

Summary
The TIM (T cell/transmembrane, immunoglobulin, and mucin) gene family plays a critical role in regulating immune responses, including allergy, asthma, transplant tolerance, autoimmunity, and the response to viral infections. The unique structure of TIM immunoglobulin variable region domains allows highly specific recognition of phosphatidylserine (PtdSer), exposed on the surface of apoptotic cells. TIM-1, TIM-3, and TIM-4 all recognize PtdSer but differ in expression, suggesting that they have distinct functions in regulating immune responses. TIM-1, an important susceptibility gene for asthma and allergy, is preferentially expressed on T-helper 2 (Th2) cells and functions as a potent costimulatory molecule for T-cell activation. TIM-3 is preferentially expressed on Th1 and Tc1 cells, and generates an inhibitory signal resulting in apoptosis of Th1 and Tc1 cells. TIM-3 is also expressed on some dendritic cells and can mediate phagocytosis of apoptotic cells and cross-presentation of antigen. In contrast, TIM-4 is exclusively expressed on antigen-presenting cells, where it mediates phagocytosis of apoptotic cells and plays an important role in maintaining tolerance. TIM molecules thus provide a functional repertoire for recognition of apoptotic cells, which determines whether apoptotic cell recognition leads to immune activation or tolerance, depending on the TIM molecule engaged and the cell type on which it is expressed.
Keywords: TIM, phosphatidylserine, asthma, galectin-9, phagocytosis, costimulation, tolerance
Introduction
The T cell/transmembrane, immunoglobulin, and mucin (TIM) gene family was positionally cloned in 2001 using a congenic mouse model of asthma (1, 2). Since that time, a great deal of evidence has accumulated indicating that this gene family plays a critical role in regulating immune responses, including transplant tolerance, autoimmunity, the regulation of allergy and asthma, and the response to viral infections (3, 4).
The TIM gene family consists of eight members (TIM-1-8) on mouse chromosome 11B1.1, and three members (TIM-1, TIM-3, and TIM-4) on human chromosome 5q33.2, located in a chromosomal region that has been repeatedly linked with asthma, allergy, and autoimmunity (2, 5, 6). The genetic organization of the TIM genes has been reviewed (7). Expression, function, and structural studies confirm that mouse TIM-1, TIM-3, and TIM-4 are the orthologues of human TIM-1, TIM-3, and TIM-4, respectively. Five other TIM genes are found only in rodent genomes and of these, only TIM-2 has been characterized and is discussed here. TIM genes encode type I cell-surface glycoproteins with common structural features including an N-terminal immunoglobulin (Ig)-like domain, a mucin domain with O-linked glycosylations and with N-linked glycosylations close to the membrane, a single transmembrane domain, and a cytoplasmic region with tyrosine phosphorylation motif(s), except in TIM-4 (2) (Fig. 1).
Fig. 1. Schematic representation of TIM protein structures.

Positions of the glycosylation sites in IgV domains are approximately positioned according to the crystal structures. Glycosylation sites in the mucin domain were predicted with NetOglyc and NetNglyc and are positioned approximately.
We and others have recently demonstrated that TIM-1, TIM-3, and TIM-4 are pattern recognition receptors specialized for recognition of phosphatidylserine (PtdSer), as shown by the highly conserved TIM-PtdSer interactions observed in TIM/PtdSer co-crystal structures (8, 9). PtdSer is normally localized to the inner leaflet of the plasma membrane but is redistributed and exposed on the outer membrane when a cell undergoes apoptosis. PtdSer on apoptotic cells provides a key signal that triggers cell engulfment (10, 11). Recognition of apoptotic cells is an essential component of tissue homeostasis and immune regulation (12).
TIM-1, TIM-3, and TIM-4 differ in molecular structure and expression patterns, suggesting that they have distinct functions in regulating T-cell responses. TIM-1, an important susceptibility gene for asthma and allergy (13), is preferentially expressed on Th2 cells and functions as a potent costimulatory molecule for T-cell activation (14). TIM-3 is preferentially expressed on T-helper 1 (Th1) and Tc1 cells and generates an inhibitory signal resulting in apoptosis of Th1 and Tc1 cells (15). Polymorphisms in TIM-1 and TIM-3 may reciprocally regulate the direction of T-cell responses, as modeled in Fig. 2. TIM-3 is also expressed on some dendritic cells (DCs) (3) and can mediate phagocytosis of apoptotic cells and cross-presentation of antigen (16). In contrast, TIM-4 is exclusively expressed on antigen-presenting cells (APCs), where it mediates phagocytosis of apoptotic cells and plays an important role in maintaining tolerance (17, 18). TIM molecules thus provide a functional repertoire for recognition of apoptotic cells, which determines whether apoptotic cell recognition leads to immune activation or tolerance, depending on the TIM molecule engaged and the cell type on which it is expressed.
Fig. 2. Reciprocal regulation of Th1 and Th2 activity by TIM-1 and TIM-3: a Yin-Yang model.
Structure of the N-terminal Ig-like domain of TIM proteins
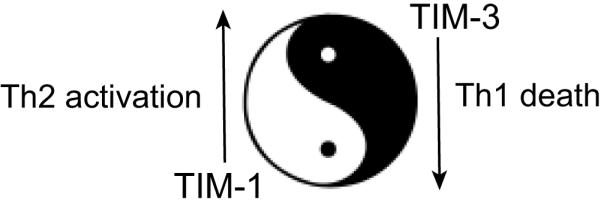
The critical feature of TIM Ig-like domains is a deep binding pocket flanked by two hydrophobic loops that can extend into a membrane (Fig. 3F). The crystal structures of the Ig-like domains of murine TIM-1, TIM-2, TIM-3, and TIM-4 have been determined (8, 19, 20), revealing that they belong to the immunoglobulin variable (IgV) set. The TIM IgV domains are composed of two antiparallel β-sheets with particularly short β-strands, B, E and D in one sheet (BED β-sheet) and A, G, F, C, C’ and C” β-strands in the other sheet (GFC β-sheet) (Fig. 3A). All TIM IgV domains contain six conserved Cys residues, and the first and last of these six Cys residues bridge the β-sheets, as in other Ig domains.
Fig. 3. Structures of the TIM Ig variable (IgV) domains and the unique conformation of the CC’ loop in each.

(A) Ribbon diagram of the mTIM-1 IgV domain (PDB 2OR8). β-strands of the GFC β-sheet are red and those in the BED face are pink, coil is orange, and helices are in light-blue. The tip of the unique loop between β-strands C and C’ (CC’ loop) is colored yellow. Cys residues and disulphide bonds are shown as green cylinders. Strands are labeled with uppercase letters and terminal ends (n and c) are in lowercase. (B-E) Detailed view of the CC’ loop in the structures of the four murine TIM IgV domains (PDB 2OR8, 2OR7, 3KAA, and 3BI9). The tip of the loop, comprising residues between the two disulfide bonds bridging the loop to the GFC β-sheet, and residues at the β-sheet interacting with the loop are shown with a ball and stick drawing and with carbons in yellow. Oxygen and nitrogen atoms are in red and blue, respectively. Hydrogen bonds between the conserved Arg and Lys residues and the tip of the loop are shown as pink dashed cylinders. (F) Surface representation of the mTIM-4 IgV domain structure determined in the absence of ligand (PDB 3BI9). Residues at the tip of the CC’ and FG loops building a narrow cavity are colored in yellow and orange, respectively.
The four additional Cys residues found in all TIM family members form two additional disulfide bonds that fix the long CC’ loop folded upwards onto the GFC β-sheet (Fig. 3A). This folded conformation of the CC’ loop onto the GFC β–sheet is a distinct structural feature of all TIM IgV domains. In homologous IgV domains of other Ig superfamily members, the CC’ loop does not cover the GFC face, and the flat, exposed GFC β-sheet is engaged in intermolecular interactions (21-24). The tip of the CC’ loop projects parallel to the FG loop at the top of the IgV domain in mTIM-1, mTIM-3, and mTIM-4 (Fig. 3B,D,E) but not in TIM-2 (Fig. 3C), generating a distinctive pocket (Fig. 3F) that is used for recognition of ligands (8, 9, 19, 20), such as PtdSer (Fig. 4). In mTIM-1, mTIM-3, and mTIM-4, interactions of the CC’ loop with two conserved basic residues (Arg and Lys) in the F and G β-strands hold the tip of the CC’ loop upwards (Fig. 3B,D,E) and make a pocket by enforcing the distance between the CC’ and FG loops (Fig. 3F). In mTIM-2, the lack of those interactions leads the CC’ loop to adopt a specific helical conformation and project away from the FG loop (Fig. 3C).
Fig. 4. Binding of PtdSer to the MILIBS in the TIM IgV domain.

Structures of mTIM-3 (PDB 3KAA) and mTIM-4 (PDB 3BIB) IgV domains in complex with PtdSer are shown in panels (A) and (B), respectively. Stick drawing of the MILIBS pocket with the bound PtdSer molecule and the metal ion (green sphere). The amino acid residues at the CC’ and FG loops building the MILIBS are shown with carbons in gray, whereas the PtdSer is shown with carbons in yellow and phosphate in orange. Oxygens are red and nitrogens blue. The fatty acid (fAc), glycerol (Gl), phosphate (Ph), and serine (S) moieties of PtdSer are labeled. Amino acid residues contacting the hydrophobic moiety of PtdSer and the conserved Asn and Asp coordinating the metal ion are labeled. Coordinations are shown as dashed red lines, whereas hydrogen bonds between the PtdSer molecule and the protein are orange. (C) Model for TIM protein binding to PtdSer in a cell membrane. Surface representation of the mTIM-4 IgV domain bound to PtdSer is shown with a phospholipid bilayer membrane. Side chains at the tip of the CC’ (Asn-Ser) and FG loops (Trp-Phe) are shown. The PtdSer and phospholipids are shown in stick representation as described above. The hydrophilic head moiety of PtdSer penetrates into the MILIBS where the phosphate coordinates with the metal ion (green sphere), whereas hydrophobic residues of the TIM-4 CC’ and FG loops penetrate the lipid bilayer. The tip of the BC loop, shown in blue, comes close to the charged head of adjacent phospholipids.
A conserved PtdSer binding mode in the TIM family
The unique pocket generated by the CC’ loop and the neighboring FG loop and revealed by high resolution analysis of TIM-4 crystals (8), contains conserved residues that coordinate with metal ions, such as calcium. This conserved binding pocket has been termed the metal ion-dependent ligand-binding site (MILIBS). Structures of mTIM-3 and mTIM-4 indicate that PtdSer binds to the MILIBS of both in a similar manner (Fig. 4A,B). The hydrophilic moiety of PtdSer penetrates into the cavity built by the CC’ and FG loops, with its acidic phosphate group coordinating with a metal ion that is linked to oxygens in the two main chains and two side chains of Asn and Asp residues in the FG loop. In the mTIM-4 structure, solved at higher resolution, a water molecule was also coordinated to the metal ion. These Asn and Asp residues are conserved in all TIM proteins binding to PtdSer and are absent in mTIM-2. The Ser residue of PtdSer fits between the metal ion and the tip of the CC’ loop (Fig. 4). The PtdSer amine group has specific interactions with the conserved Asp residue involved in metal ion coordination, whereas the carboxylate of the PtdSer Ser is hydrogen bonded to the Ser residue conserved in the CC’ loop of most TIM IgV domains. All these interactions appear specific for the L stereoisomer of PtdSer and provide specificity for phospholipid binding of the TIM proteins (8). Thus, the MILIBS pocket appears designed for the specific recognition of PtdSer.
The glycerol and fatty acid moiety that anchor the phospholipid in the lipid bilayer interact with the hydrophobic residues present in the FG loop of the mTIM proteins (Leu-Met in mTIM-3 and Phe-Trp in mTIM-4) as well as with the side chain of the Trp residue in the CC’ loop of mTIM-3 (Fig. 4A,B). It is therefore likely that the hydrophobic residues penetrate into the lipid bilayer upon TIM binding to PtdSer on the surface of apoptotic cells, such as shown in Fig. 4C for mTIM-4. The Phe-Trp residues in the FG loop are conserved in mTIM-1 and human TIM-1 and TIM-4 proteins, which all bind to PtdSer. Mutagenesis and binding studies showed that all these hydrophobic residues were critical for recognition of PtdSer (8, 16, 17). The binding model shown in Fig. 4C suggests that residues in the BC loop will be brought close to the surface of the membrane and could interact with the lipid bilayer as well. The contribution of the BC loop to the membrane binding interaction has been recently proven by mutagenesis studies of mTIM-1, mTIM-3, and mTIM-4 (9). The BC loop is variable among IgV domains of the TIM family and contains mTIM-3 polymorphic residues (1). Indeed, the observed differences in the PtdSer binding affinity between the BALB/c and HBA TIM-3 allelic variants are related to polymorphisms in the BC loop (9). Variation in the BC, CC’, and FG loops could contribute to the PtdSer binding affinities, which differ among the TIM proteins. The affinities of TIMs for PtdSer have been estimated using TIM-Ig fusion proteins and microtiter plates coated with PtdSer. Binding studies showed similar affinities of mTIM-1 and mTIM-4 for PtdSer, about 2 nM, and a lower affinity of mTIM-3, about 10 nM (9, 18).
TIM gene family: identification, linkage and association studies
Chromosome 5q23-35 has been linked to atopy (i.e., asthma, allergy, and eczema/atopic dermatitis) in many genome-wide linkage studies (5, 25). However, identification of specific asthma susceptibly genes has been difficult, because asthma is a complex trait, with multiple genes as well as environmental factors influencing the development of asthma. To simplify the genetic analysis of asthma and focus on genes located in this region, a congenic mouse model was used. C.D2 congenic lines differ from BALB/c mice (asthma susceptible), in that they contain distinct chromosomal segments derived from a DBA/2 (asthma resistant) ancestor on a BALB/c background. One of these lines, the C.D2 Es-HBA (HBA), exhibited the DBA/2 phenotype and contained a DBA/2-derived segment of chromosome 11 syntenic to human chromosome 5q23-35. Offspring from this strain were backcrossed to BALB/c mice and used to identify the novel atopy susceptibility gene locus called Tapr and within it, the TIM gene family (1).
TIM-1 polymorphisms and protection against asthma and allergy
TIM1 (HUGO designation HAVCR1) is highly polymorphic in human, monkey, and mouse, with single nucleotide polymorphisms (SNPs) as well as insertion/deletion variants primarily in the mucin domain in both humans and mice. Association analysis of the insertion/deletion variants of TIM1 in human subjects with asthma and allergy (atopic diseases) demonstrated that allelic variation of TIM1 contributed to the risk of atopy, and this association was strongest in individuals with past exposure to hepatitis A virus (HAV) (13). Specifically, the insertion of 6 amino acids in the mucin domain (157insMTTTVP) was associated with protection from atopy, but this protection was only observed in individuals who were seropositive to HAV (13). The relationship of TIM1 and atopy with HAV exposure is important, because TIM1 was discovered as the cellular receptor for HAV (HAVcr-1) in African green monkeys (26) and humans (27). Moreover, epidemiological studies in several distinct populations (Italy, Denmark, and Turkey) have shown that the prevalence of allergy and asthma was significantly lower in HAV seropositive individuals compared to HAV seronegative individuals, suggesting that infection with HAV in some way protected against the development of atopy (28, 29). The identification of TIM-1 as the receptor for HAV and as a costimulatory molecule on T cells suggest that HAV may directly affect the immune system through TIM-1, providing the strongest effects through the 157insMTTTVP polymorphism.
The association of TIM1 with atopy has been confirmed in studies of large populations in Baltimore (30), Arizona (31), Korea (32), Wuhan, China (33), and Australia (34). In one study in Australia, TIM4 was also associated with atopic dermatitis (34), and in another study TIM3 was associated with asthma (31). One study reported an absence of an association of TIM1 with atopic asthma in Japan (35), where the incidence of HAV infection is now close to zero. Another study from Shandong, China, where HAV infection is still common, showed an absence of association of TIM1 with asthma, in a patient population primarily with non-allergic asthma, in a study where patients with allergic rhinitis and atopic dermatitis were excluded (36). These results suggest that TIM1 and HAV affect the development of Th2-biased immunity, atopy, and allergic asthma, but may have limited involvement in the development of non-allergic asthma. The results of association studies showing an association of TIM1, atopy, and asthma in the context of HAV infection (13) also suggest an important interaction between genotype (TIM1 polymorphisms) and the environment (HAV infection) in regulating the development of the atopic phenotype. This idea that the environment interacts intimately with genotype is now routinely applied in most genetic studies of atopy (30, 34, 37-40).
TIM-1 in autoimmunity
Human TIM1 has been associated not only with allergy and asthma but also with autoimmune diseases, suggesting that TIM1 regulates the immune system more globally. For example, rheumatoid arthritis is associated with polymorphisms in exon 4 (mucin) (5509_5511delCAA) of TIM1 (40), and C-reactive protein or rheumatoid factor levels in patients with rheumatoid arthritis are associated with polymorphisms in the promoter region of TIM1 (41). How TIM1 regulates autoimmune disease is not known, nor is it known whether HAV infection is associated with protection from autoimmunity. Moreover, TIM1 mRNA is expressed in the cerebrospinal fluid mononuclear cells of patients with multiple sclerosis (MS), primarily in patients in remission rather than in patients in relapse, suggesting that TIM1 regulates the development of MS, possibly as a beneficial element, perhaps associated with tolerance (42).
TIM-1: expression and function
TIM-1 expression
In mice, TIM-1 is expressed on activated but not naive CD4+ T cells (1, 14). Following differentiation, TIM-1 is preferentially expressed on Th2 cells, while Th1 and Th17 cells express little or no TIM-1 (14, 43, 44). TIM-1 is expressed on mast cells (45) and at low levels on a subpopulation of B cells (46), and mRNA for TIM-1 has been shown to be present in invariant naturak killer T (iNKT) cells (42). TIM-1 is expressed on tubular epithelial cells following kidney injury (47).
TIM-1 ligands: phosphatidylserine and others
We and others have shown that both human and mouse TIM-1 are receptors for PtdSer, based on binding studies, and that TIM-1 expressing cells bound and/or engulfed apoptotic cells expressing PtdSer (8, 17, 18, 48). Before PtdSer was recognized as a TIM-1 ligand, a number of protein ligands had been proposed for TIM-1 including TIM-4 (43), TIM-1 (20, 49), TIM-3 (49), oxidized low density lipoprotein (48), and immunoglobulin α heavy chain (50). However, binding studies with highly purified proteins failed to detect a direct interaction between TIM-1 and TIM-4 (46). Miyanishi et al. (18) have shown that TIM-1 and TIM-4 can bind to separate sites on the surface of an exosome and this bridge can give the appearance of an interaction (modeled in Fig. 5). Exosomes are nanovesicles secreted by many cell types and are derived from intracellular structures of the endosomal pathway called multivesicular bodies. Exosomes expose PtdSer at their outer leaflet (51) and contain various cellular proteins, often including galectins (52). There is growing evidence that exosomes participate in cell communications and immune responses, and exosomes also appear to play an important role in tumor growth and host-tumor relationships (53). TIM proteins might serve as one of the receptors for exosomes via PtdSer.
Fig. 5. A model of TIM-1 on a T cell interacting with PtdSer.

IM-1 can interact directly with PtdSer on an apoptotic cell or alternatively, TIM-1 and TIM-4 molecules can interact with PtdSer on an exosome, forming a bridge. An exosome might bridge any two TIM proteins except TIM-2.
Mouse TIM-1 fusion protein was reported to bind specifically to cells overexpressing mTIM-4 (43); however, Wilker et al. (49) found that mTIM-1, mTIM-3, and mTIM-4 fusion proteins all bound to cells overexpressing mTIM-1, mTIM-3, and mTIM-4. Wilker interpreted these results as homotypic TIM interactions, but we believe that these interactions are a consequence of two TIM proteins binding to separate PtdSer molecules on the surface of an exosome or membrane fragment and thus being bridged. Whether these bridging interactions occur on natural cells expressing normal levels of TIM protein and have a role in the natural function of TIMs remains to be determined.
Costimulatory role of TIM-1
Cross-linking of TIM-1 on T cells with an agonist monoclonal antibody (mAb) provides a potent costimulatory signal to CD4+ T cells. TIM-1 crosslinking increases naive T-cell proliferation and cytokine production and enhanced IL-4 production by differentiated Th2 cells (14). TIM-1 also plays an important costimulatory role in vivo, since in vivo administration of agonist TIM-1 mAb 3B3 together with antigen enhanced antigen-specific T-cell responses (14), and administration of TIM-1 mAb during vaccination with influenza provided a potent adjuvant effect, enhancing the viral-specific immune response (54). Moreover, agonistic TIM-1 mAb blocked the development of respiratory tolerance (14), consistent with the idea that TIM-1 costimulation activates T cells.
Role in asthma
The Tim gene family was identified using a congenic mouse model in which polymorphisms in both TIM-1 and TIM-3 were associated with Th1-Th2 differentiation and airway hyperreactivity (AHR) between BALB/c mice and congenic HBA mice (1). Studies using TIM-1 mAbs have supported this association in that blockade of TIM-1 reduced allergen induced airway inflammation (46, 55). Interestingly, TIM-1 mAbs recognizing distinct epitopes of TIM-1 had profoundly different effects on Th1-Th2 cytokine production and airway inflammation. Thus for example, in a mouse model of asthma, TIM-1 mAbs recognizing exon 4 of the mucin domain greatly exacerbated airway inflammation and Th2 cytokine production, but another mAb reactive with the TIM-1 IgV domain blocked inflammation (46).
TIM-1 association with transplant tolerance and Tregs
The role of TIM-1 in tolerance and Treg cell development was further elucidated in transplant model studies, which noted the opposing effects of different TIM-1 mAbs on Treg generation and survival. In one study, in vivo administration of agonist TIM-1 mAb 3B3 overcame the protective effects of anti-CD154 mAb and resulted in allograft rejection (56). In this study, TIM-1 mAb was found to reduce Foxp3 expression and inhibit Treg cell development, while enhancing development of Th17 and Th1 responses (56). In contrast, an independent study found that treatment with a different TIM-1 mAb, RMT1-10, inhibited rejection of MHC-mismatched mouse cardiac allografts. Prolongation of graft survival was associated with inhibition of alloreactive Th1 responses, enhancement of Th2- type responses, and preservation of CD4+CD25+ Tregs (57). Moreover, administration of TIM-1 mAb RMT1-10 specifically inhibited IL-17-producing CD8+ T cells that mediated resistance to tolerance induction (58).
The mechanisms that determine the differential outcome of TIM-1 signaling by different TIM-1 mAbs are not understood. While TIM-1 mAbs recognizing different domains of the TIM-1 molecule have distinct functional effects, it is also possible that the strength of the signal provided by TIM-1 engagement, for example by mAbs with different affinities for TIM-1, may induce different outcomes. Thus, administration of the agonistic high affinity TIM-1 mAb 3B3 during the induction of autoimmunity enhanced pathogenic Th1 and Th17 responses and increased the severity of experimental autoimmune encephalomyelitis (EAE), whereas a lower affinity mAb, RMT1-10, increased Th2 responses and inhibited the development of EAE (59). While both antibodies bind to the TIM-1 IgV domain and induce CD3 capping, mAb 3B3 has a greater affinity and induced cytoskeletal reorganization (59). However, the differences in effects of these mAbs may also reflect their ability to block the interaction of TIM-1 with PtdSer on apoptotic cells. While mAb 3B3 is very effective at blocking the interaction of TIM-1 with PtdSer, RMT1-10 demonstrates little or no blocking capacity (R. DeKruyff, unpublished observations).
TIM-1 signaling
The molecular signal transduction mechanisms by which TIM-1 costimulates T-cell activation are beginning to be identified. Reporter assays showed that overexpression of TIM-1 in T cells results in an increase in production of IL-4 but not IFN-γ (60). Overexpression of TIM-1 resulted in increased transcription from the IL-4 promoter and activation of nuclear factor for activation of T cells (NFAT)/activator protein 1 (AP1) elements, and this was dependent on the presence of a conserved tyrosine (Y276) in the cytoplasmic tail of TIM-1 (60), suggesting that TIM-1 preferentially enhances Th2 cytokine production, consistent with the preferential expression of TIM-1 on Th2 cells. In studies utilizing Jurkat T cells that expressed TIM-1, TIM-1 was shown to colocalize on the T-cell surface with CD3 and was recruited to the T-cell receptor (TCR) signaling complex. TIM-1 cross-linking with antibody caused rapid tyrosine phosphorylation of TIM-1 as well as phosphorylation of ζ-associated protein of 70 kDa (Zap70) and IL-2-induced T-cell kinase (ITK) (61). Further insight was provided by a study that showed that the p85 subunit of phosphoinositide 3-kinase (PI3K) is recruited directly to tyrosine 276 of TIM-1 after Lck-dependent phosphorylation of the cytoplasmic tail (62).
TIM-1 function in kidney
TIM-1 is not detectable in normal kidney but is highly upregulated on kidney tubular epithelial cells following injury. In this venue, TIM-1 [also known as kidney injury molecule-1 (KIM-1)] mediates phagocytosis of apoptotic cells of the injured renal tubule and plays an important role in tissue homeostasis by facilitating clearance of apoptotic and necrotic cells of the injured tubule (48). Membrane-proximal cleavage of TIM-1 by a metalloproteinase results in shedding of a soluble form of TIM-1 in the kidney (63). We speculate that soluble TIM-1 might also bind to PtdSer in the injured kidney and inhibit coagulation. The soluble form is excreted in the urine and is a useful marker of kidney injury (64). TIM-1 is expressed on a substantial proportion of renal cell carcinomas (65, 66). TIM-1 is also expressed on monkey COS cells (26), a kidney cell line used in many transient transfection studies, and contributes to aggregation of these cells as they apoptose. TIM-1 is not expressed on 293 cells, a human kidney cell line (63).
TIM-3
TIM-3 was originally identified as a mouse Th1-specific cell surface protein that was expressed after several rounds of in vitro Th1 differentiation (67, 68) and was later shown to also be expressed on Th17 cells (44). In humans, TIM-3 is expressed on a subset of activated CD4+ T cells, on differentiated Th1 cells, on CD8+ T cells, and at lower levels on Th17 cells (69). TIM-3 is also expressed on cells of the innate immune system including mouse mast cells (45), subpopulations of macrophages and DCs (70), NK and NKT cells (42), and human monocytes (70), and on murine primary bronchial epithelial cell lines (9). TIM-3 expression is regulated by the transcription factor T-bet, since mice deficient in T-bet show reduced levels of TIM-3 expression (71).
TIM-3 ligands: galectin-9 and phosphatidylserine
Galectin-9
Zhu et al. (15) showed that galectin-9 bound to a carbohydrate structure on the IgV domain of mouse TIM-3, which has two N-linked glycosylation sites. Galectin-9 is an S-type lectin with two distinct carbohydrate recognition domains joined by a long flexible linker. While the exact structure of the target carbohydrates recognized by galectin-9 is unclear, galectin-9 has an enhanced affinity for larger poly-N-acetyllactosamine-containing structures (72). Galectin-9 does not have a signal sequence and is localized in the cytoplasm. However, it is secreted through poorly understood mechanisms and exerts its function by binding to glycoproteins on the target cell surface via their carbohydrate chains. Galectins have been linked to regulation of immune homeostasis and inflammation (73); however, the ligands for many of the galectins in the immune system are not known.
Galectin-9 is expressed broadly including in immune cells and the epithelium of the gastrointestinal tract. Galectin-9 expression is particularly high in mast cells and also found in T cells, B cells, macrophages, endothelial cells, and fibroblasts. Galectin-9 production is upregulated by IFN-γ, the major cytokine produced by Th1 cells, and thus may be part of a negative feedback loop leading to the death of TIM-3+ Th1 cells (70). Because galectin binding is relatively promiscuous, it is likely that galectin-9 has multiple target molecules. TIM-3 deficiency only reduces galectin-9-mediated Th1 death by about 40% (15), suggesting that some of the effects of in vivo administration of galectin-9 may be mediated by galectin-9 binding to receptors other than TIM-3. Galectin-9 has also been reported to exert various biological functions via interaction with CD44 (74) and IgE (75).
Engagement of TIM-3 by galectin-9 leads to Th1 cell death and a consequent decline in IFN-γ production (15). When given in vivo, galectin-9 had beneficial effects in several Th1-biased murine disease models. When administered during in vivo sensitization in an EAE model, galectin-9 was found to selectively reduce the number of IFN-γ producing cells and ameliorate disease severity (15). In a mouse model of arthritis, galectin-9 inhibited the generation of Th17 cells and suppressed collagen-induced arthritis (76). In transplant models, galectin-9 administration prolonged survival of fully mismatched cardiac allografts (77) and fully allogeneic skin grafts (78). In another study, galectin-9 inhibited contact hypersensitivity and psoriatic reactions. Galectin-9-treated mice had reduced numbers of IFN-γ and IL-17-producing T cells, suggesting a therapeutic role for galectin-9 in Th1- and/or Th17-mediated skin inflammation (79).
Recognition of PtdSer by TIM-3
We and others recently showed that both human and mouse TIM-3 are receptors for PtdSer, based on binding studies, mutagenesis, and a co-crystal structure, and that TIM-3-expressing cells bound and/or engulfed apoptotic cells expressing PtdSer (9, 16). TIM-3 has the shortest and least glycosylated mucin domain of the TIMs. The mucin domain is necessary to extend the ligand-binding IgV domain from the cell surface as tethering of the IgV domain near the cell surface by deletion of the mucin domain eliminated ligand binding (49). Ig fusion proteins containing only the IgV domain are able to bind ligand (9, 15, 19, 49). An interaction of TIM-3 with PtdSer does not exclude an interaction with Galectin-9 as the binding sites are on opposite sides of the IgV domain (Fig. 6).
Fig. 6. PtdSer and galectin-9 binding regions in the mTIM-3 IgV domain.
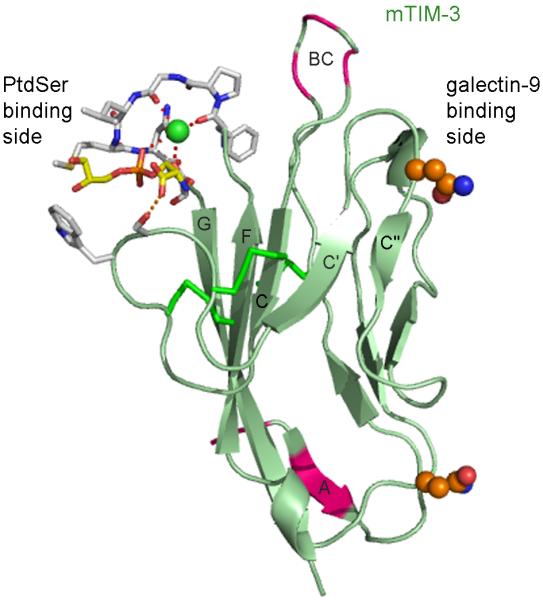
Ribbon diagram of the mTIM-3 IgV domain, with β-strands of the GFC β-sheet labeled. The opposite sides of the IgV domain engaged in PtdSer or galectin-9 binding are indicated. The residues at the FG and CC’ loops interacting with PtdSer and coordinating the metal ion (green sphere) are shown with stick drawing and with the same color coding as in Fig. 4A. The side chains of two Asn residues which can be N-glycosylated are shown as spheres and with carbons in orange, nitrogen in blue, and oxygen in red. The complex carbohydrate modifications would extend outwards from these residues. Galectin-9 can bind to some specific motif of these N-linked carbohydrates. Residues polymorphic between BALB/c and HBA in the β-strand A and the BC loop have been colored pink.
TIM-3 recognition of PtdSer was shown to be important for clearance of apoptotic cells in vivo, since mice treated with TIM-3 mAb had increased numbers of apoptotic cells in splenic follicles and increased serum levels of anti-dsDNA antibodies (16). This study also showed that TIM-3-expressing CD8+ DCs mediated phagocytosis of apoptotic cells and cross-presentation of apoptotic cell-associated antigen to CD8+ T cells (Fig. 7). This was inhibited by treatment with a TIM-3 mAb that blocked PtdSer binding, indicating an important role for TIM-3 recognition of PtdSer in this process.
Fig. 7. Model of phagocytosis of apoptotic cells by TIM-3 or TIM-4+ APCs and antigen cross-presentation.

APCs phagocytose apoptotic cells via TIM-3 or TIM-4 recognition of PtdSer. The apoptotic cells are transported to an acidic phagolysosome, and antigen is processed and presented to T cells. We believe the outcome of phagocytosis may be different for TIM-3 or TIM-4 cells, as TIM-3 has a tyrosine kinase signaling motif in the cytoplasmic domain but TIM-4 does not. Engagement of TIM-3 on APCs, particularly in conjunction with TLR stimulation, may enhance inflammatory responses by inducing inflammatory cytokine production (70).
While TIM-3+ APCs such as DCs or transfected fibroblasts phagocytized apoptotic cells, T cells expressing TIM-3 formed conjugates with but did not engulf apoptotic cells (9). Lymphocytes may lack some cellular machinery required for engulfment or alternatively have an active ‘do not eat’ receptor system (80). Cross-linking of TIM-3 on T cells by apoptotic cells may instead provide a pro-apoptotic signal to the T cell, as is induced by binding of TIM-3 on Th1 or Th17 cells by galectin-9 (15, 44). Alternatively, low levels of cross-linking may induce cytokine production (70). Indeed the interactions may synergize to crosslink TIM-3 and several possible interactions on a T-cell surface are modelled in Fig. 8.
Fig. 8. Model of TIM-3 on a T cell interacting with PtdSer and/or Galectin-9.

TIM-3 can interact directly with PtdSer on an apoptotic cell, or two TIM-3 molecules can be cross-linked by Galectin-9. TIM-3 may also interact with PtdSer on an exosome. Galectin-9 may also link TIM-3 to an N-linked glycan on another protein.
Differential recognition of PtdSer by TIM-3 polymorphic variants
Allelic variants of TIM-3 demonstrated distinct PtdSer binding affinities and phagocytic capacities. The BALB/c allele of TIM-3 bound PtdSer and mediated phagocytosis of apoptotic cells more efficiently than the HBA allele of TIM-3 (9). Polymorphisms in the BC loop of the IgV domain, which likely interact with the surface of the lipid bilayer, are responsible for these differences (Figs 4C and 6). The HBA TIM alleles originated from a DBA/2 ancestor (81) and are identical to C57BL/6 alleles. This is the first observation of a functional difference between the BALB/c and HBA alleles of TIM-3 or TIM-1 and suggests that functional differences in the TIM-3 alleles may contribute to the Th1/Th2 phenotypic differences between the strains.
TIM-3: negative regulator of T-cell responses
Several lines of evidence suggest that TIM-3 expressed on T cells functions as a co-inhibitory molecule. Administration of TIM-3 mAb (8B.2C12) during induction of EAE worsened disease progression and led to higher numbers of activated macrophages (67). Administration of TIM-3 Ig fusion protein during an immune response resulted in hyperproliferation of Th1 cells and spontaneous production of IFN-γ (82). Moreover, administration of either TIM-3 Ig fusion protein or TIM-3 mAb (8H7) to nonobese diabetic (NOD) mice accelerated the onset of diabetes (68). Identification of galectin-9 as a ligand of TIM-3 that induced death of Th1 cells and downregulated Th1 responses provided further support for TIM-3 as a negative regulator of Th1 responses (15).
TIM-3 and T-cell exhaustion
Two recent studies have implicated TIM-3 in mediating T-cell dysfunction associated with chronic viral infections (83, 84). Virus-specific T cells in chronic viral infections such as human immunodeficiency virus (HIV) and hepatitis C virus (HCV) progressively develop a range of functional impairments in cytokine production, cytotoxic activity, and proliferative capacity, termed ‘exhaustion’ (85). In progressive HIV infection, TIM-3 was expressed on about 50% of CD8+ T cells compared to 28% in healthy controls. TIM-3 expression on CD4+ T cells was elevated to a lesser extent. TIM-3 was expressed on virus-specific CD8+ T cells, and the TIM-3+ subset only partially overlapped with the PD-1+ subset. Higher levels of TIM-3 correlated positively with viral load and inversely with CD4+ T-cell counts. Virus-specific CD8+ T cells expressing high levels of TIM-3 proliferated poorly and produced much less cytokine than TIM-3-negative T cells. Targeting the TIM-3 protein with a mAb strongly enhanced cytokine production and proliferation. A TIM-3-Ig had a similar but less potent effect (84).
In chronic HCV infection, TIM-3 expression was increased on both CD4+ and CD8+ T cells and substantially overlapped with the PD-1 population. A high proportion of the TIM-3+ CD8+ population had the phenotype of central memory CD8+ T cells, while a lesser proportion had the phenotype of effector memory cells. TIM-3 expression on CD4+ T cells did not show this division. TIM-3+ CD4+ and CD8+ T cells produced less IFN-γ and TNF-α than TIM-3− cells. Treatment with TIM-3 mAb strongly enhanced T-cell proliferation and IFN- γ production and decreased IL-10 production in response to HCV peptide antigen (83). These results indicate that TIM-3 is upregulated during the course of T-cell activation and serves as a negative regulator to limit immunopathology. Therapies targeting coinhibitory molecules like TIM-3 and programmed death-1 (PD-1) are promising therapies for chronic infections.
Tumor-infiltrating TCRαβ+CD8 cells of reduced cytotoxic potential have been shown to promote chemical cutaneous carcinogenesis and are associated with malignant progression. These tumor-promoting T cells express elevated levels of TIM-3 and IL-17 and may represent a type of exhausted T cell, which can promote tumorigenesis through pro-inflammatory cytokines and inflammation (86).
TIM-3 and tolerance
TIM-3 has been shown to play an important role in peripheral tolerance in several models. In one model, long-lasting tolerance to allogeneic grafts is achieved by donor-specific transfusion (DST) and CD154 (CD40L) mAb treatment, and tolerance is mediated by enhanced donor-specific Treg suppression. Concurrent administration of TIM-3 Ig interfered with generation of functional Tregs and prevented the induction of tolerance to transplanted islets (68). Moreover, TIM-3-deficient mice were refractory to tolerance induction with DST and anti-CD154, and rapidly rejected islet allografts (68). Further support for an important role for TIM-3 in tolerance induction was shown using a different model, in which peripheral tolerance is induced by giving high doses of aqueous antigen prior to immunization. While T cells from wildtype mice were tolerized and hyporesponsive to antigen after administration of aqueous antigen, TIM-3-deficient mice were refractory to induction of tolerance in this model (82). However, the mechanisms and relative roles of TIM-3-expressing T cells or APCs that mediate tolerance induction remain to be elucidated.
TIM-3 and innate immune responses
While engagement of TIM-3 on T cells appears to downregulate T-cell responses, TIM-3 expressed on cells of the innate immune system may have a different role in immunoregulation. Recent studies indicate that TIM-3 expressed on DCs or macrophages enhances inflammatory responses, particularly in conjunction with TLR signaling (70). Thus, stimulation of splenic DCs from wildtype mice but not TIM-3-deficient mice with galectin-9 in the presence of lipopolysaccharide greatly enhanced TNF-α production. Similarly, galectin-9-mediated TNF-α production by human monocytes was blocked by TIM-3 mAb. Stimulation of the murine DC cell line D2SC1 with agonistic TIM-3 mAb led to induction of NF-κB, indicating the induction of an inflammatory transcription factor cascade. Moreover, cross-linking TIM-3 on mast cells with anti-TIM-3 enhances production of Th2 cytokines IL-4, IL-6, and IL-13 (45), which exacerbate airway inflammation.
Recent studies have shown that TIM-3 surface expression on cells of the innate immune system is upregulated following exposure to certain inflammatory stimuli. Thus, TIM-3 is undetectable on resident peritoneal macrophages, but inflammatory peritoneal exudate macrophages elicited by injection of thioglycollate express TIM-3 (16). TIM-3 surface expression is upregulated on granulocyte-macrophage colony-stimulating factor (GMCSF)-derived bone marrow-derived DCs (BMDCs) following exposure to cholera toxin (9). In a model of coxsackie virus-induced myocarditis, upregulation of TIM-3 surface expression occurred rapidly on mast cells and macrophages obtained from the peritoneum, spleen, and heart following intraperitoneal infection of BALB/c mice with virus. Administration of TIM-3 mAb at the time of infection increased cardiac inflammation, indicating a role for TIM-3 in reducing inflammation in this model (87). Together, these data suggest a circuit that promotes inflammatory responses in which TIM-3 expression on macrophages/DCs is upregulated by exposure to pathogens, and subsequent crosslinking of TIM-3 induces production of proinflammatory cytokines such as TNF-α.
TIM-3 and EAE/MS
The role of TIM-3 in modulation of autoimmune disease has been most studied in EAE, a mouse model of MS. MS pathogenesis is associated with dysregulated Th1 responses and production of the proinflammatory cytokines IFN-γ and TNF-α In vivo treatment with TIM-3 mAb during the induction of EAE accelerated disease progression and resulted in a severe form of demyelinating disease characterized by higher numbers of inflammatory foci in the central nervous system (CNS) and higher numbers of activated macrophages in demyelinating lesions (67). In contrast, treatment with the TIM-3 ligand galectin-9 was found to selectively reduce the number of IFN-γ-producing cells and ameliorate disease severity (15), suggesting a role for the TIM-3/galectin-9 pathway in regulating the pathogenic Th1 response.
In human patients with MS, T-cell clones derived from the cerebrospinal fluid produced higher levels of IFN-γ and expressed lower levels of TIM-3 compared with those from healthy control subjects (88). Treatment of normal CD4+ T cells or Th1 cells with TIM-3 small interfering RNA reduced TIM-3 expression and resulted in increased proliferation and production of IFN-γ. These results are compatible with the observation that high level expression of TIM-3 in exhausted T cells in chronic viral infections leads to less T-cell activation. Low level expression of TIM-3 in MS may indicate these cells are refractory to inhibition, thereby causing immunopathology.
In addition to the part played by Th1 cells in mediating CNS pathogenesis, resident microglia, and infiltrating monocytes contribute to CNS inflammation (89), and these cells have been shown to express TIM-3. Thus, in mice immunized for the induction of EAE, TIM-3 was upregulated on CD11b+ monocytes infiltrating the CNS and on resident microglia although TIM-3 was not expressed on activated peripheral CD11b+ cells (70). In humans, TIM-3 was found to be expressed on CD11b+ microglia in CNS white matter of normal subjects, and TIM-3 mRNA levels were higher in cells from the active border regions of MS lesions in comparison with non-inflamed tissue. In these studies, elevated expression levels of galectin-9 in astrocytes present in MS lesions relative to normal human CNS tissue suggested an immunoregulatory circuit that could terminate detrimental Th1 responses by TIM-3-expressing cells (70). However, whether TIM-3-expressing microglial cells ameliorate or exacerbate disease is not clear. Although galectin-9 may terminate Th1 responses, galectin-9 stimulates TNF-α production by human TIM-3-expressing monocytes, which could further enhance inflammatory responses. However, microglia have been suggested to regulate immunity by phagocytosing apoptotic cells and cross presenting antigen in tolerogenic pathways (90), which may be mediated in part by TIM-3 (16). Thus, the role of TIM-3 in the pathogenesis of this disease is complex and the regulatory mechanisms are not yet completely understood. Considering that myelin is a membrane with the normal compartmentalization of PtdSer (91), future studies on MS will need to clarify the role of TIM-3+ microglial cells in the phagocytosis of apoptotic myelin and cross-presentation of myelin antigens to T cells.
TIM-3 and asthma
While treatment with TIM-3 mAb exacerbated the development of EAE, TIM-3 mAb treatment was found to reduce allergen-induced AHR and airway inflammation following transfer of ovalbumin-reactive polarized Th2 cells (92). Treatment with TIM-3 mAb skewed the response to a Th1 phenotype, as the numbers of IFN-γ producing cells increased in the lung and IL-5-producing cells decreased (92).
TIM-3 signaling
Tyrosine residues in the cytoplasmic domain of TIM-3 are conserved in mouse and human and are clustered in proximal and distal regions of the cytoplasmic tail. Phosphorylation of Y235 and Y242 residues of TIM-3 appears to be responsible for potentiation of signal transduction initiated by TCR activation, upstream of NFAT/AP1 pathways, in a redundant manner, since mutation of both Y235 and Y242 but not single point mutation of either Y235 or Y242 decreased reporter activity to basal levels (93). TIM-3 appears to signal differently in DCs and T cells since ligation of TIM-3 with TIM-3 mAb resulted in different patterns of phosphorylation in these cell types (70).
TIM-4
TIM-4 was identified in the positional cloning of the TIM family and independently as SMUCKLER (spleen, mucin-containing, knockout of lymphotoxin), a gene selectively downregulated in spleens of LTα– or LTβ–deficient mice (94). In contrast to TIM-1 and TIM-3, TIM-4 is not expressed on T cells but is expressed exclusively on APCs (17, 43). In humans, TIM-4 is expressed by tingible-body macrophages located in germinal centers of tonsil and in the white pulp of the spleen (17). In mice, resident F4/80+ peritoneal macrophages express TIM-4 but not TIM-3 or MFG-E8 (9, 16-18). In contrast, thioglycollate-elicited peritoneal macrophages lose TIM-4 expression and gain expression of TIM-3 and MFG-E8 (16, 18). These results show that APCs express different sets of PtdSer receptors under different inflammatory conditions. TIM-4 is also expressed on CD169+ (MOMA-1) marginal zone metallophilic macrophages in the spleen (94, 95) and in moderate abundance by CD8α+ and CD8α− subsets of CD11c+ DCs (17, 43) but not on plasmacytoid DCs (17). TIM-4 lacks a tyrosine phosphorylation motif in its intracellular domain.
TIM-4 as a phosphatidylserine receptor
We and others have shown that both human and mouse TIM-4 are receptors for PtdSer, based on binding and mutagenesis studies and a co-crystal structure and that TIM-4-expressing cells bound and engulfed apoptotic cells expressing PtdSer (8, 17, 18). TIM-1 has also been proposed as a TIM-4 ligand but this result likely reflects bridging across an exosome as discussed in the TIM-1 section (Fig. 5).
TIM-4 and in vivo immunoregulation
Identification of TIM-4 as a receptor for phosphatidylserine prompted study of the in vivo role of TIM-4 in processing of apoptotic cells and the development of autoimmunity. Mice treated with dexamethasone to induce apoptosis in the thymus and given a TIM-4 mAb that blocks binding of PtdSer were found to develop auto-antibodies (anti-cardiolipin and anti-dsDNA antibodies) (18), demonstrating an important role for TIM-4 recognition of PtdSer in vivo as a mediator of tolerance.
The role of TIM-4 in regulating immune responses was investigated in several other models. Several studies suggest that TIM-4 may play a role in maintaining oral tolerance and prevention of food allergy. TIM-4 was upregulated in vivo on murine intestinal mucosal DCs (96) following exposure to Staphylococcus enterotoxin B (SEB). In vivo exposure to SEB and OVA resulted in Th2 responses and intestinal allergic responses which were inhibited in mice pretreated with TIM-4 or TIM-1 antibody (96), Another study showed that TIM-4 expression was enhanced on CD11c+ DCs following food antigen sensitization in the presence of cholera toxin (CT), and treatment of mice with TIM-1 antibody inhibited the allergic response to food antigen.
In one study, T cells from mice given a dual TIM-1/TIM-4 mAb at the time of antigen immunization demonstrated enhanced in vitro antigen-specific T-cell proliferation and cytokine production compared with control mice. Treatment with this mAb inhibited in vivo inflammatory responses and resulted in a significant reduction in ear swelling in a model of contact hypersensitivity and substantially ameliorated disease in an EAE model, indicating a potential role for TIM-4 in regulating inflammatory responses (95). However, these studies are somewhat difficult to interpret, since the mAb utilized in these studies cross-reacted with both TIM-4 and TIM-1 (95).
Functional studies with TIM-Ig
Administration of either TIM-4-Ig or TIM-1-Ig fusion protein in vivo with antigen increased basal T-cell proliferation and cytokine production (43). The direct effect of TIM-4 on T-cell responses was analyzed by stimulating T cells with magnetic beads coated with anti-CD3, anti-CD28, and TIM-4 Ig or isotype control. Naive T cells stimulated with beads containing TIM-4 Ig demonstrated dramatically higher levels of proliferation and higher levels of cytokine production than those stimulated with control beads (97). Moreover, stimulation with TIM-4-Ig/anti-CD3/anti-CD28 beads but not control beads induced phosphorylation of endogenously expressed TIM-1 and higher levels of ERK phosphorylation and BCl-2 expression than stimulation in the presence of control beads (97). In an independent study, the opposite effect was found with TIM-4-Ig, which was found to inhibit proliferation of naive T cells stimulated with antigen or anti-CD3 and anti-CD28, although TIM-4-Ig did not affect proliferation of activated T cells (95). We believe studies using TIM-4-Ig to engage T cells in vitro probably do not reflect the natural function of TIM-4 in phagocytosis. Similarly, TIM-Ig fusion proteins in vivo will bind to PtdSer on apoptotic cells and delay the processing of apoptotic cells, an event known to affect immune responses (98); however, this global effect of systemic administration may not reflect the natural function.
Mechanisms of TIM-mediated engulfment
The mechanism of TIM-mediated engulfment appears to be novel and involve new pathways. TIM-1 concentrates at the point of binding of an apoptotic cell on the cell surface and is part of the initial phagocytic cup. Phagocytosis of apoptotic cells is inhibited by cytochalasin D and nocodazole, inhibitors of actin filament polymerization and microtubule formation; however, binding is unaffected (48). Similarly, TIM-4-mediated engulfment is dependent on the actin cytoskeleton, myosin-II motor proteins, and ATP-dependent cellular processes (99). TIM-4-mediated engulfment did not involve ELMO1/Dock180/Rac or GULP, both well characterized engulfment signaling pathways. Surprisingly, engulfment did not require the TIM-4 transmembrane or cytoplasmic domains, suggesting that the TIM-4 extracellular domain associates with another membrane protein that engages a novel engulfment signaling pathway (99).
TIM-2 stands alone
TIM-2 is an outlier in the TIM family. The sequence of murine TIM-2 is highly homologous to murine TIM-1 but differs in that it lacks critical residues in the MILIBS (AsnAsp) that coordinate with metal ions and PtdSer (8, 20). This leads to a relatively flat surface in this region (Figs 3 and 9), and consequently, TIM-2 does not bind PtdSer (17, 18). Crystal and solution studies show that TIM-2 IgV domains form non-covalent dimers (20), exposing a large glycan-free surface on the top of the dimer for interaction with ligands (Fig. 9). The TIM-2 gene is located with other family members on mouse chromosome 11 within the TAPR locus; however, there is no structural orthologue of TIM-2 in humans (1, 7). TIM-2 appears to have arisen by gene duplication of TIM-1 and is present only in rodents (J. McIntire and G. Freeman, unpublished observations). Unlike TIM-1, TIM-3, and TIM-4, there are no polymorphic differences in TIM-2 between BALB/c and HBA strains used to identify the TAPR locus, suggesting that TIM-2 did not account for the phenotypic differences between BALB/c and HBA strains (1).
Fig. 9. The dimeric conformation of mTIM-2.
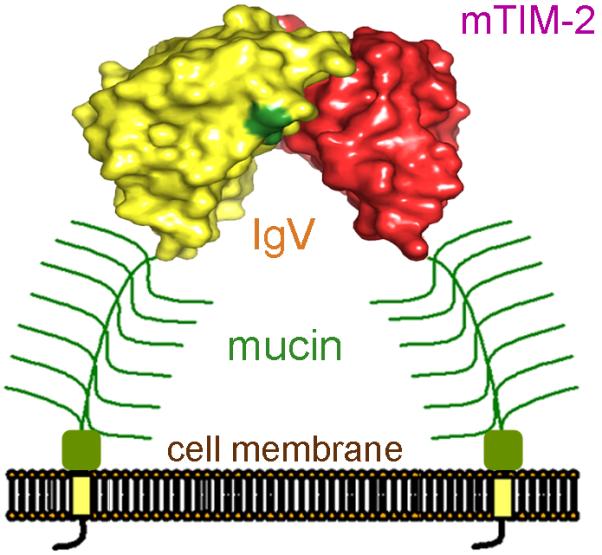
Representation of a dimeric mTIM-2 on the cell surface. The dimeric mTIM-2 IgV domain structure (PDB 2OR7) is shown with surface representation. One domain is in red and the other in yellow, with the location of the N-linked glycans marked in dark green. The glycosylated mucin domain that follows the IgV domain is shown in green.
TIM-2 ligands
TIM-2 is a receptor for H-ferritin (100), which has been reported to have immunoregulatory properties. Binding of H-ferritin to TIM-2 leads to the endocytosis of extracellular H-ferritin (100) and iron transfer. In this sense, TIM-2 has retained a ‘ligand binding and transfer across a membrane’ function common to other members of the TIM family. Human TIM-1, TIM-3, and TIM-4 do not bind H-Ferritin (V. Bailly and P. Rennert, unpublished observations), so this activity appears to be found only in rodents. Sema4A was identified as a ligand of TIM-2 by expression cloning using Sema4A Ig fusion protein (101, 102); however, other laboratories have not been able to confirm this interaction (49, 100, G. Freeman, Y.M. Hsu, and P. Rennert, unpublished observations), and further work is needed. Sema4A-Fc enhanced activation of CD4+ T cells in vitro, and mice treated with anti-Sema4A mAb had severely impaired antigen-specific responses, suggesting that Sema4A functioned as a positive T-cell costimulatory molecule. Sema4A-deficient mice demonstrate exaggerated in vivo Th2 responses, which are suggestive of the enhanced Th2 responses of TIM-2 deficient mice. However, TIM-2-deficient T cells produce enhanced levels of IFN-γ as well as Th2 cytokines while Sema4A-deficient T cells demonstrate an inability to differentiate into IFN-γ producing cells, suggesting that Sema4A and/or TIM-2 have other binding partners (102).
TIM-2 expression
mRNA analysis has shown TIM-2 expression by activated T cells, particularly following differentiation under Th2 conditions (103, 104), and in splenic B cells (104). T cells differentiated under Th2 but not Th1 conditions express cell surface TIM-2 (104), and germinal center B cells have also been reported to have high surface expression levels of TIM-2 (100).
TIM-2, T-cell activation, and Th2 responses
TIM-2 appears to function as a negative regulator of T-cell activation. Administration of TIM-2-Ig fusion protein to block TIM-2/TIM-2-ligand interaction induced high basal proliferation of splenocytes, with elevated levels of IL-2, IL-4, and IL-10. When given before disease onset in EAE, TIM-2-Ig ameliorated disease progression, suggesting a role for TIM-2 in downregulating Th2 responses (103). Another study showed that lymph node cells from TIM-2-deficient mice produced substantially higher levels of cytokines, including IL-5, IL-6, IL-10, and IFN-γ, and demonstrated higher levels of antigen-specific proliferation following immunization with antigen in comparison with wild type mice. In contrast, T-cell responses of TIM-2-deficient mice to polyclonal stimulation with anti-CD3/anti-CD28 mAb were similar to those of wildtype mice (104). In this study, TIM-2 was found to play an important role in an OVA-induced model of airway inflammation, consistent with its expression on Th2 cells. Thus, upon sensitization and challenge with OVA, TIM-2-deficient mice displayed exacerbated lung inflammation compared with wildtype counterparts (104). Overexpression of TIM-2 in Jurkat or D10 T-cell lines significantly impaired the induction of NFAT/AP1 transcriptional reporters in response to TCR stimulation (105), confirming the negative role of TIM-2 in T-cell activation.
Concluding remarks
The finding that TIM-1, TIM-3, and TIM-4 bind specifically to PtdSer establishes a new paradigm for TIM proteins as PtdSer receptors and unifies the function of the TIM gene family. Apoptotic cell death is a critical and evolutionally conserved process for elimination of unnecessary cells (106, 107), which is essential for maintenance of tissue homeostasis and self-tolerance (12). Clearance of apoptotic cells by phagocytosis can result in powerful anti-inflammatory and immunosuppressive effects (108). The recognition of PtdSer on apoptotic cells by TIM-1, TIM-3, and TIM-4 suggests that this family is critically involved in regulating immune tolerance. The defective clearance of apoptotic cells has been linked closely to autoimmune and inflammatory diseases (109-111). Phagocytes such as macrophages or DCs engulf both apoptotic and necrotic cells, and the biological response of the phagocyte differs depending on the material phagocytosed and concurrent ‘danger’ signals. Necrotic cells induce release of pro-inflammatory cytokines and stimulate upregulation of costimulatory molecules on the DC surface, which in turn induce T-cell activation and an inflammatory response. Infections will lead to increased cell death as well as engage pattern recognition receptors such as Toll-like receptors that stimulate immune responses. In contrast, engulfment of apoptotic cells in the absence of Toll-like receptor signals does not trigger the expression of proinflammatory costimulatory molecules but rather is associated with production of anti-inflammatory cytokines including TGF-β and IL-10, which prevent inappropriate inflammatory responses to self-proteins.
The finding that TIM gene family members TIM-1, TIM-3, and TIM-4, which have distinct patterns of expression on distinct cell types or on cells at specific stages of activation or differentiation, are a family of pattern recognition receptors for PtdSer suggests that the TIM proteins provide a functional repertoire for recognition of apoptotic cells. For example, TIM-1 may bind PtdSer on apoptotic cells and mediate T-cell activation, whereas TIM-3 on T cells may mediate T-cell elimination. TIM-3 and TIM-4 on APCs may mediate apoptotic cell clearance and have roles in antigen cross-presentation and tolerance. Previous work, which identified several receptors mediating apoptotic cell recognition and clearance by phagocytes (112), led to the speculation that the PtdSer receptor repertoire might provide specificity in the phagocyte response. However, receptors such as milk fat globule-EGF-factor 8 (MFG-E8) and growth arrest-specific gene 6 (GAS6) (113) are widely expressed in somatic cells and do not appear to specify phagocyte behavior following phagocytosis of apoptotic cells (12). The unique structure of TIM IgV domains and their distinctive cytoplasmic domains suggest that the TIM molecules evolved as a family of pattern recognition receptors for PtdSer that determine whether apoptotic cell recognition leads to immune activation or tolerance, depending on the TIM molecule engaged and the cell type it is expressed on.
We hypothesize that TIM-1 and TIM-3 reciprocally regulate T-cell responses (Fig. 2). Thus TIM-3 engagement may lead to Th1 death and unopposed Th2 responses whereas TIM-1 signaling would preferentially enhance Th2 responses. The allelic differences of TIM-1 and TIM-3 may together contribute to the Th2 proclivity of BALB/c versus the Th1 bias of the HBA and C57BL/6 strains. Thus, BALB/c TIM-3 binding PtdSer better than HBA TIM-3 may result in increased Th1 death and more vigorous Th2 responses. This model is consistent with studies showing that TIM-1 and TIM-3 do not direct the differentiation of T-helper cells but instead regulate the activities and survival of differentiated T cells.
The role of the TIM genes in asthma, autoimmunity, tolerance, and the response to viral infection suggests that further study of the interactions between PtdSer on apoptotic cells with TIM molecules on APCs or T cells will likely provide novel insights into the pathophysiology of these diseases. Understanding the mechanisms for the differences between TIM alleles may explain how they influence the development of disease and how a specific human TIM-1 allele can protect against the development of asthma. The critical role of these interactions in modulating disease suggests that the TIM molecules will serve as useful therapeutic targets.
Acknowledgements
This work was supported by NIH P01 AI054456 (to GJF, DTU, RDK), NIH HL069507 (RDK) and MICINN grant BFU2008-00971 (JMC).
References
- 1.McIntire JJ, et al. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol. 2001;2:1109–1116. doi: 10.1038/ni739. [DOI] [PubMed] [Google Scholar]
- 2.McIntire JJ, Umetsu DT, DeKruyff RH. TIM-1, a novel allergy and asthma susceptibility gene. Springer Semin Immunopathol. 2004;25:335–348. doi: 10.1007/s00281-003-0141-3. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. The costimulatory role of TIM molecules. Immunol Rev. 2009;229:259–270. doi: 10.1111/j.1600-065X.2009.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su EW, Lin JY, Kane LP. TIM-1 and TIM-3 proteins in immune regulation. Cytokine. 2008;44:9–13. doi: 10.1016/j.cyto.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marsh DG, et al. Linkage analysis of IL4 and other chromosome 5q31.1 markers and total serum immunoglobulin E concentrations. Science. 1994;264:1152–1156. doi: 10.1126/science.8178175. [DOI] [PubMed] [Google Scholar]
- 6.Encinas JA, Kuchroo VK. Mapping and identification of autoimmunity genes. Curr Opin Immunol. 2000;12:691–697. doi: 10.1016/s0952-7915(00)00164-3. [DOI] [PubMed] [Google Scholar]
- 7.Kuchroo V, Umetsu D, DeKruyff R, Freeman G. The TIM gene family: emerging roles in immunity and disease. Nature Rev Immunol. 2003:454–456. doi: 10.1038/nri1111. [DOI] [PubMed] [Google Scholar]
- 8.Santiago C, et al. Structures of T cell immunoglobulin mucin protein 4 show a metal-ion-dependent ligand binding site where phosphatidylserine binds. Immunity. 2007;27:941–951. doi: 10.1016/j.immuni.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeKruyff RH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184:1918–1930. doi: 10.4049/jimmunol.0903059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 11.Verhoven B, Schlegel RA, Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 13.McIntire J, et al. Immunology: hepatitis A virus link to atopic disease. Nature. 2003;425:576. doi: 10.1038/425576a. [DOI] [PubMed] [Google Scholar]
- 14.Umetsu SE, et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. 2005;6:447–454. doi: 10.1038/ni1186. [DOI] [PubMed] [Google Scholar]
- 15.Zhu C, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 16.Nakayama M, et al. Tim-3 mediates phagocytosis of apoptotic cells and the cross-presentation. Blood. 2009;113:3821–3830. doi: 10.1182/blood-2008-10-185884. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi N, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 19.Cao E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. 2007;26:311–321. doi: 10.1016/j.immuni.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 20.Santiago C, Ballesteros A, Tami C, Martinez-Munoz L, Kaplan GG, Casasnovas JM. Structures of T Cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity. 2007;26:299–310. doi: 10.1016/j.immuni.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kostrewa D, et al. X-ray structure of junctional adhesion molecule: structural basis for homophilic adhesion via a novel dimerization motif. EMBO J. 2001;20:4391–4398. doi: 10.1093/emboj/20.16.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Raaij MJ, Chouin E, van der Zandt H, Bergelson JM, Cusack S. Dimeric structure of the coxsackievirus and adenovirus receptor D1 domain at 1.7 A resolution. Structure. 2000;8:1147–1155. doi: 10.1016/s0969-2126(00)00528-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang JH, et al. Structure of a heterophilic adhesion complex between the human CD2 and CD58 (LFA-3) counterreceptors. Cell. 1999;97:791–803. doi: 10.1016/s0092-8674(00)80790-4. [DOI] [PubMed] [Google Scholar]
- 24.Jones EY, Davis SJ, Williams AF, Harlos K, Stuart DI. Crystal structure at 2.8 A resolution of a soluble form of the cell adhesion molecule CD2. Nature. 1992;360:232–239. doi: 10.1038/360232a0. [DOI] [PubMed] [Google Scholar]
- 25.Ahmadi KR, et al. Novel association suggests multiple independent QTLs within chromosome 5q21-33 region control variation in total humans IgE levels. Genes Immun. 2003;4:289–297. doi: 10.1038/sj.gene.6363968. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone SM. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 1996;15:4282–4296. [PMC free article] [PubMed] [Google Scholar]
- 27.Feigelstock D, Thompson P, Mattoo P, Zhang Y, Kaplan GG. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J Virol. 1998;72:6621–6628. doi: 10.1128/jvi.72.8.6621-6628.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matricardi PM, et al. Cross sectional retrospective study of prevalence of atopy among Italian military students with antibodies against hepatitis A virus. BMJ. 1997;314:999–1003. doi: 10.1136/bmj.314.7086.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matricardi PM, Rosmini F, Panetta V, Ferrigno L, Bonini S. Hay fever and asthma in relation to markers of infection in the United States. J Allergy Clin Immunol. 2002;110:381–387. doi: 10.1067/mai.2002.126658. [DOI] [PubMed] [Google Scholar]
- 30.Gao PS, et al. Genetic variants of the T-cell immunoglobulin mucin 1 but not the T-cell immunoglobulin mucin 3 gene are associated with asthma in an African American population. J Allergy Clin Immunol. 2005;115:982–988. doi: 10.1016/j.jaci.2005.01.035. [DOI] [PubMed] [Google Scholar]
- 31.Graves PE, et al. A cluster of five tightly linked polymorphisms in the IL-13 gene is associated with total serum IgE levels in three populations of caucasian children. Am J Resp Crit Care Med. 2000;161:A930. doi: 10.1067/mai.2000.104940. [DOI] [PubMed] [Google Scholar]
- 32.Chae S, Song J, Lee Y, Kim J, Chung H. The association of the exon 4 variations of Tim-1 gene with allergic diseases in a Korean population. Biochem Biophys Res Commun. 2003;12:346–350. doi: 10.1016/j.bbrc.2003.10.125. [DOI] [PubMed] [Google Scholar]
- 33.Wu Q, Hu L, Cai P, Li Y, Chen F, Kong L. Association analysis of TIM-1 -232G > A and 5383_5397 insertion/deletion polymorphisms with childhood asthma and total serum immunoglobulin E levels in middle China. J Investig Allergol Clin Immunol. 2009;19:146–153. [PubMed] [Google Scholar]
- 34.Page NS, Jones G, Stewart GJ. Genetic association studies between the T cell immunoglobulin mucin (TIM) gene locus and childhood atopic dermatitis. Int Arch Allergy Immunol. 2006;141:331–336. doi: 10.1159/000095459. [DOI] [PubMed] [Google Scholar]
- 35.Noguchi E, Nakayama J, Kamioka M, Ichikawa K, Shibasaki M, Arinami T. Insertion/deletion coding polymorphisms in hHAVcr-1 are not associated with atopic asthma in the Japanese population. Genes Immun. 2003;4:170–173. doi: 10.1038/sj.gene.6363935. [DOI] [PubMed] [Google Scholar]
- 36.Li JS, et al. Absence of association between two insertion/deletion coding genetic polymorphisms of TIM-1 gene and asthma in Chinese Han population. Int J Immunogenet. 2006;33:417–422. doi: 10.1111/j.1744-313X.2006.00634.x. [DOI] [PubMed] [Google Scholar]
- 37.Graves PE, Siroux V, Guerra S, Klimecki WT, Martinez FD. Association of atopy and eczema with polymorphisms in T-cell immunoglobulin domain and mucin domain-IL-2-inducible T-cell kinase gene cluster in chromosome 5 q 33. J Allergy Clin Immunol. 2005;116:650–656. doi: 10.1016/j.jaci.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, et al. A functional polymorphism in the TIM-1 gene is associated with asthma in a Chinese Han population. Int Arch Allergy Immunol. 2007;144:197–202. doi: 10.1159/000103992. [DOI] [PubMed] [Google Scholar]
- 39.Zhang CC, Wu JM, Cui TP, Wang P, Pan SX. Study on relationship between polymorphism sites of TIM-3 and allergic asthma in a population of adult Hans from Hubei province of China. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2006;23:74–77. [PubMed] [Google Scholar]
- 40.Chae SC, Song JH, Shim SC, Yoon KS, Chung HT. The exon 4 variations of Tim-1 gene are associated with rheumatoid arthritis in a Korean population. Biochem Biophys Res Commun. 2004;315:971–975. doi: 10.1016/j.bbrc.2004.01.154. [DOI] [PubMed] [Google Scholar]
- 41.Chae SC, Park YR, Song JH, Shim SC, Yoon KS, Chung HT. The polymorphisms of Tim-1 promoter region are associated with rheumatoid arthritis in a Korean population. Immunogenetics. 2005;56:696–701. doi: 10.1007/s00251-004-0743-5. [DOI] [PubMed] [Google Scholar]
- 42.Khademi M, et al. T Cell Ig- and mucin-domain-containing molecule-3 (TIM-3) and TIM-1 molecules are differentially expressed on human Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear cells in multiple sclerosis. J Immunol. 2004;172:7169–7176. doi: 10.4049/jimmunol.172.11.7169. [DOI] [PubMed] [Google Scholar]
- 43.Meyers J, et al. Tim-4 is the ligand for Tim-1, and the Tim-1-Tim-4 interaction regulates T cell expansion. Nature Immunol. 2005;6:455–464. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- 44.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 45.Nakae S, et al. TIM-1 and TIM-3 enhancement of Th2 cytokine production by mast cells. Blood. 2007;110:2565–2568. doi: 10.1182/blood-2006-11-058800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sizing ID, et al. Epitope-dependent effect of anti-murine TIM-1 monoclonal antibodies on T cell activity and lung immune responses. J Immunol. 2007;178:2249–2261. doi: 10.4049/jimmunol.178.4.2249. [DOI] [PubMed] [Google Scholar]
- 47.Ichimura T, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. 1998;273:4135–4142. doi: 10.1074/jbc.273.7.4135. [DOI] [PubMed] [Google Scholar]
- 48.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118:1657–1668. doi: 10.1172/JCI34487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilker PR, Sedy JR, Grigura V, Murphy TL, Murphy KM. Evidence for carbohydrate recognition and homotypic and heterotypic binding by the TIM family. Int Immunol. 2007;19:763–773. doi: 10.1093/intimm/dxm044. [DOI] [PubMed] [Google Scholar]
- 50.Tami C, et al. Immunoglobulin A (IgA) is a natural ligand of hepatitis A virus cellular receptor 1 (HAVCR1), and the association of IgA with HAVCR1 enhances virus-receptor interactions. J Virol. 2007;81:3437–3446. doi: 10.1128/JVI.01585-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zakharova L, Svetlova M, Fomina AF. T cell exosomes induce cholesterol accumulation in human monocytes via phosphatidylserine receptor. J Cell Physiol. 2007;212:174–181. doi: 10.1002/jcp.21013. [DOI] [PubMed] [Google Scholar]
- 52.Klibi J, et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood. 2009;113:1957–1966. doi: 10.1182/blood-2008-02-142596. [DOI] [PubMed] [Google Scholar]
- 53.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 54.Soo Hoo W, et al. Vaccination with cell immunoglobulin mucin-1 antibodies and inactivated influenza enhances vaccine-specific lymphocyte proliferation, interferon-gamma production and cross-strain reactivity. Clin Exp Immunol. 2006;145:123–129. doi: 10.1111/j.1365-2249.2006.03107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Encinas JA, et al. Anti-T-cell Ig and mucin domain-containing protein 1 antibody decreases TH2 airway inflammation in a mouse model of asthma. J Allergy Clin Immunol. 2005;116:1343–1349. doi: 10.1016/j.jaci.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 56.Degauque N, et al. Immunostimulatory Tim-1-specific antibody deprograms Tregs and prevents transplant tolerance in mice. J Clin Invest. 2008;118:735–741. doi: 10.1172/JCI32562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ueno T, et al. The emerging role of T cell Ig mucin 1 in alloimmune responses in an experimental mouse transplant model. J Clin Invest. 2008;118:742–751. doi: 10.1172/JCI32451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuan X, et al. Targeting Tim-1 to overcome resistance to transplantation tolerance mediated by CD8 T17 cells. Proc Natl Acad Sci USA. 2009;106:10734–10739. doi: 10.1073/pnas.0812538106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao S, et al. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J Exp Med. 2007;204:1691–1702. doi: 10.1084/jem.20062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Souza AJ, Oriss TB, O’Malley KJ, Ray A, Kane LP. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc Natl Acad Sci USA. 2005;102:17113–17118. doi: 10.1073/pnas.0508643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Binne LL, Scott ML, Rennert PD. Human TIM-1 associates with the TCR complex and up-regulates T cell activation signals. J Immunol. 2007;178:4342–4350. doi: 10.4049/jimmunol.178.7.4342. [DOI] [PubMed] [Google Scholar]
- 62.de Souza AJ, Oak JS, Jordanhazy R, DeKruyff RH, Fruman DA, Kane LP. T cell Ig and mucin domain-1-mediated T cell activation requires recruitment and activation of phosphoinositide 3-kinase. J Immunol. 2008;180:6518–6526. doi: 10.4049/jimmunol.180.10.6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV. Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J Biol Chem. 2002;277:39739–39748. doi: 10.1074/jbc.M200562200. [DOI] [PubMed] [Google Scholar]
- 64.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62:237–244. doi: 10.1046/j.1523-1755.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 65.Vila MR, et al. Hepatitis A virus receptor blocks cell differentiation and is overexpressed in clear cell renal cell carcinoma. Kidney Int. 2004;65:1761–1773. doi: 10.1111/j.1523-1755.2004.00601.x. [DOI] [PubMed] [Google Scholar]
- 66.Han WK, et al. Human kidney injury molecule-1 is a tissue and urinary tumor marker of renal cell carcinoma. J Am Soc Nephrol. 2005;16:1126–1134. doi: 10.1681/ASN.2004070530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monney L, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 68.Sanchez-Fueyo A, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–1101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 69.Hastings WD, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39:2492–2501. doi: 10.1002/eji.200939274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson AC, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318:1141–1143. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 71.Anderson AC, et al. Th1 transcription factor T-bet regulates the expression of Tim-3. Eur J Immunol. doi: 10.1002/eji.200939842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nagae M, et al. Structural analysis of the recognition mechanism of poly-N-acetyllactosamine by the human galectin-9 N-terminal carbohydrate recognition domain. Glycobiology. 2009;19:112–117. doi: 10.1093/glycob/cwn121. [DOI] [PubMed] [Google Scholar]
- 73.Rabinovich GA, et al. Galectins and their ligands: amplifiers, silencers or tuners of the inflammatory response? Trends Immunol. 2002;23:313–320. doi: 10.1016/s1471-4906(02)02232-9. [DOI] [PubMed] [Google Scholar]
- 74.Katoh S, et al. Galectin-9 inhibits CD44-hyaluronan interaction and suppresses a murine model of allergic asthma. Am J Respir Crit Care Med. 2007;176:27–35. doi: 10.1164/rccm.200608-1243OC. [DOI] [PubMed] [Google Scholar]
- 75.Niki T, et al. Galectin-9 is a high affinity IgE-binding lectin with anti-allergic effect by blocking IgE-antigen complex formation. J Biol Chem. 2009;284:32344–32352. doi: 10.1074/jbc.M109.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seki M, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008;127:78–88. doi: 10.1016/j.clim.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 77.He W, et al. Galectin-9 significantly prolongs the survival of fully mismatched cardiac allografts in mice. Transplantation. 2009;88:782–790. doi: 10.1097/TP.0b013e3181b47f25. [DOI] [PubMed] [Google Scholar]
- 78.Wang F, et al. Activation of Tim-3-Galectin-9 pathway improves survival of fully allogeneic skin grafts. Transpl Immunol. 2008;19:12–19. doi: 10.1016/j.trim.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 79.Niwa H, et al. Stable form of galectin-9, a Tim-3 ligand, inhibits contact hypersensitivity and psoriatic reactions: a potent therapeutic tool for Th1- and/or Th17-mediated skin inflammation. Clin Immunol. 2009;132:184–194. doi: 10.1016/j.clim.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 80.Kinchen JM, Ravichandran KS. Phagocytic signaling: you can touch, but you can’t eat. Curr Biol. 2008;18:R521–524. doi: 10.1016/j.cub.2008.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ruscetti S, Matthai R, Potter M. Susceptibility of BALB/c mice carrying various DBA/2 genes to development of Friend murine leukemia virus-induced erythroleukemia. J Exp Med. 1985;162:1579–1587. doi: 10.1084/jem.162.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sabatos CA, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 83.Golden-Mason L, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009;83:9122–9130. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones RB, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med. 2006;203:2223–2227. doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kwong BY, et al. Molecular analysis of tumor-promoting CD8(+) T cells in two-stage cutaneous chemical carcinogenesis. J Invest Dermatol. 2010 doi: 10.1038/jid.2009.362. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Frisancho-Kiss S, et al. Cutting edge: T cell Ig mucin-3 reduces inflammatory heart disease by increasing CTLA-4 during innate immunity. J Immunol. 2006;176:6411–6415. doi: 10.4049/jimmunol.176.11.6411. [DOI] [PubMed] [Google Scholar]
- 88.Koguchi K, Anderson DE, Yang L, O’Connor KC, Kuchroo VK, Hafler DA. Dysregulated T cell expression of TIM3 in multiple sclerosis. J Exp Med. 2006;203:1413–1418. doi: 10.1084/jem.20060210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202:13–23. doi: 10.1016/s0022-510x(02)00207-1. [DOI] [PubMed] [Google Scholar]
- 90.Chan A, et al. Phagocytosis of apoptotic inflammatory cells by microglia and its therapeutic implications: termination of CNS autoimmune inflammation and modulation by interferon-beta. Glia. 2003;43:231–242. doi: 10.1002/glia.10258. [DOI] [PubMed] [Google Scholar]
- 91.Ohler B, et al. Role of lipid interactions in autoimmune demyelination. Biochim Biophys Acta. 2004;1688:10–17. doi: 10.1016/j.bbadis.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 92.Kearley J, McMillan SJ, Lloyd CM. Th2-driven, allergen-induced airway inflammation is reduced after treatment with anti-Tim-3 antibody in vivo. J Exp Med. 2007;204:1289–1294. doi: 10.1084/jem.20062093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee J, Su EW, Phuah J, Hainline S, Kane LP. Determination of cytoplasmic tyrosine residues of TIM-3 involved in regulation of TCR signaling [abstract] J Immunol. 2009;182:35. [Google Scholar]
- 94.Shakhov AN, et al. SMUCKLER/TIM4 is a distinct member of TIM family expressed by stromal cells of secondary lymphoid tissues and associated with lymphotoxin signaling. Eur J Immunol. 2004;34:494–503. doi: 10.1002/eji.200324590. [DOI] [PubMed] [Google Scholar]
- 95.Mizui M, et al. Bimodal regulation of T cell-mediated immune responses by TIM-4. Int Immunol. 2008;20:695–708. doi: 10.1093/intimm/dxn029. [DOI] [PubMed] [Google Scholar]
- 96.Yang PC, et al. TIM-4 expressed by mucosal dendritic cells plays a critical role in food antigen-specific Th2 differentiation and intestinal allergy. Gastroenterology. 2007;133:1522–1533. doi: 10.1053/j.gastro.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 97.Rodriguez-Manzanet R, et al. TIM-4 expressed on APCs induces T cell expansion and survival. J Immunol. 2008;180:4706–4713. doi: 10.4049/jimmunol.180.7.4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jinushi M, et al. Milk fat globule epidermal growth factor-8 blockade triggers tumor destruction through coordinated cell-autonomous and immune-mediated mechanisms. J Exp Med. 2009;206:1317–1326. doi: 10.1084/jem.20082614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19:346–351. doi: 10.1016/j.cub.2009.01.042. [DOI] [PubMed] [Google Scholar]
- 100.Chen TT, et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med. 2005;202:955–965. doi: 10.1084/jem.20042433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumanogoh A, Kikutani H. Immune semaphorins: a new area of semaphorin research. J Cell Sci. 2003;116:3463–3470. doi: 10.1242/jcs.00674. [DOI] [PubMed] [Google Scholar]
- 102.Kumanogoh A, et al. Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature. 2002;419:629–633. doi: 10.1038/nature01037. [DOI] [PubMed] [Google Scholar]
- 103.Chakravarti S, et al. Tim-2 regulates T helper type 2 responses and autoimmunity. J Exp Med. 2005;202:437–444. doi: 10.1084/jem.20050308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rennert PD, et al. T cell, Ig domain, mucin domain-2 gene-deficient mice reveal a novel mechanism for the regulation of Th2 immune responses and airway inflammation. J Immunol. 2006;177:4311–4321. doi: 10.4049/jimmunol.177.7.4311. [DOI] [PubMed] [Google Scholar]
- 105.Knickelbein JE, de Souza AJ, Tosti R, Narayan P, Kane LP. Cutting edge: inhibition of T cell activation by TIM-2. J Immunol. 2006;177:4966–4970. doi: 10.4049/jimmunol.177.8.4966. [DOI] [PubMed] [Google Scholar]
- 106.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 107.Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96:245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 108.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 109.Botto M, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 110.Taylor PR, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hanayama R, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 112.Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest. 2001;108:957–962. doi: 10.1172/JCI14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Scott RS, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]