine higher than endogenous Stat92E but below the level of detection of the PY-20 antibody used in this experiment
We determined that Stat92EΔNΔC acts epistatically to upd because its mis-expression specifically in the eye rescued the small-eye phenotype of outstretched (os), a viable upd allele that has small eyes and outstretched wings (Lindsley and Zimm, 1992). upd is an X-linked genes, and os/Y males or os/os females manifest these eye and wing phenotypes. We used ey-Gal4 (Hauck et al., 1999) to drive expression of Stat92EΔNΔC in os/Y males and compared the size of the eye in os/Y; Stat92EΔNΔC/CyO and os/Y; Stat92EΔNΔC/ey-Gal4 males (n=80 eyes for both genotypes). We found that os/Y; Stat92EΔNΔC/CyO flies had adult eyes that were identical in size to os/Y males, indicating that the presence of the transgene alone does not generate a phenotype (Fig. 5I and data not shown). In contrast, os/Y; Stat92EΔNΔC/ey-Gal4 males had eyes that were nearly identical to those in wild type +/Y males and were significantly larger than those in os/Y; Stat92EΔNΔC/CyO males, an observation that is statistically significant (p<10-34, Student's T-test) (Fig. 5J,K). Notably, ey>Stat92EΔNΔC did not rescue the outstretched wing phenotype of os/Y flies.
The dominant-active properties of Stat92EΔNΔC require Y711
Tyrosine phosphorylation on conserved C-terminal tyrosine residues is required for the function of mammalian STATs and for the constitutive activity of Cys mutants STAT3-C and STAT1-C (Li and Shaw, 2006; Liddle et al., 2006). To examine whether this was also the case for Stat92EΔNΔC, we generated a Stat92EΔNΔC transgene that has a Tyr711 to Phe substitution (Stat92EΔNΔCY711F). We found that either in the presence or in the absence of Hop, Stat92EΔNΔCY711F could not activate socs1-luc above background levels (Fig. 4C, Lanes 7,8). This is similar to the transcriptional co-activation abilities of a Stat92E that only contains a substitution of Tyr711 to Phe (Fig. 4A,B, Lanes 8). Therefore, the dominant-active abilities of Stat92EΔNΔC require phosphorylation of Tyr711 and by inference the formation of a phosphorylated, activated dimer.
Stat92EΔNΔC forms dimers with endogenous Stat92E following activation
To address whether the dominant-active behavior of Stat92EΔNΔC resulted from dimer formation with a functional Stat92E, we determined whether of Stat92EΔNΔC caused melanotic tumors in the absence of endogenous Stat92E. We generated positively-marked Stat92E85C9 clones that over-expressed Stat92EΔNΔC (see Materials and Methods). We were unable to find any melanotic tumors despite the examination of hundreds of larvae harboring these clones (data not shown). These data indicate that Stat92EΔNΔC causes tumors and overgrowths only in tissues that contain a functional Stat92E protein. This conclusion is supported by the observations that Stat92EΔNΔC never caused over-growths in Stat92E85C9 M+ or Stat92E397 M+ mutant eye discs.
To address if there is a physical interaction between endogenous Stat92E and Stat92EΔNΔC, we performed co-immunoprecipitation experiments in S2 cells. We found that in the absence of pervandate stimulation, endogenous Stat92E did not co-immunoprecipitate with either Stat92EFL or Stat92EΔNΔC (Fig. 5L, Lanes 1,3,5,7). In contrast, after pervandate stimulation, endogenous Stat92E was detected in immuno-precipitates of both Stat92EFL and Stat92EΔNΔC (Fig. 5L, Lanes 2,4,6,8). Taken together, these data indicate that endogenous Stat92E:Stat92EΔNΔC dimers are required for dominant-active behavior of Stat92EΔNΔC.
Arg442 is required for Stat92E-induced transcriptional activation
The Stat92E85C9 allele results from a substitution of Pro for Arg at residue 442 (Fig. 1B) (Silver and Montell, 2001). This Arg residue is conserved in the C. elegans STAT, STA-1, and in mammalian STATs 2, 3, 5, and 6 (Fig. 7C and (Wang and Levy, 2006b; Yan et al., 1996)). To determine the functional importance of Arg at position 442 in Stat92E, we generated these constructs: Stat92ER442P, which mimics the 85C9 mutation; Stat92ER442K, which contains a conservative substitution of Arg442 to Lys; and Stat92ER442A, which contains a non-conservative substitution of Arg442 to Ala. We found that Stat92ER442P cannot rescue the Stat92E small-eye phenotype, shift the 85C9 lethal phase or activate transcription (Fig. 3J,M and Fig. 4A, B, Lanes 9), which is consistent with Stat92E85C9 being a strong hypomorph. We reasoned that if a positive charge were required at position 442, then Stat92ER442K should be functional, while Stat92ER442A should not. However, we found that neither of these Stat92E variants could rescue the Stat92E small-eye phenotype, shift the lethal phase or induces socs1-luc transcription (Fig. 3K,L,M; Fig. 4A, B, Lanes 10 and 11). Moreover, all of three Arg mutants had dominant-negative effects in a Stat92E hypomorphic background. The embryonic lethality of 85C9 zygotic homozygotes rose to more than 70% with the expression of these constructs, and the R442P, R442K and R442A (as well as the Y711F) crosses had to be set up for one year to collect 20 adult females for quantification of area of the eye (Figs. 3S and Fig. S1). We ruled out the possibility that the inactivity of the R442 mutants was due to aberrant expression (Fig. 4D,E, Lanes 7-9 and Fig. S2H-J). We also ruled out the possibility that R442 was required for nuclear translocation. Stat92ER442P was cytosolic in the absence of stimulation, and translocated to the nucleus upon treatment with pervanadate, similar to endogenous Stat92E and Stat92EFL (Fig. 6A-D,G,H). By contrast, Stat92EY711F remained cytoplasmic in both the presence and absence of stimulation, as has been observed for mammalian STATs (Fig. 6E,F and (Levy and Darnell, 2002)). Lastly, the R442 mutant proteins behaved like Stat92EFL with regards to tyrosine phosphorylation: they contained undetectable levels of phospho-tyrosine in the absence of stimulation and high levels following pervanadate treatment (Fig. 4D,E, Lanes 7-9). We conclude that Arg442 is not required for Stat92E activation or for nuclear translocation but that it is indispensible for transcriptional co-activation by Stat92E.
Figure 7. Model of action of Stat92EΔNΔC and of Stat92ER442.
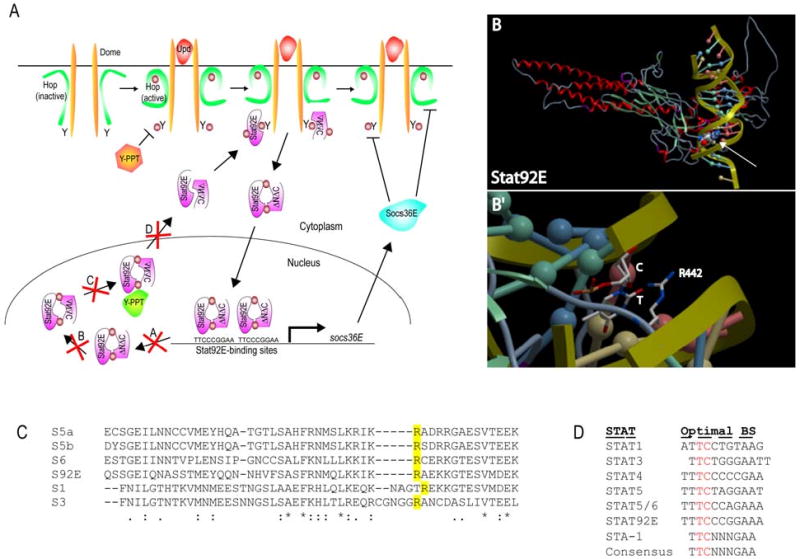
(A) Basal levels of tyrosine phosphorylation of Dome, Hop and/or Stat92E are kept in check by cellular tyrosine phosphatases (orange Y-PPT). Inactive Stat92EFL:Stat92EΔNΔC dimers exist at low levels in the cytoplasm of unstimulated cells and can bind to the basally-actiavted phosphorylated receptor via their intact SH2 domains. Once bound to the activated receptor, these Stat92E proteins are themselves phosphorylated, leading to the formation of activated Stat92EFL:Stat92EΔNΔC dimers that translocate to the nucleus, bind consensus Stat92E binding elements on DNA and modulate gene transcription. Possible molecular mechanism underlying the dominant active behavior of the Stat92EFL:Stat92EΔNΔC dimers. They could (A) stay bound longer to DNA that endogenous dimers; (B) be more resistant to conformational change to an unphosphorylated dimer; (C) be more resistant to dephosphorylation by a nuclear tyrosine phosphatase (green Y-PPT) and/or (D) resist export from the nucleus. (B) Homology model of Stat92E based on the crystallographic structure of a phosphorylated STAT1 dimer bound to DNA (Chen et al., 1998). Arg442 is displayed as spherical space-filled atoms and contacts DNA in the minor groove (white arrow). (B′) Zoom of (A). Arg442 is shown in stick figure form. Arg442 appears to be interacting with a thymine (T, shown in stick figure form) and with a cytosine (C, shown as a red ball) located in the minor groove of the DNA helix. (D) The optimal DNA binding sites for mammalian STATs 1-6, Stat92E and STA-1.
Figure 6. Stat92ER442P can translocate to the nucleus after activation.
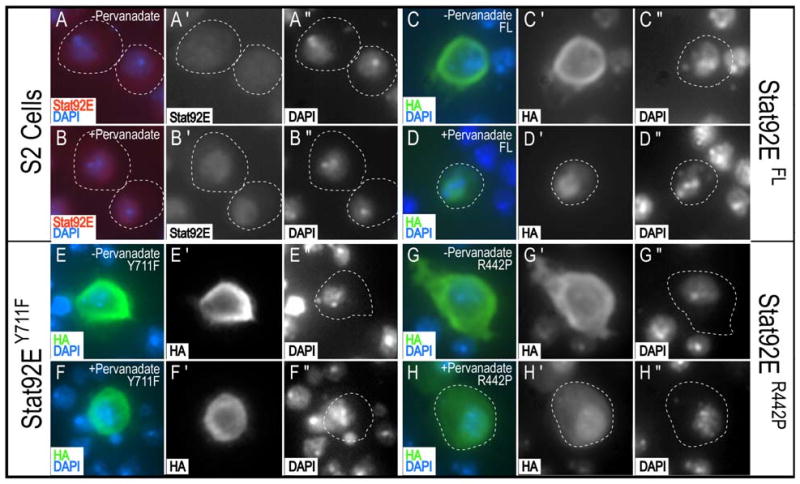
Untransfected S2 cells (A,B), or S2 cells transiently transfected with Stat92EFL (C,D), Stat92EY711F (E,F), or Stat92ER442P(G,H). Cells were unstimulated (A,C,E,G) or stimulated with pervanadate (B,D,F,H). Stat92E (red), HA (green), and DAPI (blue). In the absence of stimulation, endogenous Stat92E (A), Stat92EFL (C), Stat92EY711F (E), and Stat92ER442P (G) are cytoplasmic. Upon stimulation, endogenous Stat92E (B), Stat92EFL (D) and Stat92ER442P (H) translocate to the nucleus. Stat92EY711F (F) does not translocate to the nucleus upon stimulation.
Arg442 may directly contact the minor groove of the DNA helix
We reasoned that Arg442 may be important for maintaining a STAT-DNA interface. To test this hypothesis by observing where Arg442 is located on the 3D structure of Stat92E relative to bound DNA, we generated a homology model of Stat92E based on the crystallographic structure of a phosphorylated STAT1 dimer bound to DNA (Chen et al., 1998). This model revealed that Arg442 likely contacts DNA directly, presumably by accessing the minor groove of the DNA helix (Fig. 7B). The model comfortably positions the Arg442 side chain with 484 A2 surface area of contact with the DNA bases in the minor groove. Contact with the major groove or with other Stat92E binding partners is unlikely from this location as long as DNA is bound to Stat92E, since the loop on which Arg442 sits is nestled deeply in the minor groove and should not extend out of the minor groove even if it takes on a different conformation than that seen in the model. Interestingly, the location of Arg442 and the observation that Lys is not functional at this position in Stat92E suggest that only protein side chain:DNA interactions which depend exclusively on the Arg guanidinium group are operative here for Stat92E. The list of canonical candidate interactions of this type have been previously published (Luscombe et al., 2001), and only one is possible with the sequence of bases recognized by Stat92E, namely a complex, stacked base minor groove interaction with a cytosine and thymine. This complex would be lost with the Stat92ER422 mutation to Pro, which eliminates the side-chain guanidinium group. Thus, our model and the observed data suggests that Arg442 interacts with the T and C of the TTCnnnGAA that is present in the optimal binding sites of nearly all STAT proteins, including Stat92E and STA-1 (Fig. 7D and (Decker et al., 1997)). These data suggest that Arg442 contributes to Stat92E's ability to recognize its target DNA binding site.
Discussion
In this study we use in vivo and in vitro assays to perform a structure-function analysis of Stat92E. We find that neither the N- terminus (residues 1-133) nor the C-terminus (residues 725-761) is required for Stat92E function in vivo and in vitro. However, when both N- and C-termini are removed, the resulting protein Stat92EΔNΔC has dominant-active properties, including oncogenesis. Furthermore, we demonstrate that Stat92EΔNΔC with a Tyr to Phe substitution at residue 711 (Stat92EΔNΔCY711F) is non-functional. We also showed that both Stat92EY711F and Stat92ER442P are non-functional and manifested dominant-negative activities in vivo. The lack of function of the Stat92EY711F variant is likely due to an inability to form an activated dimer, as has been previously reported (Yan et al., 1996). Our data suggest that Stat92ER442P cannot function because it does not maintain normal interactions between activated Stat92E dimers and cognate DNA.
Our study is important for several reasons, perhaps foremost of which is that we are the first group to identify a dominant-active Stat92E. The simultaneous removal of the N-terminal 133 and the C-terminal 36 amino acids results in a truncated protein that has constitutive activity, which causes melanotic tumors when mis-expressed in wild type larvae and transactivates a Stat92E reporter genes in vitro and in vivo in the absence of stimulation. We show that the activity of Stat92EΔNΔC is dependent on the presence of Y711, as is that of Stat92EFL. These data suggest that Stat92EΔNΔC is activated in a manner similar to full length Stat92E (i.e., by reciprocal interactions between SH2 domains and phosphorylated Y711 residues on adjacent Stat92E protein). Consistent with this, we show that Stat92EΔNΔC forms a dimer with endogenous Stat92E (i.e., a Stat92E: Stat92EΔNΔC dimer) after pervanadate stimulation. Therefore, Stat92EΔNΔC appears to behave like wild type Stat92E that has lost a negative regulatory component. One possibility is that removal of the N- and C-termini prolongs the interaction between the Stat92E: Stat92EΔNΔC dimer and DNA, leading to increased gene activation. Another is that Stat92E:Stat92EΔNΔC dimers cannot be efficiently dephosphorylated and/or return to a parallel dimer conformation, which may be required for normal export of Stat92E dimers to the cytoplasm (Chen et al., 2003). Constitutive activity is not observed by the removal of either the N or C-terminus domain individually, and therefore, deletion of the both domains must contribute to this activity. The constitutive activity of Stat92EΔNΔC in vitro is significantly robust that it could be used for RNAi screens to identify enhancers and suppressors of Stat92E activity, which may have conserved functions in restricting tumorigenesis in mammals. In addition, the high rate of melanotic tumor formation observed in clones mis-expressing Stat92EΔNΔC will make this mutation an extremely useful in vivo reagent as well.
One unresolved issue is how the constitutive activity of Stat92EΔNΔC is initiated in the first place. The model described above predicts that in the absence of stimulation (1) Stat92E:Stat92EΔNΔC dimers should be detected, if only at very low levels, and/or (2) Stat92EΔNΔC should be tyrosine phosphorylated, again if only at very low levels. Unexpectedly neither result is observed. To rectify these results, we invoke a model in which a few activated STAT dimers (in this case Stat92E: Stat92EΔNΔC dimers) are constantly generated within a cell, and their activity is quenched by tyrosine phosphatases (Fig. 7A). Although we have not been able to detect these dimers, we know that this is a plausible model because treatment of a cell with vanadate, a pan-phosphatase inhibitor, activates STATs in a ligand-independent manner in vivo and in vitro, presumably by inhibiting the enzymes that keep their low level of activation in check (Duff et al., 1997; Sweitzer et al., 1995; Tourkine et al., 1995). We hypothesize that Stat92E:Stat92EΔNΔC dimers may better evade the actions of tyrosine phosphatases, resulting in a steady state of increased STAT signaling in a cell and ultimately leading sustained increased expression of STAT targets and, in certain cell types like the blood, oncogenesis.
As mentioned above, we also discovered that Stat92E lacking either the N- or the C-terminus functioned similarly to Stat92EFL. They rescued the eye/antennal/head phenotypes, as well as the hatching rate, and they shifted the lethality in either 85C9/85C9 or 397/397 zygotic mutants. The fact that a transgene encoding a Stat92E lacking the last 36 amino acids could rescue the phenotypes associated with homozygosity for the 397 allele, which is predicted to encode a protein lacking all amino acids after Trp594, suggest that the transactivation domain of Stat92E does not residue at the C-terminus, since a Stat92EΔC: Stat92E397 dimer would lack any residue after 724. Alternatively, there could be more than one transactivation domain in Stat92E, one at the C-terminus and the other located elsewhere in the protein. The proteins encoded by the Stat92E85C9 and Stat92E397 alleles are predicted to have N-terminal domains (this work and (Silver and Montell, 2001)). Previous work has shown that two N-termini are required for the formation of non-phosphorylated dimers, which are the preferred substrated of receptor/JAK complexes and are required for cytokine-dependent activation of STAT4 (Chang et al., 2003; Chen et al., 2003; Murphy et al., 2000; Ota et al., 2004; Shuai et al., 1996). As such, the single N-terminus present in any potential dimers between Stat92EΔN and Stat92E85C9 or between Stat92EΔN and Stat92E397 is unlikely to support dimer formation/activation in Stat92E85C9 and Stat92E397 homozygous mutant cells, respectively.
Our results showing that Stat92EΔN can function like Stat92EFL differ from those reported by two other groups (Henriksen et al., 2002; Karsten et al., 2006). The former authors reported that a Stat92E lacking the first 133 residues was generated by transcription of an alternative promoter in response to pathway activation and acted as dominant negative in vivo. Mis-expression of their Stat92EΔN construct in wild type embryos led to the same phenotype as complete loss of Stat92E: the loss of expression of even-skipped stripes 3, 5 and 7 (Henriksen et al., 2002; Hou et al., 1996). The latter group reported that mis-expression of a Stat92EΔN protein tagged at the C-terminal by GFP (Stat92EΔN–GFP) in the embryo abrogated expression of trachealess, which is also lost in embryos null for dome gene (Brown et al., 2001; Chen et al., 2002; Karsten et al., 2006). The reason for the discrepacy between our and their results is not clear at present.
Our study is also noteworthy in that it is the first analysis of Stat92E structure-function variants that employs in vivo rescue assays in a Stat92E homozygous mutant background. We previously reported that 90% of animals carrying large patches of Stat92E homozygous mutant tissue in the eye-antennal disc fail to hatch but that these phenotypes were rescued when these animals expressed a full-length version of Stat92E in the eye-antennal disc (this study and (Ekas et al., 2006)). We now show that mis-expression of Stat92EFL in 85C9/85C9 zygotic mutants significantly shifted the lethal phase, from embryonic/first larval instar to third larval instar/early pupa. Currently we do not understand why the UASp-3HA-Stat92EFL transgene did not rescue to 85C9/85C9 homozygotes to adulthood, but one possibility is low expression. Transgenes driven by UASp, which are designed for expression in the germ-line (Rorth, 1996), are expressed at significantly lower levels of than those driven by UAST, which are expressed in the soma (Brand and Perrimon, 1993), and the levels of Stat92E required to rescue to 85C9 or 397 zygotic mutants to adulthood simply might not have been achieved.
We are the first to demonstrate that Tyr711 is required for Stat92E function in vivo, which is consistent with in vitro data that a substitution of Tyr711 to Phe abolished the ability of Stat92E to bind DNA (Karsten et al., 2006; Yan et al., 1996). However, we do not observe a reduction in eye size when we over-express this Stat92E construct in a wild type fly eye. We believe that this is due to our inability to over-express this construct at high enough levels to inhibit the function of endogenous Stat92E. Finally, we have also shown that an Arg residue is specifically required at position 442 for Stat92E function. Our homology model of Stat92E suggests that Arg442 is involved in DNA binding and may be a recognition element for the binucleotide TTCnnnGAA in the Stat92E binding site, suggesting that this interaction may be important for Stat92E recognition of its DNA target site. The Arg442 of Stat92E is conserved in the C. elegans STAT, STA-1, and in all mammalian STAT2,3,5a,5b and 6. Therefore, it will be interesting in the future to determine if this residue is also required for the function of STAT proteins in which this Arg residue is conserved.
Supplementary Material
Acknowledgments
We are indebted to Kent Nybakken for providing the C5HA3 vector prior to publication (Nybakken et al., 2005), to Uwe Vinkemeier for several key reagents, to Steve Hubbard for help with the Arg442 mutations and to Tempei Ikegame for making socs1-luc. We thank Ram Dasgupta, Jessica Treisman, Norbert Perrimon and the Bloomington Stock Center for fly stocks and reagents. Several mAbs used in this study were obtained from the DSHB, Department of Biological Sciences, Iowa City, IA 52242. We thank David Levy, Mark Chong, Lawrence Gardner and Yaming Wang and Ines Carrera, and members of the Bach and Dasgupta labs for helpful comments and guidance.
This project was support in part by a Basil O'Connor Starter Scholar Research Award (Grant No.: 5-FY06) from the March of Dimes Foundation to E.A.B; by an NIH institutional training grant (T32 GM066704-03) to L.A.E.; by a Research Scholar Grant (RSG-DDD-115829) from the American Cancer Society to E.A.B.; and a National Institutes of Health grant from NIGMS (1R01GM085075) to E.A.B.
Footnotes
The authors state no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agaisse H, Petersen UM, Boutros M, Mathey-Prevot B, Perrimon N. Signaling Role of Hemocytes in Drosophila JAK/STAT-Dependent Response to Septic Injury. Dev Cell. 2003;5:441–450. doi: 10.1016/s1534-5807(03)00244-2. [DOI] [PubMed] [Google Scholar]
- Arbouzova NI, Zeidler MP. JAK/STAT signalling in Drosophila: insights into conserved regulatory and cellular functions. Development. 2006;133:2605–2616. doi: 10.1242/dev.02411. [DOI] [PubMed] [Google Scholar]
- Ayala-Camargo A, Ekas LA, Flaherty MS, Baeg GH, Bach EA. The JAK/STAT pathway regulates proximo-distal patterning in Drosophila. Dev Dyn. 2007;236:2721–2730. doi: 10.1002/dvdy.21230. [DOI] [PubMed] [Google Scholar]
- Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, Perrimon N, Baeg GH. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr Patterns. 2007;7:323–331. doi: 10.1016/j.modgep.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Bach EA, Tanner JW, Marsters S, Ashkenazi A, Aguet M, Shaw AS, Schreiber RD. Ligand-induced assembly and activation of the gamma interferon receptor in intact cells. Mol Cell Biol. 1996;16:3214–3221. doi: 10.1128/mcb.16.6.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Vincent S, Zeidler MP, Perrimon N. A sensitized genetic screen to identify novel regulators and components of the Drosophila janus kinase/signal transducer and activator of transcription pathway. Genetics. 2003;165:1149–1166. doi: 10.1093/genetics/165.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeg GH, Zhou R, Perrimon N. Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 2005;19:1861–1870. doi: 10.1101/gad.1320705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- Binari R, Perrimon N. Stripe-specific regulation of pair-rule genes by hopscotch, a putative Jak family tyrosine kinase in Drosophila. Genes Dev. 1994;8:300–312. doi: 10.1101/gad.8.3.300. [DOI] [PubMed] [Google Scholar]
- Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–1279. [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem. 2003;278:34133–34140. doi: 10.1074/jbc.M304531200. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Brown S, Hu N, Hombria JC. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr Biol. 2001;11:1700–1705. doi: 10.1016/s0960-9822(01)00524-3. [DOI] [PubMed] [Google Scholar]
- Brown S, Hu N, Hombria JC. Novel level of signalling control in the JAK/STAT pathway revealed by in situ visualisation of protein-protein interaction during Drosophila development. Development. 2003;130:3077–3084. doi: 10.1242/dev.00535. [DOI] [PubMed] [Google Scholar]
- Callus BA, Mathey-Prevot B. SOCS36E, a novel Drosophila SOCS protein, suppresses JAK/STAT and EGF-R signalling in the imaginal wing disc. Oncogene. 2002;21:4812–4821. doi: 10.1038/sj.onc.1205618. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins. 1995;23:403–414. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
- Chang HC, Zhang S, Oldham I, Naeger L, Hoey T, Kaplan MH. STAT4 requires the N-terminal domain for efficient phosphorylation. J Biol Chem. 2003;278:32471–32477. doi: 10.1074/jbc.M302776200. [DOI] [PubMed] [Google Scholar]
- Chao JL, Tsai YC, Chiu SJ, Sun YH. Localized Notch signal acts through eyg and upd to promote global growth in Drosophila eye. Development. 2004;131:3839–3847. doi: 10.1242/dev.01258. [DOI] [PubMed] [Google Scholar]
- Chen HW, Chen X, Oh SW, Marinissen MJ, Gutkind JS, Hou SX. mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes Dev. 2002;16:388–398. doi: 10.1101/gad.955202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bhandari R, Vinkemeier U, Van Den Akker F, Darnell JE, Jr, Kuriyan J. A reinterpretation of the dimerization interface of the N-terminal domains of STATs. Protein Sci. 2003;12:361–365. doi: 10.1110/ps.0218903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, Karras JG, Levy DE, Inghirami G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11:623–629. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005;11:595–596. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Kaykas A, Moon RT, Perrimon N. Functional genomic analysis of the Wnt-wingless signaling pathway. Science. 2005;308:826–833. doi: 10.1126/science.1109374. [DOI] [PubMed] [Google Scholar]
- Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res. 1997;17:121–134. doi: 10.1089/jir.1997.17.121. [DOI] [PubMed] [Google Scholar]
- Dominguez M, Casares F. Organ specification-growth control connection: new in-sights from the Drosophila eye-antennal disc. Dev Dyn. 2005;232:673–684. doi: 10.1002/dvdy.20311. [DOI] [PubMed] [Google Scholar]
- Duff JL, Quinlan KL, Paxton LL, Naik SM, Caughman SW. Pervanadate mimics IFNgamma-mediated induction of ICAM-1 expression via activation of STAT proteins. J Invest Dermatol. 1997;108:295–301. doi: 10.1111/1523-1747.ep12286465. [DOI] [PubMed] [Google Scholar]
- Ekas LA, Baeg GH, Flaherty MS, Ayala-Camargo A, Bach EA. JAK/STAT signaling promotes regional specification by negatively regulating wingless expression in Drosophila. Development. 2006;133:4721–4729. doi: 10.1242/dev.02675. [DOI] [PubMed] [Google Scholar]
- Flaherty MS, Salis P, Evans CJ, Ekas LA, Marouf A, Zavadil J, Banerjee U, Bach EA. chinmo is a functional effector of the JAK/STAT pathway that regulates eye development, tumor formation and stem cell self-renewal in Drosophila. Developmental Cell. doi: 10.1016/j.devcel.2010.02.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty MS, Zavadil J, Ekas LA, Bach EA. Genome-wide expression profiling in the Drosophila eye reveals unexpected repression of notch signaling by the JAK/STAT pathway. Dev Dyn. 2009;238:2235–2253. doi: 10.1002/dvdy.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MM, Weaver BK, Gergen JP, Reich NC. A novel functional activator of the Drosophila JAK/STAT pathway, unpaired2, is revealed by an in vivo reporter of pathway activation. Mech Dev. 2005;122:939–948. doi: 10.1016/j.mod.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Hanratty WP, Dearolf CR. The Drosophila Tumorous-lethal hematopoietic oncogene is a dominant mutation in the hopscotch locus. Mol Gen Genet. 1993;238:33–37. doi: 10.1007/BF00279527. [DOI] [PubMed] [Google Scholar]
- Harrison DA, Binari R, Nahreini TS, Gilman M, Perrimon N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. Embo J. 1995;14:2857–2865. doi: 10.1002/j.1460-2075.1995.tb07285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DA, McCoon PE, Binari R, Gilman M, Perrimon N. Drosophila unpaired encodes a secreted protein that activates the JAK signaling pathway. Genes Dev. 1998;12:3252–3263. doi: 10.1101/gad.12.20.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck B, Gehring WJ, Walldorf U. Functional analysis of an eye specific enhancer of the eyeless gene in Drosophila. Proc Natl Acad Sci U S A. 1999;96:564–569. doi: 10.1073/pnas.96.2.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen MA, Betz A, Fuccillo MV, Darnell JE., Jr Negative regulation of STAT92E by an N-terminally truncated STAT protein derived from an alternative promoter site. Genes Dev. 2002;16:2379–2389. doi: 10.1101/gad.1020702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Chen Z, Scherr H, Lackey M, Bolduc C, Bergmann A. vps25 mosaics display non-autonomous cell survival and overgrowth, and autonomous apoptosis. Development. 2006;133:1871–1880. doi: 10.1242/dev.02356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombria JC, Brown S, Hader S, Zeidler MP. Characterisation of Upd2, a Drosophila JAK/STAT pathway ligand. Dev Biol. 2005;288:420–433. doi: 10.1016/j.ydbio.2005.09.040. [DOI] [PubMed] [Google Scholar]
- Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci. 2000;25:496–502. doi: 10.1016/s0968-0004(00)01624-8. [DOI] [PubMed] [Google Scholar]
- Hou XS, Melnick MB, Perrimon N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell. 1996;84:411–419. doi: 10.1016/s0092-8674(00)81286-6. [DOI] [PubMed] [Google Scholar]
- Issigonis M, Tulina N, de Cuevas M, Brawley C, Sandler L, Matunis E. JAK-STAT signal inhibition regulates competition in the Drosophila testis stem cell niche. Science. 2009;326:153–156. doi: 10.1126/science.1176817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Awano W, Suzuki K, Hiromi Y, Yamamoto D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development. 1997;124:761–771. doi: 10.1242/dev.124.4.761. [DOI] [PubMed] [Google Scholar]
- James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- Janody F, Lee JD, Jahren N, Hazelett DJ, Benlali A, Miura GI, Draskovic I, Treisman JE. A mosaic genetic screen reveals distinct roles for trithorax and polycomb group genes in Drosophila eye development. Genetics. 2004;166:187–200. doi: 10.1534/genetics.166.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL, Cario H, Pahl HL, Collins A, Reiter A, Grand F, Cross NC. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41:446–449. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juni N, Awasaki T, Yoshida K, Hori SH. The Om (1E) mutation in Drosophila ananassae causes compound eye overgrowth due to tom retrotransposon-driven overexpression of a novel gene. Genetics. 1996;143:1257–1270. doi: 10.1093/genetics/143.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsten P, Hader S, Zeidler MP. Cloning and expression of Drosophila SOCS36E and its potential regulation by the JAK/STAT pathway. Mech Dev. 2002;117:343–346. doi: 10.1016/s0925-4773(02)00216-2. [DOI] [PubMed] [Google Scholar]
- Karsten P, Plischke I, Perrimon N, Zeidler MP. Mutational analysis reveals separable DNA binding and trans-activation of Drosophila STAT92E. Cell Signal. 2006;18:819–829. doi: 10.1016/j.cellsig.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, Bass A, Marubayashi S, Heguy A, Garcia-Manero G, Kantarjian H, Offit K, Stone RM, Gilliland DG, Klein RJ, Levine RL. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41:455–459. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klueg KM, Alvarado D, Muskavitch MA, Duffy JB. Creation of a GAL4/UAS-coupled inducible gene expression system for use in Drosophila cultured cell lines. Genesis. 2002;34:119–122. doi: 10.1002/gene.10148. [DOI] [PubMed] [Google Scholar]
- Kretzschmar AK, Dinger MC, Henze C, Brocke-Heidrich K, Horn F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J. 2004;377:289–297. doi: 10.1042/BJ20030708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- Lambertsson A. The minute genes in Drosophila and their molecular functions. Adv Genet. 1998;38:69–134. doi: 10.1016/s0065-2660(08)60142-x. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D'Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Li L, Shaw PE. Elevated activity of STAT3C due to higher DNA binding affinity of phosphotyrosine dimer rather than covalent dimer formation. J Biol Chem. 2006;281:33172–33181. doi: 10.1074/jbc.M606940200. [DOI] [PubMed] [Google Scholar]
- Liddle FJ, Alvarez JV, Poli V, Frank DA. Tyrosine phosphorylation is required for functional activation of disulfide-containing constitutively active STAT mutants. Biochemistry. 2006;45:5599–5605. doi: 10.1021/bi0525674. [DOI] [PubMed] [Google Scholar]
- Lindsley DL, Zimm GG. The Genome of Drosophila melanogaster 1992 [Google Scholar]
- Luo H, Hanratty WP, Dearolf CR. An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. Embo J. 1995;14:1412–1420. doi: 10.1002/j.1460-2075.1995.tb07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JE, Jr, Chen X. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell. 2005;17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Mertens C, Zhong M, Krishnaraj R, Zou W, Chen X, Darnell JE., Jr Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. 2006;20:3372–3381. doi: 10.1101/gad.1485406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev Cell. 2005;9:699–710. doi: 10.1016/j.devcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–221. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- Mukherjee T, Hombria JC, Zeidler MP. Opposing roles for Drosophila JAK/STAT signalling during cellular proliferation. Oncogene. 2005;24:2503–2511. doi: 10.1038/sj.onc.1208487. [DOI] [PubMed] [Google Scholar]
- Murphy TL, Geissal ED, Farrar JD, Murphy KM. Role of the Stat4 N domain in receptor proximal tyrosine phosphorylation. Mol Cell Biol. 2000;20:7121–7131. doi: 10.1128/mcb.20.19.7121-7131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neculai D, Neculai AM, Verrier S, Straub K, Klumpp K, Pfitzner E, Becker S. Structure of the unphosphorylated STAT5a dimer. J Biol Chem. 2005;280:40782–40787. doi: 10.1074/jbc.M507682200. [DOI] [PubMed] [Google Scholar]
- Novak U, Ji H, Kanagasundaram V, Simpson R, Paradiso L. STAT3 forms stable homodimers in the presence of divalent cations prior to activation. Biochem Biophys Res Commun. 1998;247:558–563. doi: 10.1006/bbrc.1998.8829. [DOI] [PubMed] [Google Scholar]
- Nybakken K, Vokes SA, Lin TY, McMahon AP, Perrimon N. A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signaling pathway. Nat Genet. 2005;37:1323–1332. doi: 10.1038/ng1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcaydu D, Harutyunyan A, Jager R, Berg T, Gisslinger B, Pabinger I, Gisslinger H, Kralovics R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41:450–454. doi: 10.1038/ng.341. [DOI] [PubMed] [Google Scholar]
- Ota N, Brett TJ, Murphy TL, Fremont DH, Murphy KM. N-domain-dependent nonphosphorylated STAT4 dimers required for cytokine-driven activation. Nat Immunol. 2004;5:208–215. doi: 10.1038/ni1032. [DOI] [PubMed] [Google Scholar]
- Rawlings JS, Rennebeck G, Harrison SM, Xi R, Harrison DA. Two Drosophila suppressors of cytokine signaling (SOCS) differentially regulate JAK and EGFR pathway activities. BMC Cell Biol. 2004;5:38. doi: 10.1186/1471-2121-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–612. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- Reynolds-Kenneally J, Mlodzik M. Notch signaling controls proliferation through cell-autonomous and non-autonomous mechanisms in the Drosophila eye. Dev Biol. 2005;285:38–48. doi: 10.1016/j.ydbio.2005.05.038. [DOI] [PubMed] [Google Scholar]
- Rorth P. A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc Natl Acad Sci U S A. 1996;93:12418–12422. doi: 10.1073/pnas.93.22.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kroeger KM, Volk HD, Eidne KA, Grutz G. Preassociation of nonactivated STAT3 molecules demonstrated in living cells using bioluminescence resonance energy transfer: a new model of STAT activation? J Leukoc Biol. 2004;75:792–797. doi: 10.1189/jlb.1003496. [DOI] [PubMed] [Google Scholar]
- Sefton L, Timmer JR, Zhang Y, Beranger F, Cline TW. An extracellular activator of the Drosophila JAK/STAT pathway is a sex-determination signal element. Nature. 2000;405:970–973. doi: 10.1038/35016119. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liao J, Song MM. Enhancement of antiproliferative activity of gamma interferon by the specific inhibition of tyrosine dephosphorylation of Stat1. Mol Cell Biol. 1996;16:4932–4941. doi: 10.1128/mcb.16.9.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DL, Montell DJ. Paracrine signaling through the JAK/STAT pathway activates invasive behavior of ovarian epithelial cells in Drosophila. Cell. 2001;107:831–841. doi: 10.1016/s0092-8674(01)00607-9. [DOI] [PubMed] [Google Scholar]
- Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM. The Berkeley Drosophila Genome Project gene disruption project: Single P-element insertions mutating 25% of vital Drosophila genes. Genetics. 1999;153:135–177. doi: 10.1093/genetics/153.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancato LF, David M, Carter-Su C, Larner AC, Pratt WB. Preassociation of STAT1 with STAT2 and STAT3 in separate signalling complexes prior to cytokine stimulation. J Biol Chem. 1996;271:4134–4137. doi: 10.1074/jbc.271.8.4134. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1639. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D, Sebti SM. Cucurbitacin Q: a selective STAT3 activation inhibitor with potent antitumor activity. Oncogene. 2005;24:3236–3245. doi: 10.1038/sj.onc.1208470. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Calvo S, Kraus MH, Finbloom DS, Larner AC. Characterization of a Stat-like DNA binding activity in Drosophila melanogaster. J Biol Chem. 1995;270:16510–16513. doi: 10.1074/jbc.270.28.16510. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev Cell. 2005;9:711–720. doi: 10.1016/j.devcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Tourkine N, Schindler C, Larose M, Houdebine LM. Activation of STAT factors by prolactin, interferon-gamma, growth hormones, and a tyrosine phosphatase inhibitor in rabbit primary mammary epithelial cells. J Biol Chem. 1995;270:20952–20961. doi: 10.1074/jbc.270.36.20952. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Y JG, C PH, Posakony JW, Barolo S, Kim J, Henry Sun Y. Upd/Jak/STAT signaling represses wg transcription to allow initiation of morphogenetic furrow in Drosophila eye development. Dev Biol. 2007;306:760–771. doi: 10.1016/j.ydbio.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Sun YH. Long-range effect of upd, a ligand for Jak/STAT pathway, on cell cycle in Drosophila eye development. Genesis. 2004;39:141–153. doi: 10.1002/gene.20035. [DOI] [PubMed] [Google Scholar]
- Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell. 2005;9:687–698. doi: 10.1016/j.devcel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Vinkemeier U, Moarefi I, Darnell JE, Jr, Kuriyan J. Structure of the amino-terminal protein interaction domain of STAT-4. Science. 1998;279:1048–1052. doi: 10.1126/science.279.5353.1048. [DOI] [PubMed] [Google Scholar]
- Wang Y, Levy DE. C. elegans STAT cooperates with DAF-7/TGF-beta signaling to repress dauer formation. Curr Biol. 2006a;16:89–94. doi: 10.1016/j.cub.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Wang Y, Levy DE. C. elegans STAT: evolution of a regulatory switch. Faseb J. 2006b;20:1641–1652. doi: 10.1096/fj.06-6051com. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Xu X, Sun YL, Hoey T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. Science. 1996;273:794–797. doi: 10.1126/science.273.5276.794. [DOI] [PubMed] [Google Scholar]
- Yan R, Small S, Desplan C, Dearolf CR, Darnell JE., Jr Identification of a Stat gene that functions in Drosophila development. Cell. 1996;84:421–430. doi: 10.1016/s0092-8674(00)81287-8. [DOI] [PubMed] [Google Scholar]
- Yang E, Henriksen MA, Schaefer O, Zakharova N, Darnell JE., Jr Dissociation time from DNA determines transcriptional function in a STAT1 linker mutant. J Biol Chem. 2002;277:13455–13462. doi: 10.1074/jbc.M112038200. [DOI] [PubMed] [Google Scholar]
- Zeidler MP, Bach EA, Perrimon N. The roles of the Drosophila JAK/STAT pathway. Oncogene. 2000;19:2598–2606. doi: 10.1038/sj.onc.1203482. [DOI] [PubMed] [Google Scholar]
- Zeidler MP, Perrimon N, Strutt DI. Polarity determination in the Drosophila eye: a novel role for unpaired and JAK/STAT signaling. Genes Dev. 1999;13:1342–1353. doi: 10.1101/gad.13.10.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler MP, Tan C, Bellaiche Y, Cherry S, Hader S, Gayko U, Perrimon N. Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat Biotechnol. 2004;22:871–876. doi: 10.1038/nbt979. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wrzeszczynska MH, Horvath CM, Darnell JE., Jr Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol Cell Biol. 1999;19:7138–7146. doi: 10.1128/mcb.19.10.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Henriksen MA, Takeuchi K, Schaefer O, Liu B, ten Hoeve J, Ren Z, Mao X, Chen X, Shuai K, Darnell JE., Jr Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc Natl Acad Sci U S A. 2005;102:3966–3971. doi: 10.1073/pnas.0501063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.