

Abstract
Human hepatocellular carcinoma (HCC) is a heterogeneous disease of distinct clinical subgroups. A principal source of tumor heterogeneity may be cell type of origin which in liver includes hepatocyte and/or adult stem/progenitor cells. To address this issue, we investigated the molecular mechanisms underlying the fate of the enzyme-altered preneoplastic lesions in the resistant hepatocyte (RH) model. Sixty samples classified as focal lesions, adenoma, early and advanced HCCs were micro-dissected after morphological and immunohistochemical evaluation and subjected to global gene expression profiling. The analysis of progression of the persistent GSTP+ focal lesions to fully developed HCC showed that about 50% of persistent nodules and all HCCs expressed CK19 whereas 14% of remodeling nodules were CK19+. Unsupervised hierarchical clustering of the expression profiles also grouped the samples according to CK19 expression. Further, supervised analysis using the differentially expressed genes in each cluster combined with the gene connectivity tools identified 1308 unique genes and a predominance of the AP-1/JUN network in the CK19+ lesions. In contrast, the CK19-negative cluster exhibited only limited molecular changes (156 differentially expressed genes vs. normal liver) consistent with remodeling towards differentiated phenotype. Finally, comparative functional genomics revealed a stringent clustering of CK19+ early lesions and advanced HCCs with human HCCs characterized by poor prognosis. Furthermore, the CK19 associated gene expression signature accurately predicted the patient survival (P<0.009) and tumor recurrence (P<0.006).
Conclusion
Our data establish CK19 as a prognostic marker of early neoplastic lesions and strongly suggest the progenitor derivation of HCC in the rat RH model. The capacity of CK19 associated gene signature to stratify HCC patients according to clinical prognosis indicates the usefulness of the RH model for studies of stem/progenitor-derived HCC.
It is well recognized that hepatocellular carcinoma (HCC) is a serious global health problem (1–3). Although HCC frequency is highest in Asia and sub-Saharan Africa, the incidence and mortality rates are increasing in the United States in recent years and are anticipated to double over the next decade(4). Despite the current improvements in treatment(5) and diagnostics, only 30–40% of patients with HCC are eligible for curative therapy (6).
Insights into the molecular pathogenesis of HCC have revealed a substantial heterogeneity of the malignancy (7). In most instances, HCC develops in a liver compromised by chronic hepatitis and/or cirrhosis(8). There is extensive evidence that under these conditions of tissue injury the normally quiescent adult liver stem cells are activated (9) and thus become a potential target cell population in liver cancer(10–12). This notion is supported by the data demonstrating a progressive upregulation of hepatic progenitor cell (HPC) markers in cirrhosis(13) as well as in dysplastic nodules in human liver(14) and adenoma(15). In rodents, hepatic stem/progenitor cell origin of HCC has been also postulated(16, 17).
In the adult liver, hepatocytes express CK8 and 18 whereas biliary epithelial cells express CK7 and 19 in addition to CK8 and 18. Abnormal expression of CK19 in the hepatic parenchyma has been attributed to remodeling of cirrhotic nodules and HPC proliferation(18). We have recently identified a subclass of human HCC which is enriched for the genes expressed in fetal hepatoblasts(19), including the progenitor cell markers, CK7 and 19. The CK19+ HCC subtype was characterized by the worst clinical prognosis among all HCC subclasses, suggesting that CK19 may be a potential prognostic marker for HCC(19, 20).
In an attempt to establish an experimental system to examine the potential contribution of HPC to liver cancer, we have employed the well characterized rat model of resistant hepatocytes (RH)(21). In this model, tumors are initiated by a single dose of a chemical carcinogen (e.g. diethylnitrosamine, DENA) and promoted by a brief treatment with 2-acetylaminofluorene (AAF) combined with partial hepatectomy (PH)(21). In the past, a modification of the RH model (i.e. omission of DENA initiation that eliminates cancer formation) has been extensively used in studies of activation, expansion and differentiation of adult hepatic stem cells(22). The advantage of the RH model is that the expansion of both DENA-initiated cell populations and adult stem cells can be examined at the same time during AAF+PH driven promotion. Furthermore, both cell populations share a number of the same early markers (e.g. GGT, GSTP and CK19). We therefore hypothesized that these markers would be lost as the progeny of the adult liver stem cells differentiates towards hepatocytes but retained in the preneoplastic lesions progressing to liver cancer. This hypothesis is supported in part by the fact that very few of the GSTP+ early preneoplastic lesions progress to HCC in the RH model(23).
To investigate the molecular changes underlying progression of preneoplastic lesions to HCC, we performed a comprehensive genomic analysis of laser-capture micro-dissected early persistent GSTP+ lesions as well as fully developed HCC. Gene expression profiling revealed two distinct gene clusters that significantly differentiated early lesions and advanced carcinomas based on the CK19 expression. Further analysis showed extensive molecular changes in the CK19+/GSTP+ lesions including a significant enrichment in the functional gene networks driven by AP-1/JUN consistent with their progression towards HCC. In contrast, the CK19−/GSTP+ lesions displayed only limited changes in gene expression as compared to normal liver parenchyma, suggesting a reversal of the early neoplastic phenotypes. Finally, we used a comparative functional genomics approach to demonstrate that the CK19-associated gene expression signature can successfully predict the clinical outcome of human HCC.
Materials and Methods
Animals and Experimental Protocol
Male F-344 rats (100–125g) purchased from Charles River (Milan, Italy) were kept on a laboratory diet (Ditta Mucedola, Milan, Italy) and given food and water ad libitum with a 12-hour light/dark daily cycle. The animals were housed in an AAALAC facility and cared for in accordance with guidelines from the Animal Care and Use Committee, NIH. The hepatic carcinogenesis was induced according to the RH model(21). Rats were injected intraperitoneally with diethylnitrosamine (DENA, Sigma, MO) at a dose of 150 mg/kg body weight. After a 2-week recovery, rats were fed a diet containing 0.02% 2-acetylaminofluorene (2-AAF, Sigma, MO) for 1 week followed by a two-thirds partial hepatectomy (PHx), and an additional week of 2-AAF diet. The animals were then returned to the basal diet, and euthanized at 10 weeks, 9, and 14 months (Supplementary Fig. 1). Rats which received DENA alone or were exposed to 2-AAF and PHx without carcinogen were used as controls.
Microarray Analysis
RNA was extracted from 60 micro-dissected samples using manufactures’ protocol (Qiagen). RNAs from 53 human HCCs were obtained from Caucasian and Chinese patients described by Lee et al.(7) (Supplementary Table 1). The RNA integrity was A280/A260>2 (ND1000, Thermo Scientific) and RIN≥6 (Agilent 2100 Bioanalyzer, Agilent Technologies)). 100 ng RNA were amplified and incubated for 16 hours at 37°C according to manufactures’ specification (Ambion, Austin TX). The efficiency of amplification was quantified using RiboGreen RNA kit (Invitrogen, Carlsbad, CA). Hybridization, washing, labeling (Cy3-streptavidin, Amersham Biosciences, Piscataway, NJ), and scanning were performed on BeadStation500 using reagents and protocols supplied by the manufacturer (illumina, San Diego, CA). 750 ng biotinylated cRNA were hybridized to RatRef-12 expression beadchips (illumina, San Diego, CA) for 18 hours at 58°C. The human HCC samples were hybridized to humanRef-8v2 beadchips. Image analysis and data extraction were automated (BeadScanv3.2, illumina).
Statistics
Data collection was performed in BeadStudiov3.3 (illumina)(23, 24). The detection score for a gene was computed from the z-value relative to that of negative controls. The technical error was estimated by iterative robust least squares fit and the data set normalized using quantile and background subtraction. FDR-adjusted p-values were calculated using the Benjamini-Hochberg procedure(25). The illumina error model was used to identify genes differentially expressed at P≤ 0.001 between focal lesions and normal liver. Analysis of network connectivity was completed using Ingenuity Pathway Analysis (IPA7.1). The significance of each network and the connectivity was estimated in IPA.
Integration of the human HCC and rat data sets was performed by z-transformation. The probability of overall survival and time to recurrence were estimated according to Kaplan-Meier and Mantel-Cox statistics (GraphPad Prism5.01). A class random variance model using 7 prediction algorithms was applied to build a prediction model and evaluate the prognostic value of the CK19 signature (α≤0.001). To estimate the accuracy of the prediction models, random permutations and leave-one-out cross-validation (LOOCV) were repeated 1,000 times. A Cox proportional hazards model and Wald statistics were used to identify genes significantly associated with survival (P≤0.01). To estimate the accuracy, univariate permutation tests were repeated 10,000 times.
Additional Methodology
Detailed methods for histology, immunohistochemistry, classification of hepatic lesions, and laser-capture micro-dissection (LMD) are described in the Supplementary Methods.
Results
Morphology of GSTP+ Focal Lesions
Two types of focal lesions could be identified at 10 weeks after DENA-initiation based on the GSTP staining: persistent (P) nodules with a strong, uniform GSTP staining and remodeling (R) nodules characterized by a faint and irregular shaped staining, indicating a progressive loss of GSTP (Table 1, Fig. 1A). The R nodules were composed of the hepatocytic cells with eosinophilic ground-glass cytoplasm, enlarged nuclei, and prominent central nucleoli. Nine months after DENA administration, the majority of the lesions progressed to adenomas with some showing signs of neoplastic transformation, such as nuclear atypia (early HCCs, eHCC) (Supplementary Fig. 2A,B). The latter lesions were GSTP+ although staining was not always uniformly distributed (Fig. 1B). Fourteen months after initiation, all rats presented multiple tumors resulting in liver weight increase up to 60–70 grams. Histopathological evaluation revealed that tumors were of the trabecular type with hepatocyte-like cells arranged in multiple-cell thick plates (Supplementary Fig. 2C). Apoptotic bodies and mitoses were commonly observed. Control groups did not show any signs of neoplastic transformation.
Table 1.
Measurement of GSTP+ and CK19+ focal lesions
| GSTP+ nodules (No./cm2) | GSTP+ area (% of total liver area) | CK19+ nodules (% of GSTP+ nodules) | CK19+ area (% of total liver area) | ||
|---|---|---|---|---|---|
| BN 190 | P | 10,89 | 21,70% | 36,36% | 4,11% |
| R | 12,87 | 14,33% | 0,00% | 0,00% | |
| BN 191 | P | 6,57 | 17,18% | 50,00% | 4,51% |
| R | 31,57 | 36,15% | 10,42% | 1,51% | |
| BN 192 | P | 4,66 | 5,27% | 57,14% | 2,18% |
| R | 36,00 | 12,24% | 5,56% | 0,61% | |
| BN 193 | P | 11,53 | 15,54% | 53,33% | 5,11% |
| R | 24,61 | 25,23% | 21,88% | 1,06% | |
| BN 194 | P | 13,91 | 26,93% | 43,75% | 10,81% |
| R | 14,78 | 23,35% | 29,41% | 2,47% | |
| BN 195 | P | 7,33 | 11,35% | 37,50% | 1,52% |
| R | 19,26 | 18,47% | 19,05% | 2,00% | |
| MEAN | P | 9,148 | 16,33% | 46,35% | 4,71% |
| R | 23,181 | 21,63% | 14,38% | 1,27% | |
P: Persistent, R: Remodeling
Figure 1.
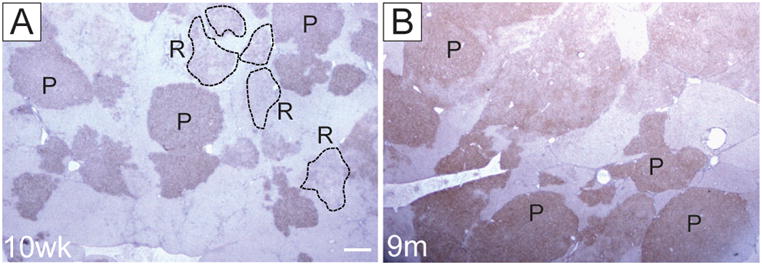
The pattern of GSTP immunostaining in livers of F344 rats submitted to RH protocol. (A) 10 weeks after DENA initiation; (B) 9 months after DENA initiation. P, persistent; R, remodelling GSTP+ foci. Sections were counterstained with hematoxylin. Original magnification, ×10.
Classification of Early Focal Lesions and Adenomas by GSTP and CK19 Staining
Recent molecular analysis of human HCC identified a prognostic subclass of patients with HCC potentially derived from HPC and characterized by the progenitor cell markers, CK7 and 19(19). Thus, we sought to answer the following questions: i) is there a subset of persistent GSTP+ lesions which is characterized by CK19 staining; ii) if so, is the transcriptomic profile of this subpopulation different from that seen in the remodelling GSTP+ lesions, and iii) are these profiles similar/dissimilar to the HPC gene expression profile in human HCC.
Immunohistochemical analysis of the livers performed at 10 weeks following the RH protocol revealed the different pattern of CK19 staining within the preneoplastic GSTP+ lesions ranging from a strong uniform to a weaker heterogeneous or no staining (Fig. 2A,B and Supplementary Fig. 3A–F). Approximately 50% of GSTP+ persistent nodules showed some degree of CK19 expression, while only 14% of nodules that stained faintly for GSTPdisplayed CK19 staining, suggesting that CK19 expression may be lost as hepatocytes revert to a more differentiated phenotype (Table 1). This observation is supported by the fact that GSTP-negative lesions were also negative for CK19. Interestingly, the hepatocyte nuclear factor 4 (HNF4), a well known marker of hepatocytic differentiation, showed an inverse expression pattern with CK19 staining (Fig. 2C,D). Also, GSTP+ adenomas were both CK19-positive and negative (Fig. 3). However, all but one early HCC displayed a strong CK19 staining indicating that progression of CK19-negative lesions to HCC is a rare event. Consistently, all HCCs developed by 14 months were uniformly CK19+.
Figure 2.
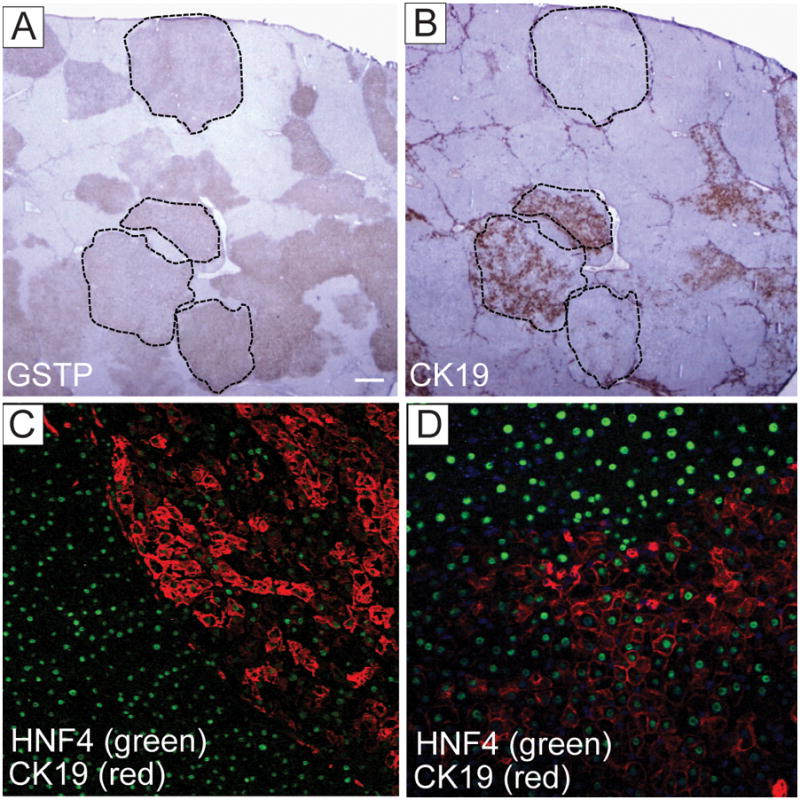
Expression of CK19 and HNF4 in persistent focal lesions. (A) GSTP and (B) CK19 immunostaining performed on parallel liver sections at 10 weeks after DENA initiation. Dashed areas demarcate the same focal lesions shown in A and B. Original magnification ×10. (C, D) Dual-color immunofluorescence staining with HNF4 (green) and CK19 (red) in liver section at 10 weeks after DENA initiation. Original magnification ×100 (C); ×200 (D).
Figure 3.

Expression of GSTP and CK19 in adenomas. (A, B) GSTP staining; (C,D) CK19 staining and (E,F) H&E staining on serial sections from two micro-dissected adenomas. Note the presence of CK19+ and CK19− persistent GSTP+ adenomas in livers at 9 months after DENA initiation. Original magnification, ×40.
Microarray Classification of Neoplastic Liver Lesions in the Rat
To generate a gene expression signature specific to the early focal lesions, we micro-dissected 19 foci and analyzed the molecular changes by high-precision transcriptomics (Fig. 4). In addition to the early foci, we dissected 20 adenomas, 13 eHCC, and 8 fully developed HCCs, representing consecutive steps in hepatocarcinogenesis. In order to focus the analysis on the persistent nodules, all selected lesions were uniformly GSTP+.
Figure 4.

Development of the CK19-associated gene signature. (A) Unsupervised hierarchical cluster analysis of 60 micro-dissected hepatic lesions including 19 focal lesions at 10 weeks, 20 adenomas at 9 months, 13 early HCCs at 9 months and 8 advanced HCCs at 14 months following DENA administration. A list of 469 differentially expressed genes was computed at P≤0.001 using normal rat liver as a reference. The position of two well characterized progenitor/cancer stem cell markers, Epcam and Aldh1a1, are shown. (B) Unsupervised clustering of the rat lesions. The dendrogram demonstrates a significant separation of the rat lesions according to the CK19 expression profile. The CK19+ lesions belong to cluster R1 (red). (C, D) Class prediction. The probability of correct classification was estimated using a Bayesian compound covariate prediction model for (C) the gene signature (R1 and R2 classification), and (D) the CK19 expression profile during leave-one-out cross-validation (LOOCV). To ensure the accuracy in the prediction method, random permutations were repeated 1,000 times. The degree of the correct classification is represented by the area under the ROC curve (AUC).
First, we applied an unsupervised approach to identify the differentially expressed genes between the early foci and normal rat livers. A list of 469 significantly regulated genes was found at P≤0.001. Hierarchical cluster analysis grouped all the rat lesions into two major clusters (R1 and R2). The probability of correct sub-classification was estimated by class prediction with an accuracy of 0.98 (Fig. 4C). In cluster R1, a subgroup of the early focal lesions and adenomas was clustered together with the eHCC and advanced HCC, suggesting the likelihood of their progression to HCC (Fig. 4B). The remaining foci (10/19) were grouped with adenomas (12/20) consistent with the delayed progression to HCC or remodeling into the surrounding liver parenchyma.
Next, we integrated the unsupervised analysis together with the information obtained from immunohistochemical staining against CK19. Significantly, we found a separation of the preneoplastic and malignant lesions based on the CK19 expression with estimated accuracy of correct classification of 0.95 (P<0.0001) (Fig. 4B,D). The majority of eHCC (12/13) and all advanced HCC were positive for CK19+ and clustered together with CK19+ foci and adenomas whereas the CK19-negative focal lesions belonged to the subcluster R2 together with CK19-negative adenomas.
Characteristics of CK19-Positive Focal Lesions in Hepatocarcinogenesis
We evaluated the transcriptomic differences between CK19+ and CK19− foci using a supervised analysis, selecting unique genes in each cluster (P≤0.001). A total of 2638 genes were identified as differentially regulated compared to the normal liver with 156 genes and 1308 genes being unique to CK19− and CK19+ foci, respectively. Applying pathway analysis tools, several connectivity maps were constructed based on the previously reported interactions between the members of the significant gene set. The connectivity of the top regulatory networks showed a dominance of AP-1/JUN and MAPK14/JNK (Supplementary Fig. 4). These networks are known to control inflammation, stress responses and tumorigenesis (26). In the CK19+ foci, both MAPK14 and JNK were downregulated whereas expression of AP-1/JUN was increased compared to the CK19-negative focal lesions. Importantly, we previously showed that the HB signature is driven by the downstream targets of AP-1(19). Furthermore, the increase in JUN expression was attributed to an increase in JUB/Ajuba which is known to promote activation of murine c-Jun(27) and the phoshorylation of β-catenin(28).
Based on the list of homologous rat and human genes, we matched the expression profile unique to CK19+ foci with the human HCC data set. We then assessed the enrichment of the CK19+ gene signature in HCC subtype A and B by using the nonparametric gene set enrichment analysis (GSEA)(29) (Fig. 5A). The CK19+ gene signature was significantly enriched (P=0.002) and positively correlated with the poor survival subclass A and progenitor-derived HCCs. An enrichment analysis of the CK19+ gene signature with the Molecular Signatures Database (MSigDB) demonstrated a significant overlap with several stem cell-related genes sets (Supplementary Table 2). Notably, among the top 10 gene sets, an overlap was found with several liver-specific gene sets. Also, assessment of the human stem cell module map(30) revealed a significant overlap between the gene expression signature unique to CK19+ foci and genes associated with stem cell derivation (Fig. 5B). Together, these data show that CK19+/GSTP+ persistent nodules exhibit an HPC-like expression profile.
Figure 5.

Gene set enrichment analysis of the CK19+ gene signature. (A) The analysis demonstrated a significant positive correlation between the CK19+ gene signature and the gene expression signature of subclass A and hepatoblast-like (HB) human HCCs (Red, Class A). The HB subclass was previously shown to have a fetal hepatoblast-like and progenitor-type expression signature. This subtype has the worst clinical prognosis. Several stem cell and liver specific gene sets in the Molecular Signatures Database were found among the top 10 gene sets which positively correlated with the CK19+ gene signature (Supplementary Table 2). (B) Association of the CK19+ gene signature and the stem cell module map. In this comparison, we used the genes given in the human embryonic stem cell-like module. More than 15% of the genes in the CK19+ gene expression signature overlapped with the module map.
Next, we examined the functional connectivity among the significant genes specific for the CK19-negative focal lesions. The most predominant feature was the overexpression of KLF10/TIEG (TGF-β inducible early transcription factor gene), previously described as a tumor suppressor gene in breast cancer(31). KLF10 expression has been shown to be sufficient to induce apoptosis in Hep3B cells(32). Moreover, loss of KLF10 interfered with TGFβ-induced apoptosis and promoted carcinogenesis(32). Thus, KLF10 may be an early response gene responsible for TGFβ-dependent apoptosis thereby contributing to the remodeling phenotype.
The Prognostic Power of the CK19 Gene Signature
We next examined the potential usefulness of the RH model to gain insight into the relevance of HPC for human HCC. For this purpose we used a comparative functional genomics approach(33, 34). This approach is based on the hypothesis that since regulatory elements of evolutionarily related species are conserved, gene expression signatures reflecting similar phenotypes in different species could be also conserved. The hypothesis has been supported by numerous studies demonstrating that cross-compared gene expression data from human HCC and rodent HCC models can identify the aberrant phenotypes reflecting the evolutionarily conserved molecular pathways(35, 36). Here we applied the signature of 276 orthologous genes found between human and rat, to integrate the rat lesions with a data set of 53 human HCCs (Fig. 6 and Supplementary Table 1). The hierarchical clustering of CK19+ and negative lesions coincided with the previously described human HCC subtypes A and B characterized by a poor or better prognosis, respectively(37) (Fig. 6B). Notably, cluster C2 that encompassed subtype A HCC and CK19+ neoplastic lesions in the rat, also included human HCC defined by the progenitor-type hepatoblast-like (HB) signature and the worst clinical prognosis(19). An optimized gene classifier from the CK19-associated genes contained 110 genes which demonstrated a highly significant prognostic power with a probability of correct class prediction of 0.98 (P<0.001) (Fig. 6C and Supplementary Table 3). The overall performance of the classifier in 7 different prediction models ranged from 89% to 98% (Supplementary Table 4A,B). Accordingly, the gene signature efficiently predicted both survival of patients (P<0.009) and time to recurrence (P<0.006) (Fig. 6D,E).
Figure 6.

The prognostic power of the CK19 gene signature. (A) The HCC data set was generated from a cohort of 53 HCCs obtained from Caucasian and Chinese patients and hybridized to illumina bead chips. A list of 276 orthologous genes between the two species presented on both platforms was identified. (B) Comparative functional genomics. The CK19 gene signature separates the human HCC according to the previous A and B sub-classification. Individual human and rat samples are shown in red and green, respectively. (C) Development of a gene classifier for prediction of the prognostic value of the CK19 gene signature. A Bayesian compound covariate prediction analysis showed a probability of 0.98 for correct classification. The probability is represented by the area under the ROC curve (AUC). To build a classifier, a class random variance model was used to identify genes univariately significant at α≤0.001. Seven different algorithms were used in the prediction of the correct classification during LOOCV with a prediction rate 89 to 98%. The performance of the classifier estimating the correct classification in each of the prediction models is given in supplementary table 4. (D, E) Cumulative survival. Integration and cluster analysis identified clinically relevant distinct subgroups of human HCC based on the CK19 gene expression signature. Kaplan-Meier plots and Mantel-Cox statistical analysis were applied in the survival analysis.
A Cox proportional hazard model applied to test the prognostic utility of the gene classifier discriminated the patients according to the clinical prognosis (Fig. 7). Using Wald statistics, 29 significant genes were then identified (P<0.01) with at least a 2-fold difference in expression ratio. This signature successfully differentiated the patients according to survival thus strengthening the prognostic power of the CK19-associated gene signature.
Figure 7.
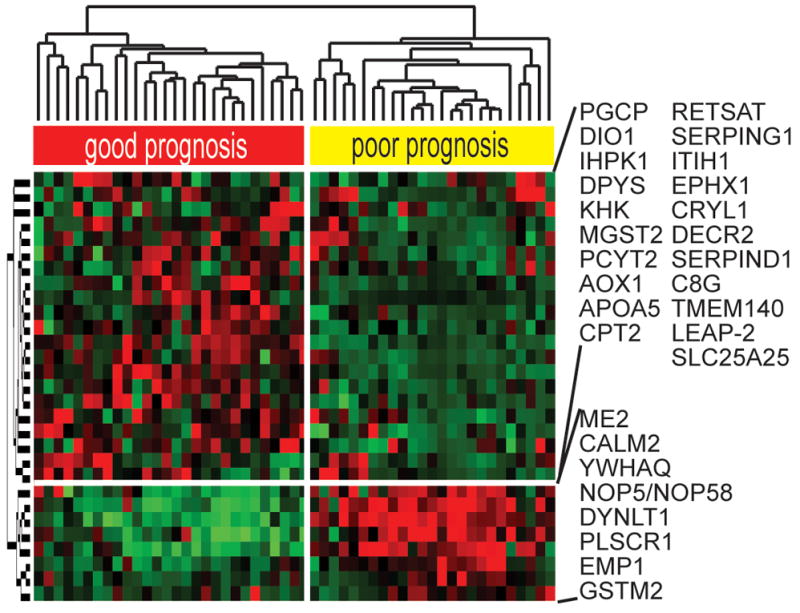
Prognostic survival genes. Hierarchical cluster analysis of the HCC data set. Genes which were significantly (P≤0.01) associated with the disease outcome were identified by applying a Cox proportional hazards model and Wald statistics. 29 genes independently demonstrated a prognostic ability. To estimate the accuracy, univariate permutation tests were repeated 10,000 times.
Discussion
In this study, we report a comprehensive characterization of the neoplastic development induced in the rat liver by the RH protocol. To investigate evolution of the early preneoplastic lesions, the persistent GSTP+ lesions (Fig. 1) ranging from foci to fully developed HCC were examined for the expression of the HPC marker CK19 and subjected to global gene expression analysis (Fig. 4). A subset of the early focal lesions (9/19) as well as adenomas (8/20), eHCC (12/13), and all HCC (8/8) were CK19+ (Figs. 2–3 and Supplementary Fig. 3). Significantly, unsupervised clustering of hepatic lesions without prior knowledge of CK19 staining clearly differentiated the CK19+ from CK19-negative lesions (Fig. 4).
Assessment of the translational value of animal models of human cancer which are generated under conditions far different from those seen in humans, poses a major challenge. As an attempt to meet this challenge, we earlier established a comparative functional genomics approach to evaluate the usefulness of mouse models for human liver cancer(34, 35). This approach has been successfully used for other cancers as well(33, 36). Here we applied this approach to show a co-segregation of the CK19+ rat lesions with human HCC of the subclass A and HB subtypes (Fig. 6). Recently, we determined that the gene expression profiles of HB subtype and fetal rat hepatoblasts are closely related(19) suggesting that the CK19+ foci may be of HPC origin. Further, the CK19+ foci clustered together with the more advanced HCC indicating that they might progress to full blown HCC. The CK19 expression is unlikely to be etiology-dependent since it has been described in different stages of hepatitis (HBV or HCV) and alcohol-associated HCC (13–15, 38), but rather “cell of origin”-related. Supervised analysis of the CK19+ foci and negative lesions further suggested a HPC derivation of the CK19+ lesions as evidenced by a significant overlap with the human stem cell module map (Fig. 5B). A multivariate gene enrichment analysis demonstrated that the CK19+ gene list positively correlates (P=0.002) with human HCC classified in subclass A and HB (Fig. 5A). Also, the GSEA indentified a statistically significant overlap with several liver-specific and stem cell-like gene sets (Supplementary Table 2). Indeed, it was shown that patients with HCC expressing biliary markers similar to CK19 presented a more aggressive disease with both clinical and pathobiological implications(20).
Applying pathway analysis tools, several connectivity maps were constructed which showed a specific down-regulation of the serine and threonine protein kinases MAPK8 (JNK) and MAPK14 (p38α) among the CK19+ lesions (Supplementary Fig. 4). MAPK14 functions as a tumor-suppressor by inhibiting Ras signaling(39). Similarly, decreased MAPK14 activity was described to promote Ras-dependant transformation (40). Also, MAPK14 has been shown to antagonize c-Jun signaling, suggesting that decreased MAPK14 expression may promote hepatocarcinogenesis(41). Indeed, hepatocyte-specific knockout of MAPK14 resulted in a major increase in c-Jun(42). In our study, we demonstrate an upregulation of the key transcription factor AP-1 (JUN/FOS gene network) in the CK19+ rat HCC. This observation concurs with the HB signature characterized by activation of the AP-1 downstream signaling(19). Consistently, a role for c-Jun in promoting proliferation was shown during progression of preneoplastic hepatocytes in a mouse model(43). Livers deficient in c-Jun displayed a p53-dependent increase in p21 protein which correlated with higher p38α activity(44). Additionally, upregulation of c-Jun expression has been found to reduce expression of transcription factor HNF4 during acute-phase response and liver regeneration similar to our observations in the CK19+ foci (Fig. 2C)(45). AP-1 activity was also associated with the increase in Kruppel-like factor 6 (KLF6) expression reported to be involved in the protection against apoptosis in HCC(46). Moreover, expression of ITGAV which is known to promote angiogenesis was enhanced in the CK19+ foci compared to the CK19 negative lesions, suggesting the proliferative advantage of the former. In contrast, the CK19-negative foci demonstrated a more “benign” gene profile which was consistent with normal liver parenchyma. The only significant network identified among the CK19-negative lesions showed an increase of TGF-β inducible early growth response gene (TIEG/KLF10) described as a tumor-suppressor gene(31), implying a potential mechanism for regression of the early neoplastic lesions.
In conclusion, our data demonstrate the progenitor derivation of the majority of the HCC developed in the RH rat model and identify CK19 as a prognostic marker of the early persistent preneoplastic lesions in the liver. Furthermore, by applying comparative functional genomics we show that the CK19-associated gene signature can robustly stratify HCC patients according to clinical outcome, indicating the usefulness of the RH rat model for reproducing stem/progenitor-derived human HCC.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and by funds from Associazione Italiana Ricerca sul Cancro (AIRC), Italy.
Abbreviations
- CK
Cytokeratin
- DENA
Diethylnitrosamine
- HCC
Hepatocellular carcinoma
- eHCC
early HCC
- HPC
Hepatic progenitor cell
- GSTP
Glutathione S-transferase
- PHx
Partial hepatectomy
- RH model
Resistant hepatocyte model
References
- 1.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Sherman M. Hepatocellular carcinoma: epidemiology, risk factors, and screening. Semin Liver Dis. 2005;25:143–154. doi: 10.1055/s-2005-871194. [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HB. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology. 2004;127:S27–34. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 7.Lee JS, Thorgeirsson SS. Genome-scale profiling of gene expression in hepatocellular carcinoma: classification, survival prediction, and identification of therapeutic targets. Gastroenterology. 2004;127:S51–55. doi: 10.1053/j.gastro.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Grisham JW. Interspecies comparison of liver carcinogenesis: implications for cancer risk assessment. Carcinogenesis. 1997;18:59–81. doi: 10.1093/carcin/18.1.59. [DOI] [PubMed] [Google Scholar]
- 9.Hsia CC, Evarts RP, Nakatsukasa H, Marsden ER, Thorgeirsson SS. Occurrence of oval-type cells in hepatitis B virus-associated human hepatocarcinogenesis. Hepatology. 1992;16:1327–1333. doi: 10.1002/hep.1840160604. [DOI] [PubMed] [Google Scholar]
- 10.Komuta M, Spee B, Vander Borght S, De Vos R, Verslype C, Aerts R, Yano H, et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology. 2008;47:1544–1556. doi: 10.1002/hep.22238. [DOI] [PubMed] [Google Scholar]
- 11.Libbrecht L, Bielen D, Verslype C, Vanbeckevoort D, Pirenne J, Nevens F, Desmet V, et al. Focal lesions in cirrhotic explant livers: pathological evaluation and accuracy of pretransplantation imaging examinations. Liver Transpl. 2002;8:749–761. doi: 10.1053/jlts.2002.34922. [DOI] [PubMed] [Google Scholar]
- 12.Theise ND, Saxena R, Portmann BC, Thung SN, Yee H, Chiriboga L, Kumar A, et al. The canals of Hering and hepatic stem cells in humans. Hepatology. 1999;30:1425–1433. doi: 10.1002/hep.510300614. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka S, Yamamoto T, Tanaka H, Kodai S, Ogawa M, Ichikawa T, Hai S, et al. Potentiality of combined hepatocellular and intrahepatic cholangiocellular carcinoma originating from a hepatic precursor cell: Immunohistochemical evidence. Hepatol Res. 2005;32:52–57. doi: 10.1016/j.hepres.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Libbrecht L, Desmet V, Van Damme B, Roskams T. The immunohistochemical phenotype of dysplastic foci in human liver: correlation with putative progenitor cells. J Hepatol. 2000;33:76–84. doi: 10.1016/s0168-8278(00)80162-2. [DOI] [PubMed] [Google Scholar]
- 15.Libbrecht L, De Vos R, Cassiman D, Desmet V, Aerts R, Roskams T. Hepatic progenitor cells in hepatocellular adenomas. Am J Surg Pathol. 2001;25:1388–1396. doi: 10.1097/00000478-200111000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Maronpot RR, Montgomery CA, Jr, Boorman GA, McConnell EE. National Toxicology Program nomenclature for hepatoproliferative lesions of rats. Toxicol Pathol. 1986;14:263–273. doi: 10.1177/019262338601400217. [DOI] [PubMed] [Google Scholar]
- 17.Sell S, Dunsford HA. Evidence for the stem cell origin of hepatocellular carcinoma and cholangiocarcinoma. Am J Pathol. 1989;134:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 18.Su Q, Fu Y, Liu YF, Zhang W, Liu J, Wang CM. Laminin induces the expression of cytokeratin 19 in hepatocellular carcinoma cells growing in culture. World J Gastroenterol. 2003;9:921–929. doi: 10.3748/wjg.v9.i5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 20.Wu PC, Fang JW, Lau VK, Lai CL, Lo CK, Lau JY. Classification of hepatocellular carcinoma according to hepatocellular and biliary differentiation markers. Clinical and biological implications. Am J Pathol. 1996;149:1167–1175. [PMC free article] [PubMed] [Google Scholar]
- 21.Solt D, Farber E. New principle for analysis of liver carcinogenesis. Am J Pathol. 1976;88:595–618. [PMC free article] [PubMed] [Google Scholar]
- 22.Grisham JW, Thorgeirsson SS. Liver Stem Cells. In: Potten CS, editor. Stem Cells. New York: Academic Press; 1997. pp. 234–282. [Google Scholar]
- 23.Kuhn K, Baker SC, Chudin E, Lieu MH, Oeser S, Bennett H, Rigault P, et al. A novel, high-performance random array platform for quantitative gene expression profiling. Genome Res. 2004;14:2347–2356. doi: 10.1101/gr.2739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan JB, Yeakley JM, Bibikova M, Chudin E, Wickham E, Chen J, Doucet D, et al. A versatile assay for high-throughput gene expression profiling on universal array matrices. Genome Res. 2004;14:878–885. doi: 10.1101/gr.2167504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini YHY. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 26.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 27.Kanungo J, Pratt SJ, Marie H, Longmore GD. Ajuba, a cytosolic LIM protein, shuttles into the nucleus and affects embryonal cell proliferation and fate decisions. Mol Biol Cell. 2000;11:3299–3313. doi: 10.1091/mbc.11.10.3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haraguchi K, Ohsugi M, Abe Y, Semba K, Akiyama T, Yamamoto T. Ajuba negatively regulates the Wnt signaling pathway by promoting GSK-3beta-mediated phosphorylation of beta-catenin. Oncogene. 2008;27:274–284. doi: 10.1038/sj.onc.1210644. [DOI] [PubMed] [Google Scholar]
- 29.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 30.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramaniam M, Hefferan TE, Tau K, Peus D, Pittelkow M, Jalal S, Riggs BL, et al. Tissue, cell type, and breast cancer stage-specific expression of a TGF-beta inducible early transcription factor gene. J Cell Biochem. 1998;68:226–236. [PubMed] [Google Scholar]
- 32.Ribeiro A, Bronk SF, Roberts PJ, Urrutia R, Gores GJ. The transforming growth factor beta(1)-inducible transcription factor TIEG1, mediates apoptosis through oxidative stress. Hepatology. 1999;30:1490–1497. doi: 10.1002/hep.510300620. [DOI] [PubMed] [Google Scholar]
- 33.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 34.Lee JS, Thorgeirsson SS. Comparative and integrative functional genomics of HCC. Oncogene. 2006;25:3801–3809. doi: 10.1038/sj.onc.1209561. [DOI] [PubMed] [Google Scholar]
- 35.Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- 36.Sweet-Cordero A, Mukherjee S, Subramanian A, You H, Roix JJ, Ladd-Acosta C, Mesirov J, et al. An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat Genet. 2005;37:48–55. doi: 10.1038/ng1490. [DOI] [PubMed] [Google Scholar]
- 37.Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, Durnez A, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- 38.Durnez A, Verslype C, Nevens F, Fevery J, Aerts R, Pirenne J, Lesaffre E, et al. The clinicopathological and prognostic relevance of cytokeratin 7 and 19 expression in hepatocellular carcinoma. A possible progenitor cell origin. Histopathology. 2006;49:138–151. doi: 10.1111/j.1365-2559.2006.02468.x. [DOI] [PubMed] [Google Scholar]
- 39.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–215. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 40.Qi X, Pohl NM, Loesch M, Hou S, Li R, Qin JZ, Cuenda A, et al. p38alpha antagonizes p38gamma activity through c-Jun-dependent ubiquitin-proteasome pathways in regulating Ras transformation and stress response. J Biol Chem. 2007;282:31398–31408. doi: 10.1074/jbc.M703857200. [DOI] [PubMed] [Google Scholar]
- 41.Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Komnenovic V, et al. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 42.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, Wagner EF. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–192. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 44.Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, Hui L, et al. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 2006;20:2306–2314. doi: 10.1101/gad.390506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qian X, Samadani U, Porcella A, Costa RH. Decreased expression of hepatocyte nuclear factor 3 alpha during the acute-phase response influences transthyretin gene transcription. Mol Cell Biol. 1995;15:1364–1376. doi: 10.1128/mcb.15.3.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sirach E, Bureau C, Peron JM, Pradayrol L, Vinel JP, Buscail L, Cordelier P. KLF6 transcription factor protects hepatocellular carcinoma-derived cells from apoptosis. Cell Death Differ. 2007;14:1202–1210. doi: 10.1038/sj.cdd.4402114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.