

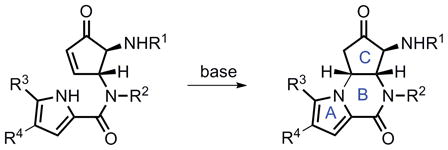
Abstract
In this article, we report a full account of our recent development of pyrroles and N-alkoxyamides as new classes of nucleophiles for palladium-catalyzed AAA reactions, along with application of these methodologies in the total synthesis of agelastatin A, a marine natural product with exceptional anti-cancer activity and other biological properties. Our method allows for access to either regioisomer of the pyrrolopiperazinones (6 and 19) with high efficiency and enantioselectivity. Note that isomer 19 was obtained via a cascade reaction through a double allylic alkylation pathway. From regioisomer 6, the total synthesis of (+)-agelastatin A was completed in a very short fashion (four steps from 6), during the course of which we developed a new copper catalyst for aziridination and an In (OTf)3-DMSO system to oxidatively open an N-tosyl aziridine. Starting with the other pyrrolopiperazinone 19, a five-step sequence has been developed to furnish a formal total synthesis of (−)-agelastatin A. A unique feature of our syntheses is the use of two rather different strategies for the total syntheses of both enantiomers of agelastatin A using the same enantiomer of a chiral palladium catalyst.
Keywords: alkaloid, asymmetric catalysis, alkylation, pyrrole, total synthesis
Introduction
During the last several decades, asymmetric catalysis has dramatically improved the synthetic efficiency of producing biologically important molecules. Among various catalytic asymmetric methods, metal-catalyzed asymmetric allylic alkylation (AAA) constitutes an incredibly feasible reaction, largely attributed to its broad substrate scope and its capability to asymmetrically form different bonds, including carbon-carbon, carbon-oxygen, carbon-nitrogen, and carbon-sulfur bonds, etc.[1] For those AAA reactions, however, the scope of the potential nucleophiles is still unknown. Recently, due to the increased medicinal significance of nitrogen containing bioactive molecules[2], especially those with a bromopyrrole moiety[3] such as the agelastatins (Figure 1), we began to explore pyrroles and N-alkoxyamides as nucleophiles in AAA reactions. In this article, we report a full account of our recent development of Pd-catalyzed pyrrole- and N-alkoxyamide-based AAA reactions, along with a detailed description of our enantioselective total synthesis of both (+) and (−) agelastatin A via two rather different strategies that rely on the use of the same enantiomer of a chiral palladium catalyst.
Figure 1.

Agelastatin A–D.
Agelastatins A–D, which possess a highly fused tetracyclic ring structure, comprise a family of oroidin alkaloids. Agelastatin A (1) along with agelastatin B (2), were first isolated in 1993 by Pietra et al. from a deep water marine sponge Agelas dendromorpha collected in the Coral Sea near New Caledonia.[4] Later in 1998, agelastatin C (3) and D (4) were isolated from the West Australia sponge Cymbastela sp. by Molinski and co-workers.[5] The agelastatins exhibit exceptional biological activities; most notable of these is their nanomolar activity against a broad range of cancer cell lines that include human KB nasopharyngeal cancer cells (IC50 = 0.075 μg/mL), L1210 murine tumor cell line, RT112/84 bladder carcinoma cells, SK-MEL-5 melanoma cells, HCT-116 colon carcinoma cells, and MDA-MB-435s breast cancer cells.6 In many cases, agelastatin A was shown to be 1.5 to 16 times more potent than the frontline chemotherapeutic agent cisplatin at inhibiting cell growth.[6c] Very recently, agelastatin A has been found to inhibit osteopontin-mediated adhesion, invasion, and colony formation.[7] It also inhibits glycogen synthase kinase-3 β (GSK-3β) at low concentration, suggesting a potential approach for the treatment of Alzheimer’s disease.[4] Hale et al. has also suggested that agelastatin A might also function as a novel insulin mimetic.[8] In addition, agelastin A has been reported to possess potent activity against brine shrimp (LC50 = 1.7 ppm), larvae of beet army worm and corn rootworm.[5]
Besides these appealing biological activities, the structure of agelastain A has also been attractive to the synthetic community. First, this natural product contains an unusual 5-6-5-5 fused tetracylic system, including a bromopyrrole motif. Second, it contains four contiguous stereocenters, all of which are located in a 5-membered ring. In addition, each of these stereogenic centers bears a nitrogen atom. All of these features constitute significant challenges for the total synthesis of agelastatin A. To date, several elegant total syntheses of this natural product have been reported.[9] In 1999, Weinreb et al. completed the first total synthesis of racemic agelastatin A, highlighted by a novel N-sulfinyl dienophile Diels–Alder reaction followed by a tandem N-S bond cleavage, [2,3] sigmatropic rearrangement, and oxazolidinone formation to efficiently construct a [3.3.0] bicycle from cyclopentadiene.[10] Feldman and Saunders in 2002 reported the first enantioselective synthesis of (−)-agelastatin A and B, in which a unique approach using alkynyliodonium salts was described.[11] Starting from the Hough-Richardson aziridine, Hale and co-workers completed a formal total synthesis of (−)-agelastatin A in 2003 along with a new total synthesis in 2004.[12] The Davis group then published an asymmetric synthesis of 1 in 2005 using a chiral sulfinimine.[13] Shortly after, Wehn and Du Bois completed a total synthesis of (−)-agelastatin A by application of a Rh-catalyzed intramolecular aziridination reaction.[14] A recent synthesis by the Ichikawa group in 2007 featured the double usage of a [3,3] sigmatropic rearrangement of allyl cyanate.[15] In 2008, Yoshimitsu–Tanaka disclosed a new asymmetric synthesis of agelastatin A using an azidoformate-mediated aziridination reaction.[16] Most recently, a racemic synthesis was reported by Dickson and Wardrop, employing a strategy based on multiple usage of the trichloroacetamide functionality.[17] While most previous total syntheses relied on an intramolecular Michael addition to provide the ABC-ring system of 1 (eq 1), we devised a new strategy to construct this key tricycle by dual usage of Pd AAA reactions. Herein, we provide a full report of our work directed towards the total synthesis of agelastatin A.[18]
 |
(1) |
Results and Discussion
Synthesis Plan
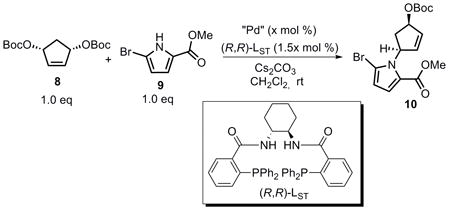
From a retrosynthetic viewpoint (Scheme 1), agelastatin A (1) could be accessed from tricyclic amino ketone 5 that in turn could be derived from tricyclic olefin 6. An intramolecular Pd-catalyzed allylic alkylation of amide 7 was envisioned to provide pyrrolopiperazinone 6. Amide 7 could be ultimately synthesized from an intermolecular Pd-catalyzed desymmetrization AAA between bisallylic carbonate 8 and pyrrole 9. The most attractive feature of this sequence was that a complex tricyclic structure such as 6 could be efficiently and enantioselectively obtained from two consecutive AAA reactions.
Scheme 1.

Retrosynthetic analysis.
Pd-Catalyzed AAA Reactions with Pyrroles and N-Methoxyamides as Nucleophiles Leading to the Formation of Tricycle Pyrrolopiperazinone 6
Throughout the initial studies for the Pd-catalyzed AAA between 8 and 9, Cs2CO3 and CH2Cl2 were found to be the best base and solvent combination (eq 2). The use of other solvents, such as THF or DMF, or other bases, such as NaH, Hünig’s base or DBU, gave either low conversion or decomposition of the starting materials. The yield and enantioselectivity of this desymmetrization reaction were further optimized by varying the palladium source, catalyst loading, base loading, and concentration (Table 1). Lowering the amount of base increased the enantioselectivity from 84% to 92% ee, but reduced the yield (entries 1 and 2). Entry 3 indicated that lowering the catalyst loading at the same substrate concentration afforded the same yield for this transformation, but a reduction in ee value (66%) was obtained. When the catalyst loading was decreased while keeping the catalyst concentration the same as in entry 1, almost the same enantioselectivity was obtained but with lower yield (entry 4). Upon changing the palladium source from Pd2(dba)3·CHCl3 to [Pd(π-C3H5)Cl]2, both the yield and the enantioselectivity were significantly enhanced. From these studies emerged the most practical set of conditions, as shown in entry 6, which provided the N-alkyl pyrrole 10 in 83% yield and 92% ee. At this point, the absolute configuration was tentatively assigned by analogy to other AAA reactions of substrate 8.[1]
Table 1.
Selected optimization studies.
| Entry | Pd source (mol %) | Cs2CO3 (equiv) | Substrate Conc. (M) | Yield (%)[a] | ee (%)[b] |
|---|---|---|---|---|---|
| 1 | Pd2(dba)3·CHCl3 (5) | 1.0 | 0.02 | 90 | 84 |
| 2 | Pd2(dba)3·CHCl3 (5) | 0.7 | 0.02 | 75 | 92 |
| 3 | Pd2(dba)3·CHCl3 (2.5) | 1.0 | 0.02 | 89 | 66 |
| 4 | Pd2(dba)3·CHCl3 (1.25) | 1.0 | 0.08 | 75 | 85 |
| 5 | [Pd(π-C3H5)Cl]2 (2) | 1.0 | 0.17 | 88–93 | 87 |
| 6 | [Pd(π-C3H5)Cl]2 (1.25) | 1.0 | 0.08 | 83 | 92 |
Isolated yield
Enantioselectivities were determined by chiral HPLC.
 |
(2) |
Direct transformation of the carboxylate ester 10 to N-methoxyamide 7 was next attempted, however, treatment with a variety of reagents[19] resulted in either no product or cleavage of the t-butyl carbonate (Boc) group. A two-step process was then developed involving careful hydrolysis of the methyl ester with lithium hydroxide, followed by amide formation via the acyl chloride to give N-methoxyamide 7 in high yield (Scheme 2). Notably, the N-methoxyamide was chosen because of the increased nucleophilicity relative to a simple amide, which should facilitate the subsequent cyclization.
Scheme 2.

Synthesis of compound 7.
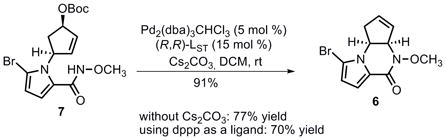
The intramolecular Pd-catalyzed AAA with N-methoxyamide as the nucleophile proceeded successfully (eq 3). Although a chiral ligand is not necessary for this cyclization, when (R,R)-LST (LST: standard Trost ligand) was used as ligand,[20] piperazinone 6 was obtained in 91% yield. Use of achiral dppp proved to be less efficient and gave 6 in 70% yield. Cs2CO3 was not required for this cyclization, but in the absence of base, tricycle 6 was obtained in only 77% yield.
 |
(3) |
Alkene Elaboration
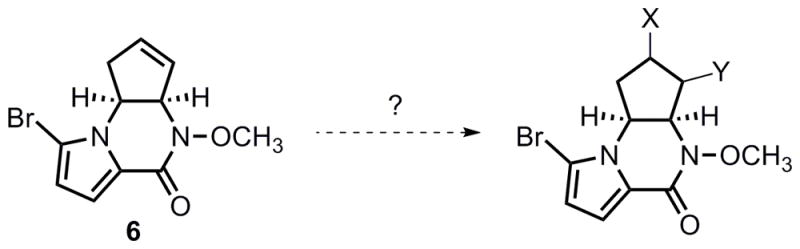
Elaboration of intermediate 6 to the natural product required dual functionalization of the cyclopentene olefin (Scheme 3). Hydroxybromination and hydroxyiodination of 6 only yielded the pyrrole-brominated or iodinated products, while the alkene functionality remained intact.
Scheme 3.
Advancement of compound 6.
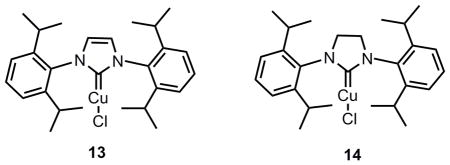
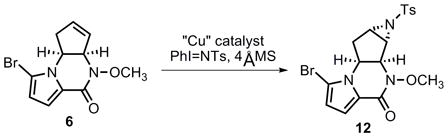
Thus, an aziridination route to transform pyrrolopiperazinone 6 was eventually sought. Although no reaction occurred either using either Sharpless’ method[21] with bromine as catalyst or using tosyl azide under thermal or photochemical conditions[22], a copper-catalyzed aziridination with PhI=N-Ts as the nitrene source[23] proved to be more promising (eq 4). Due to the rather electron-deficient character of the olefin moiety in 6, a number of different copper catalysts, solvents, and other conditions were examined. Selected optimization results are summarized in Table 2. Molecular sieves have been found to be a critical additive to secure consistent results. Among the various catalyst systems employed, copperI thiophene-2-carboxylate (CuTc, entry 2) in acetonitrile and copperI triflate (CuOTf) in benzene (entries 6 and 7) exhibited good reactivity. A more effective catalyst was found when using complex 13 or 14, and the desired aziridine product 12 was obtained in 51% and 52 % yield respectively (entries 10 and 11). Although N-heterocyclic carbene (NHC) metal complexes have been known to enable carbene transfer to olefins,[24] to our knowledge, these examples represent the first NHC-copper catalyzed aziridination reactions.[25]
Table 2.
Selected optimization of aziridination reaction.
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol %) | PhI=NTs (equiv) | Solvent | Temp.(°C) | Yield (%)[a] |
| 1 | Cu(acac)2 (10) | 2 | CH3CN | 0 to rt | 28 |
| 2 | CuTc (20) | 2 | CH3CN | 0 to rt | 29–40 |
| 3 | Cu(OTf)2 (20) | 2 | CH3CN | rt | trace |
| 4 | CuOTf (10) | 2 | DME | rt | 0 |
| 5 | CuOTf (30) | 3 | CH3CN | rt | 16 |
| 6 | CuOTf (10) | 2 | benzene | 0 to rt | 30 |
| 7 | CuOTf (50) | 2 | benzene | rt | 41 |
| 8 | CuOTf (100) | 2 | benzene | rt | 47 |
| 9 | Cu(py)2Cl2 (100) | 2 | DCM | rt | <30 |
| 10 | Catalyst 13 (50) | 5 | benzene[b] | 0 to rt | 52 |
| 11 | Catalyst 14 (50) | 5 | benzene | 0 to rt | 51 |
Isolated yield.
Decomposition was observed when CH3CN was used as solvent.
 |
(4) |
Hydrolytic ring opening of the un-activated tosyl aziridine functionality in 12 proved non-trivial. When treating aziridine 12 either with CAN (10 mol % up to 1 equiv)[26] or BF3·Et2O (0.2 equiv) in aqueous acetonitrile,[27] or with a catalytic amount of TFA in aqueous acetone at room temperature, only recovered starting material was provided. Reaction of 12 with excess TFA upon conventional heating (97 °C) in aqueous dioxane ultimately delivered the desired α-amino alcohol 15 in 97% yield. However, this result is difficult to reproduce. Moreover, this reaction required a long reaction time (ca. 48 h) with continuous refilling of TFA every 8 h. To increase the rate of this transformation, the effect of microwave heating was investigated. To our delight, the microwave assisted hydrolytic ring opening was completed in only 2.5 h at 150 °C, and amino alcohol 15 was obtained reproducibly in 84% yield (Scheme 4). The α-amino ketone 5 was then secured in 70–80% yield by treatment of 15 with Dess–Martin periodinane.[28]
Scheme 4.

Synthesis of compound 5.
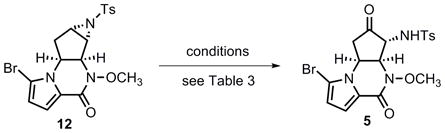
With access to 5, a direct oxidative ring opening of aziridine 12 was then pursued (eq 5), and the explored conditions are summarized in Table 3. When using IBX as oxidant in combination with β-cyclodextrin[29] in aqueous acetone at room temperature (entry 1), only the starting material 12 was isolated. Higher reaction temperatures (50–60 °C) using aqueous dioxane as solvent led to decomposition of the substrate (entry 2). DMSO has been known to be able to oxidatively open epoxides to give α-hydroxy ketones[30], however, few cases existed using DMSO in oxidative aziridine ring opening.[31] Heating 12 in dry DMSO at 80 °C did provide the desired α-amino ketone 5 but in only 42% yield (entry 3). A substantial amount of the direct hydrolytic ring opening product 15 was observed as the major product. As shown in entry 4, addition of molecular sieves did not help minimize the formation of hydrolysis byproducts. On the contrary, it seemed to be detrimental for this reaction, and no desired product could be isolated. Although microwave conditions were demonstrated to be efficient in the hydrolytic ring opening of aziridine 12, significant decomposition was detected after heating 12 in DMSO for 1 h at 150 °C in a microwave apparatus (entry 5). A broad range of additives were also surveyed to improve the reaction yield. For example, addition of a stoichiometric amount of Hünig’s base or sodium chloride generated a complex mixture of products, and addition of water yielded more hydrolytic product 15. On the other hand, addition of a Lewis acid, In(OTf)3, was found to be most effective for this transformation. Treatment of 12 with 0.7 equiv In(OTf)3[32] in dry DMSO at 80 °C for 6 h cleanly produced α-amino ketone 5 in 91% yield (entry 6), and the direct hydrolytic ring opening only occurred to a very limited amount (ca. 5%). This observation can be plausibly explained by the mild Lewis acidity of In(OTf)3 to chemoselectively activate the aziridine functionality,[33] thus allowing facile attack by DMSO (A plausible mechanism is shown in Scheme 5). To the best of our knowledge, this reaction represents the first InIII-catalyzed oxidative ring opening of a tosyl aziridine with DMSO.
Table 3.
Oxidative ring-opening of aziridine 12.
| Entry | Conditions | Results |
|---|---|---|
| 1 | IBX, β-cyclodextrin, aqueous acetone, rt | recovered 12 only |
| 2 | IBX, β-cyclodextrin, aqueous dioxane, 50–60 °C | decomposition of 12 |
| 3 | DMSO, 80 °C | 5 (42% yield[a]) |
| 4 | DMSO, molecular sieves, 80 °C | decomposition of 12 |
| 5 | DMSO, microwave, 150 °C | decomposition of 12 |
| 6 | DMSO, In(OTf)3 (0.7 equiv), 80 °C | 5 (91% yield[a]) |
Isolated yield.
Scheme 5.

Plausible mechanism for the Oxidative ring-opening of aziridine 12.
 |
(5) |
End Game
Advancement of α-amino ketone 5 to agelastatin A required installation of the D-ring urea. After an extensive study, Cs2CO3 was found to most efficiently catalyze the N-acylation of sulfonamide with methyl isocyanate (Scheme 6). Treatment of 5 with 20 mol % Cs2CO3 in methylene chloride at 0 °C followed by slow addition of a benzene solution of methyl isocyanate at room temperature successfully provided tetracycle 16 in 53% yield. Using a weaker base such as K2CO3 or using CuCl as catalyst[34] proved less effective.
Scheme 6.

End game.
It is not totally unexpected that removal of the N-Ts group of 16 needed a great extent of experimentation. A summary of attempted conditions is summarized in Table 4. Dissolving metal reduction of 16 with lithium in the presence of a catalytic amount of naphthalene only yielded the debrominated products (entry 1). Attempts to remove the N-Ts group either with magnesium metal in methanol[35] or with TBAF[36] also failed (entries 2, 3 and 4). Photochemical conditions have been demonstrated to cleave nitrogen-sulfone bonds,[37] however, irradiation of 16 with UV light (254 nm) in several solvents (entries 5–7) did not afford any useful amount of product. In the end, we turned to the SmI2 mediated radical reduction method.[38] Gratifyingly, treatment of 16 with freshly prepared SmI2-THF solution selectively removed both the N-Ts and N-OMe groups without reduction of the pyrrolyl bromide, which ultimately provided (+)-agelastatin A (1) in 88% yield. Synthetic (+)-agelastatin A displayed identical spectroscopic data (1H NMR, 13C NMR, IR, MS, and Rf value) to those reported for the natural product. The optical rotation of synthetic (+)-agelastatin A was [α]D: + 53.2 (c 0.13, MeOH), which compared well to the rotation of naturally isolated (−)-agelastatin A [α]D: −59.3 (c 0.13, MeOH)[5] reported under identical conditions. This also established the absolute configuration of the Pd AAA as shown in eq 2.
Table 4.
Removal of the N-Ts and N-OMe groups.
| Entry | Conditions | Results |
|---|---|---|
| 1 | Li, cat. naphthlene, −78 °C | debrominated products |
| 2 | Mg, MeOH, sonication | no reaction |
| 3 | Mg, MeOH, reflux | debrominated products |
| 4 | TBAF, THF | decomposition of 16 |
| 5 | hv (254 nm), CH3CN | 1 (trace) + decomposition of 16 |
| 6 | hv (254 nm), dry THF | decomposition of 16 |
| 7 | hv (254 nm), THF-H2O | decomposition of 16 |
| 8 | SmI2 (8 equiv), THF, 0 °C to rt | 1 (88% yield[a]) |
Isolated yield.
Development of Pd-Catalyzed One-Pot Cascade AAA to Enable a More Efficient Tricyclic Pyrrolopiperazinone Formation
With successful application of both the pyrrole and the N-methoxyamide as nucleophiles respectively in the Pd-catalyzed AAA, a cascade cyclization reaction was next designed to streamline the synthesis (Scheme 7). For a nucleophile containing both pyrrole and N-methoxyamide (17), a one-pot double AAA could be envisioned, in which a tricyclic pyrrolopiperazinone (6 or 19) could be obtained with high efficiency. A further intriguing question arises—which nitrogen will serve as the kinetically faster nucleophile?
Scheme 7.

A cascade Pd-catalyzed AAA.
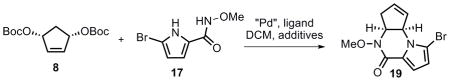
Toward this end, the bifunctional nucleophile 17 was synthesized in two steps from commercially available 2-(trichloroacetyl)pyrrole 20 (Scheme 8). To effect the Pd-catalyzed cascade AAA transformation, one challenge was to chemoselectively differentiate the two nitrogen nucleophiles in substrate 17, enabling one to react at a much faster rate than the other. Attempts to promote initial attack by the pyrrole nitrogen were unsuccessful.[39] Given that the nitrogen of the N-methoxyamide should be more nucleophilic relative to the pyrrole, extensive effort was then conducted to allow a tandem cyclization to access pyrrolopiperazinone 19 (Table 5). Surprisingly, almost no reaction occurred in the presence of a base (entries 1, 2 and 4). We hypothesized that after deprotonation, 17 could act as a bidentate ligand for palladium (Figure 2), thereby preventing the first ionization. On the basis of this hypothesis, 10 mol % of acetic acid was added to the reaction. To our delight, piperazinone 19 was obtained in 51% yield when (R,R)-LST and Pd2(dba)3·CHCl3 were used as catalyst (entry 6). As shown in entry 7, one equiv of N,O-bis(trimethylsilyl) acetamide (BSA) also proved to be effective (50% yield), but given the higher cost and moisture sensitivity of BSA, further optimization was pursued on employing a catalytic amount of acetic acid. Higher yield (88%) was obtained when using racemic standard Trost lignad (LST), presumably because the rate of the second allylic alkylation (from 18 to 19) could likely be accelerated by using the other enantiomer of the Pd catalyst as in a matched case. Based on this hypothesis, the identified optimum conditions involved addition of a portion of racemic catalyst after the completion of the first AAA, and piperazinone 19 could be obtained in up to 82% yield and 97.5% ee (entry 11). Thus, using the same ligand [(R,R) standard Trost ligand] as in eq 2 and 3, but with a different pyrrole nucleophile (17), the other pyrrolopiperazinone regioisomer (19) could be efficiently accessed. Another way to look at the tricycle 19 is that it can be considered as a pseudoenantiomer of tricycle 6, for example, they would be enantiomers if the location of the double bond was the same.
Scheme 8.

Synthesis of compound 17.
Table 5.
Cascade AAA for the synthesis of piperazinone 19[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Pd source (mol %) | Ligand (mol %) | Additive (mol %) | Temp. (°C) | Yield (%)[b] | Ee (%)[c] |
| 1 | [Pd(π-C3H5)Cl]2 (2) | (R,R)-LST (6) | LiHMDS (100) | rt | 0 | NA |
| 2 | [Pd(π-C3H5)Cl]2 (2) | (R,R)-LST (6) | LiHMDS (200) | rt | 0 | NA |
| 3 | [Pd(π-C3H5)Cl]2 (2) | (R,R)-LST (6) | none | rt | trace | NA |
| 4 | [Pd(π-C3H5)Cl]2 (2) | (R,R)-LST (6) | Cs2CO3 (100) | rt | 0 | NA |
| 5 | [Pd(π-C3H5)Cl]2 (5) | (R,R)-LST (15) | HOAc (10) | rt | trace [d] | NA |
| 6 | Pd2(dba)3·CHCl3 (5) | (R,R)-LST (15) | HOAc (10) | rt | 51 | NA |
| 7 | Pd2(dba)3·CHCl3 (5) | (R,R)-LST (15) | BSA (100) | rt | 50 | NA |
| 8 | Pd2(dba)3·CHCl3 (5) | (R,R)-LST (15) | HOAc (10) | 0 to rt | 65[e] | 89 |
| 9 | Pd2(dba)3·CHCl3 (5) | Rac-LST (15) | HOAc (10) | rt | 88[e] | NA |
| 10 | Pd2(dba)3·CHCl3 (5) | (R,R)-LST (15) | HOAc (10) | 0 to rt | 80[e,f] | 96 |
| 11 | Pd2(dba)3·CHCl3 (5) | (R,R)-LST (15) | HOAc (10) | 0 to rt | 82[e,g] | 97.5 |
Unless otherwise indicated, all reactions were performed with 1.0 equiv of 8 and 1.0 equiv of 17 at 0.2 M in DCM.
Isolated yield.
Enantioselectivities were determined by chiral HPLC.
Single alkylation product was the main product.
The reaction was performed with 1.5 equiv of 8 and 1.0 equiv of 17.
Another portion of Pd2(dba)3CHCl3 (5 mol %), (R,R)-LST (15 mol %) was added after 1 h.
Another portion of Pd2(dba)3CHCl3 (5 mol %), rac- LST (15 mol %) was added after 3.5 h.
Figure 2.
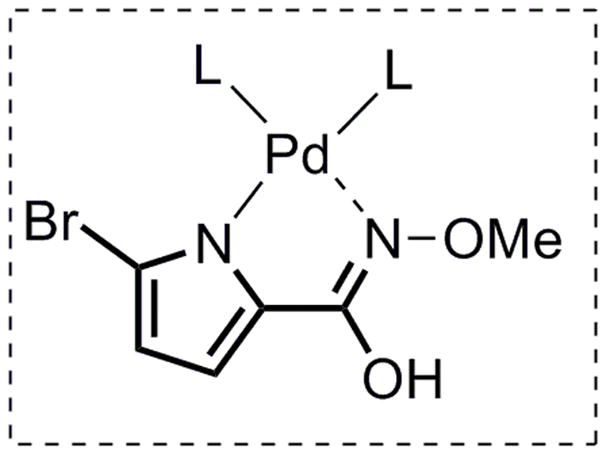
Plausible structure of Pd-17 complex.
Formal Total Synthesis of (−)-Agelastatin A
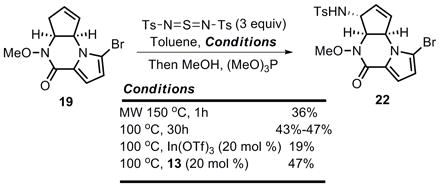
With tricycle 19 in hand, we turned to examine the feasibility of transforming this advanced intermediate to the natural (−)-enantiomer of agelastatin A. Inspired by the work of Weinreb,[10],[40] pyrrolopiperazinone 19 was subjected to allylic amination conditions.[41] As shown in eq 6, several modifications were attempted in order to improve the yield for this Kresze reaction, but the original thermal conditions gave the best results (43–47% yield). Microwave conditions or Lewis acid additives [20 mol % In(OTf)3] caused increased decomposition, and addition of NHC-copper complex 13 neither harmed nor benefited this transformation. Although the yield of this allylic amination is moderate, we were pleased to find that a single regio- and diastereomer was obtained given the complexity of substrate 19, which was assigned as tricycle 22 by analogy.
 |
(6) |
Based on the precedent from Weinreb’s synthesis, it seemed to be straightforward to advance 22 to the natural product 1. However, any attempts to functionalize the cyclopentene olefin in 22 remained unfruitful (Scheme 9). For example, under several hydroboration conditions (BH3, 9-BBN-H, Rh-catalyzed hydroboration, etc) and epoxidation conditions (mCPBA with or without a buffer, MTO-UHP, Mo(CO)6-catalyzed epoxidation, DMDO, etc), either a complicated reaction mixture was obtained, or no reaction occurred.
Scheme 9.

Unsuccessful advancement of compound 22.
After extensive experimentation, we eventually found that the presence of the acidic sulfonamide N-H hydrogen seemed to be detrimental for the subsequent functionalization of the adjacent olefin. The problem was solved by altering the reaction sequence between hydroboration of the olefin and acylation of the sulfonamide (Scheme 10). Reaction of 22 with methyl isocyanate and 0.2 equiv Cs2CO3 in DCM provided urea 23 in 90% yield. When treating 23 with BH3 in THF followed by oxidative treatment with hydrogen peroxide and sodium hydroxide, secondary alcohol (−)-15 was isolated as the major product, however, surprisingly, the urea group was cleaved, presumably by the basic oxidative workup. Alcohol (−)-15 is spectrosopically identical to compound (+)-15, an intermediate in our synthesis of (+)-agelastatin A. The optical rotation of (−)-15 was [α]D: − 35.6 (c 0.34, DCM), which compared well to the rotation of (+)-15 [α]D: +36.2 (c 0.90, DCM). This hydrolysis problem could be overcome by changing to a less-basic workup. Indeed, upon working up the hydroboration reaction with aqueous sodium perborate, alcohol 24 was obtained as the major regio- and diastereomer, and the relative stereochemistry of 24 was tentatively assigned by analogy to (−)-15. Subsequent Dess–Martin oxidation of 24 provided tetracycle (−)-16 in 84% yield, which displayed identical spectroscopic data (1H NMR, 13C NMR, IR and Rf value) to those of (+)-16. The optical rotation of (−)-16 was [α]D: −11.1 (c 0.17, CHCl3), which compared well to the rotation of (+)-16 [α]D: +9.7 (c 0.73, CHCl3). Given that the final N-Ts and N-OMe cleavage of (−)-16 has been demonstrated on its enantiomer (Scheme 6), therefore, we completed the formal total synthesis of (−)-agelastatin A starting from pyrrolopiperazinone 19 in five steps.
Scheme 10.

Formal total synthesis of (−)-agelastatin A.
Conclusion
Two new classes of nucleophiles, pyrroles and N-alkoxyamides (hydroxamic esters), have been employed in palladium-catalyzed AAA reactions. By varying the functional groups at the 2-position of pyrroles, we were able to efficiently and enantioselectively access either regioisomer of the pyrrolopiperazinones 6 and 19 (Scheme 11), among which 19 was obtained via a cascade reaction through a double allylic alkylation pathway. Using regioisomer 6, we completed the total synthesis of (+)-agelastatin A in 8 steps from pyrrole 9, during the course of which, we developed a new copper catalyst for aziridination, and an In(OTf)3-DMSO system to oxidatively open an N-tosylaziridine. Starting from the other pyrrolopiperazinone (19), a five-step sequence has been developed to furnish a formal total synthesis of (−)-agelastatin A, requiring just the reductive cleavage of the N-Ts and N-OMe bonds as performed on the enantiomer. In either of the two syntheses, although we use reagents that require substituents for the appropriate reactivity, which need to be removed, it is worth noting that no protecting groups have been employed in the sense of putting on such a group to impart chemoselectivity that then must be removed. For example, the O-Boc and N-OMe groups were used as activating groups, and the N-Ts groups were inherent from the C-N bond forming reactions.
Scheme 11.

Total synthesis of (+) and (−)-agelastatin A with two different routes.
Therefore, we have developed two distinctive strategies for the enantioselective total synthesis of marine alkaloid agelastatin A. As a rather unique feature of our syntheses, two complementary routes differing only in the choice of nucleophile in the Pd AAA reactions provide either enantiomer of this natural product.
Experimental Section
Selected experimental procedures for the preparation of 9, 6, 12, 5, 19, and 1 (agelastatin A) appear below. Full experimental details for all new compounds are given in the Supporting Information.
Compound 9
To a solution of pyrrole-2-carboxylate methyl ester (2.0 g, 16 mmol) in THF (160 ml) and MeOH (80 ml) was added NBS (recrystalized, 0.49 g, 2.8 mmol) at 0 °C. The reaction was stirred at 0 °C for 30 min, before another portion of NBS (recrystalized, 0.63 g, 3.5 mmol) was added. After 40 min, more NBS (recrystalized, 0.51 g, 2.9 mmol) was added to the reaction mixture. After another 30 min, another portion of NBS (recrystalized, 1.27 g, 7.1 mmol) was added. The resulting solution was then stirred for another 2 h, before the solvent was removed under vacuum. Compound 9 was purified via silica gel flash column chromatography (petroleum ether/diethyl ether = 20/1, then 8/1) to give a white floppy solid (1.63 g, 50%): mp: 101–103 °C; Rf: 0.35 (petroleum ether/ether = 4/1); 1H NMR (CDCl3, 400 MHz): δ 9.1 (br s, 1H), 6.82 (dd, J = 3.0, 3.5 Hz, 1H), 6.21 (dd, J = 3.0, 3.5 Hz, 1H), 3.86 (s, 3H); 13C NMR (CDCl 3, 100 MHz) δ 160.8, 123.7, 116.8, 112.7, 105.2, 51.8; IR (film): 3250, 2924, 2853, 1702, 1552, 1449, 1415, 1389, 1327, 1207 cm−1; HRMS (C6H6NO2Br): Calc’d. 202.958190 (M+), Found 202.958096.
Compound 6
A solution of Pd2(dba)3·CHCl3 (2.29 mg, 0.00025 mmol) and (R,R)-LST (5.18 mg, 0.00075 mmol) in CH2Cl2 (1 ml), which had been stirred at rt for 10 min, was added to a mixture of compound 7 (20 mg, 0.05 mmol) and Cs2CO3 (16.3 mg, 0.05 mmol) under Ar. The mixture was stirred at rt for 12 h, and then filtered through a celite cake. The solvent was removed under vacuum, and compound 6 was purified via silica gel flash column chromatography (petroleum ether/ether = 8/1, then CH2Cl2/MeOH = 40/1) to give a white solid (12.9 mg, 91.5%): mp: 107–109 °C; Rf: 0.6 (CH2Cl2/MeOH = 9/1); [α]D: +164.7 (CH2Cl2, c 0.34); 1H NMR (CDCl3, 500 MHz) δ 6.94 (dd, J = 1, 4 Hz, 1H), 6.30 (dd, J = 1, 4 Hz, 1H), 6.22-6.20 (m, 1H), 6.13-6.12 (m, 1H), 4.84-4.79 (m, 2H), 3.85 (s, 3H), 3.13-3.07 (m, 1H), 2.63-2-58 (m, 1H); 13C NMR (CDCl3, 125 MHz) δ 156.3, 135.5, 128.6, 123.7, 116.1, 106.8, 101.6, 64.1, 64.0, 57.2, 38.2; IR (film): 2927, 2853, 1668, 1545, 1418, 1358, 1334, 1310, 1027 cm−1: HRMS (C11H11N2O2Br): Calc’d 282.000389 (M+), Found 281.999714.
Compound 12
Benzene (1 ml) was added to a mixture of 4 Å molecular sieve (75 mg), compound 6 (20 mg, 0.071 mmol), PhI=NTs (132 mg, 0.355 mmol) and catalyst 13 (17.3 mg, 0.0355 mmol) under N2. The resulting mixture was stirred at rt for 4 h, before it was filtered through a silica gel cake and rinsed with ethyl acetate. The solvent was removed under vacuum, and compound 12 was purified via alumina (neutral, activity 3) flash column chromatography (petroleum ether/ethyl acetate = 8/1, 4/1, then 3/1) to give a colorless foam (16.6 mg, 52%): Rf: 0.5 (petroleum ether/ethyl acetate=3/2); [α]D: +21.8 (c 0.85, CHCl3); 1H NMR (CDCl3, 500 MHz): δ 7.86 (dd, J = 1.5, 6.5 Hz, 2H), 7.40 (dd, J = 0.5, 8.5 Hz, 2H), 6.94 (d, J = 4.0 Hz, 1H), 6.26 (d, J = 4.5 Hz, 1H), 4.50-4.44 (m, 2H), 3.97 (s, 3H), 3.88 (d, J = 5 Hz, 1H), 3.61 (dd, J = 2.5, 5.0 Hz, 1H), 2.77 (dd, J = 7.0, 14 Hz, 1H), 2.48 (s, 3H), 1.93 (ddd, J = 14.5, 9.0, 2.5 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 158.2, 145.5, 134.8, 130.3, 128.2, 122.8, 116.2, 114.1, 106.2, 64.5, 61.3, 53.3, 44.7, 44.1, 32.9, 22.0; IR (film): 2925, 2855, 1682, 1543, 1433, 1416, 1325, 1163 cm−1; HRMS (C18H18N3O4SBr): Calc’d. 451.020139 (M+), Found 451.020921.
Compound 5
From Compound 15: To a solution of compound 15 (10 mg, 0.0213 mmol) and NaHCO3 (5.4 mg, 0.0639 mmol) in CH2Cl2 (0.25 ml) was added Dess-Martin periodinane (13.5 mg, 0.032 mmol). The resulting mixture was stirred at rt for 30 min before quenched with sat. aq. Na2S2O3. Compound 5 was purified via silica gel flash column chromatography (petroleum ether/ethyl acetate = 3/1, then 3/2) as a white solid (ca. 7 mg 70–80%).
From Compound 12: DMSO (1 ml) was added to a mixture of compound 12 (30 mg, 0.0665 mmol) and In(OTf)3 (26 mg, 0.0462 mmol) at rt under N2. The resulting solution was heated at 80 °C for 6 h, before diluted with ethyl acetate (30 ml). The solution was washed with brine. The aqueous phases were combined, and extracted with ethyl acetate three times. The combined the organic phase was dried over MgSO4, and compound 5 was purified via silica gel flash column chromatography (petroleum ether/ethyl acetate = 3/1, then 3/2) to give a white solid (28.3 mg, 91%): mp: 100–102 °C, Rf: 0.3 (petroleum ether/ethyl acetate = 1/1); [α]D: +57.8 (c 0.97, CH2Cl2); 1H NMR (CDCl3, 500 MHz) δ 7.82 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 4.0 Hz, 1H), 6.35 (d, J = 4.0 Hz, 1H), 5.63 (d, J = 7.5 Hz, 1H), 5.23-5.18 (m, 1H), 5.00 (d, J = 2.5 Hz, 1H), 4.98 (d, J = 5.5 Hz, 1H), 4.00 (br, 1H), 3.84 (s, 3H), 3.02 (dd, J = 7.5, 18 Hz, 1H), 2.68 (dd, J = 11, 18 Hz, 1H), 2.46 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 206.3, 157.9, 144.7, 134.7, 130.0, 127.7, 122.6, 116.7, 114.3, 106.4, 64.1, 61.1, 51.9, 41.2, 29.7, 21.6; IR (film): 3252, 2924, 2854, 1768, 1652, 1548, 1416, 1338, 1162, 1093, 1025 cm−1; HRMS (C18H18N3O5SBr): Calc’d. 469.013008 (M+), Found 469.013016.
Compound 19
(eg. Table 5, entry 11) A solution of Pd2(dba)3CHCl3 (5.2 mg, 0.005 mmol) and (R,R)-LST (10.4 mg, 0.015 mmol) in degassed CH2Cl2 (0.5 ml), which had been stirred for 10 min at 0 °C, was added to a mixture of compound 17 (21.9 mg, 0.1 mmol), compound 8 (45 mg, 0.15 mmol) and HOAc (10 μl, 1M solution in CH2Cl2) under Ar. The mixture was stirred at rt for 3.5 h, and then was added a solution of Pd2(dba)3CHCl3 (5.2 mg, 0.005 mmol) and Rac-LST (10.4 mg, 0.015 mmol) in degassed CH2Cl2 (0.5 ml), which had been stirred for 10 min at rt. The resulting solution was stirred at rt for 3 h. Compound 19 was purified via silica gel flash column chromatography (petroleum ether/ethyl acetate = 4/1, then 3/1) as a colorless solid (23.0 mg, 82%, 97.5% ee by HPLC OD column, 90:10 heptane: i-propanol, 0.8 ml/min): mp: 111–113 °C; Rf: 0.2 (petroleum ether/ethyl acetate = 4/1); [α]D: −120.2 (c 1.0, CH2Cl2); 1H NMR (CDCl3, 500 MHz) δ 6.91 (d, J = 5.0 Hz, 1H), 6.28 (d, J = 5.0 Hz, 1H), 6.02 (m, 1H), 5.95 (m, 1H), 5.29 (d, J = 8.0 Hz, 1H), 4.65 (ddd, J = 8.0, 6.5, 2.5 Hz, 1H), 3.85 (s, 3H), 2.92 (d, J = 21.5 Hz, 1H), 2.71 (ddd, J = 21.5, 6.5, 2.5 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 157.3, 132.0, 129.0, 123.8, 114.7, 113.6, 104.8, 63.0, 61.6, 60.7, 36.4; IR (film): 2925, 2854, 1667, 1545, 1417, 1320, 1028 cm−1; HRMS (C11H11N2O2Br): Calc’d. 282.000389 (M+), Found 281.999491.
(+)-Agelastatin A (1)
Freshly made SmI2 (1.6 ml, 0.1 M in THF) was added to compound 16 (10.0 mg, 0.019 mmol) under argon at 0 °C. The resulting blue solution was allowed to gradually warm to rt and stirred for 2 h before another 0.5 ml SmI2 (0.1 M in THF) was added. After the solution was stirred at rt for 15 min, THF was removed under vacuum. The residue was first purified by silica gel chromatography (10% to 15% MeOH in CH2Cl2). A yellow solid was obtained, which was then dissolved in 15% MeOH in CH2Cl2 and filtered through charcoal. The (+)-Agelastatin A was further purified by another silica gel chromatography (10% to 15% MeOH in CH2Cl2) to give an off-white solid (6.0 mg, 88%): mp: 195 °C (decomposed); Rf: 0.25 (10% MeOH in CH2Cl2); [α]D: + 53.2 (c 0.13, MeOH); 1H NMR (CD3OD, 500 MHz): δ 6.91 (d, J = 4.0 Hz, 1H), 6.33 (d, J = 4.0 Hz, 1H), 4.60 (m, 1H), 4.09 (d, J = 5.5 Hz, 1H), 3.89 (s, 1H), 2.81 (s, 3H), 2.65 (dd, J = 6.5, 13 Hz, 1H), 2.10 (dd, J = 13, 13 Hz, 1H); 13C NMR (CD3 OD, 150 MHz) δ 161.4, 161.1, 124.1, 116.0, 113.8, 107.2, 95.7, 67.4, 62.2, 54.4, 40.0, 24.2; IR (film): 3346 (br), 2923, 1685, 1644, 1555, 1424, 1380 cm−1; ESI (C12H13N4O3Br): 341.0 [M+H]+, 363.0 [M+Na]+.
Acknowledgments
We thank the National Science Foundation and the National Institutes of Health (GM33049) for their generous support of our programs. Mass spectra were provided by the Mass Spectrometry Regional Center for the University of California-San Francisco, supported by the NIH Division of Research Resources. GD is a Stanford graduate fellow. Palladium salts were generously supplied by Johnson-Matthey. We thank Prof. Du Bois and Dr. Wehn for a TLC sample of agelastatin A. We also thank Prof. Weinreb for providing an experimental procedure of the allylic amination reaction.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.For a recent review: Trost BM, Crawley ML. Chem Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w.Also see Trost BM. Chem Pharm Bull. 2004;50:1–14. doi: 10.1248/cpb.50.1.
- 2.(a) Cordell GA, editor. The Alkaloids Chemistry and Biology. Vol. 62. Academic Press; San Diego: 2005. and earlier volumes in the series. [Google Scholar]; (b) Hesse M, editor. Alkaloids: Nature’s Curse or Blessing. Wiley-VCH; New York: 2002. [Google Scholar]
- 3.Gribble GW. J Nat Prod. 1992;55:1353–1395.Also see: Faulkner DJ. Nat Prod Rep. 2002;19:1–48. doi: 10.1039/b009029h.Berlinck RGS, Kossuga MH. Nat Prod Rep. 2005;22:516–550. doi: 10.1039/b209227c. and references therein.
- 4.(a) D’Ambrosio M, Guerriero A, Debitus C, Ribes O, Pusset J, Leroy S, Pietra F. J Chem Soc, Chem Commun. 1993:1305–1306. [Google Scholar]; (b) D’Ambrosio M, Guerriero A, Chiasera G, Pietra F. Helv Chim Acta. 1994;77:1895–1902. [Google Scholar]; (c) D’Ambrosio M, Guerriero A, Ripamonti M, Debitus C, Waikedre J, Pietra F. Helv Chim Acta. 1996;79:727–735. [Google Scholar]
- 5.Hong TW, Jimenez DR, Molinski TF. J Nat Prod. 1998;61:158–161. doi: 10.1021/np9703813. [DOI] [PubMed] [Google Scholar]
- 6.(a) Maijer L, Thunnissen AM, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkov EM, Kim SH, Pettit GR. Chem Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]; (b) Pettit GR, Ducki S, Herald DL, Doubek DL, Schmidt JM, Chapuis JC. Oncol Res. 2005;15:11–20. doi: 10.3727/096504005775082075. [DOI] [PubMed] [Google Scholar]; (c) Hale KJ, Domostoj MM, El-Tanani M, Campbell FC, Mason CK. Total synthesis and mechanism of action studies on the antitumour alkaloid, (−)-agelastatin A. In: Harmata M, editor. Strategies and tactics in organic synthesis. Academic Press; London: 2005. pp. 352–394. [Google Scholar]
- 7.Mason CK, McFarlane S, Johnston PG, Crowe P, Erwin PJ, Domostoj MM, Campbell FC, Manaviazar S, Hale KJ, El-Tanai M. Mol Cancer Ther. 2008;7:548–558. doi: 10.1158/1535-7163.MCT-07-2251. [DOI] [PubMed] [Google Scholar]
- 8.Hale KJ, Domostoj MM, Tocher DA, Irving E, Scheinmann F. Org Lett. 2003;5:2927–2930. doi: 10.1021/ol035036l. [DOI] [PubMed] [Google Scholar]
- 9.For a review, see: Weinreb SM. Nat Prod Rep. 2007;24:931–948. doi: 10.1039/b700206h.
- 10.Stien D, Anderson GT, Chase CE, Koh YH, Weinreb SM. J Am Chem Soc. 1999;121:9574–9579. [Google Scholar]
- 11.(a) Feldman KS, Saunders JC. J Am Chem Soc. 2002;124:9060–9061. doi: 10.1021/ja027121e. [DOI] [PubMed] [Google Scholar]; (b) Feldman KS, Saunders JC, Wrobleski ML. J Org Chem. 2002;67:7096–7109. doi: 10.1021/jo026287v. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hale KJ, Domostoj MM, Tocher DA, Irving E, Scheinmann F. Org Lett. 2003;5:2927–2930. doi: 10.1021/ol035036l. [DOI] [PubMed] [Google Scholar]; (b) Domostoj MM, Irving E, Scheinmann F, Hale KJ. Org Lett. 2004;6:2615–2618. doi: 10.1021/ol0490476. [DOI] [PubMed] [Google Scholar]
- 13.Davis FA, Deng J. Org Lett. 2005;7:621–623. doi: 10.1021/ol047634l. [DOI] [PubMed] [Google Scholar]
- 14.Wehn PA. Ph.D. Thesis. Stanford University; 2005. [Google Scholar]
- 15.Ichikawa Y, Yamaoka T, Nakano K, Kotsuki H. Org Lett. 2007;9:2989–2992. doi: 10.1021/ol0709735. [DOI] [PubMed] [Google Scholar]
- 16.Yoshimitsu T, Ino T, Tanaka T. Org Lett. 2008;10:5457–5460. doi: 10.1021/ol802225g. [DOI] [PubMed] [Google Scholar]
- 17.Dickson DP, Wardrop DJ. Org Lett. 2009;11:1341–1344. doi: 10.1021/ol900133v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.For a preliminary report of a portion of our work, see: Trost BM, Dong G. J Am Chem Soc. 2006;128:6054–6055. doi: 10.1021/ja061105q.
- 19.Reagents that have been tried: AlMe3, ClMgiPr, KCN, MgCl2, Zr(OBut)4, N-heterocyclic carbene, Sn[N(TMS)2]2, etc.
- 20.The reason of using (R,R) ligand in this cyclization was initially to examine the possibility of cascading two AAA reactions in one reaction vessel, although here is a mismatched situation.
- 21.Jeong JU, Tao B, Sagasser I, Henniges H, Sharpless KB. J Am Chem Soc. 1998;120:6844–6845. [Google Scholar]
- 22.Andrade ES, Nunes RJ, Uieara M. Syn Commun. 2004;34:3073–3081. [Google Scholar]
- 23.Evans DA, Bilodeau MT, Faul MM. J Am Chem Soc. 1994;116:2742–2153. [Google Scholar]
- 24.(a) Fructos MR, Belderrain TR, Nicasio MC, Nolan SP, Kaur H, Díaz-Requejo MM, Pérez PJ. J Am Chem Soc. 2004;126:10846–10847. doi: 10.1021/ja047284y. [DOI] [PubMed] [Google Scholar]; (b) Fructos MR, Belderrain TR, Frément P, Scott NM, Nolan SP, Kaur H, Díaz-Requejo MM, Pérez PJ. Angew Chem Int Eng. 2005;44:5284–5288. doi: 10.1002/anie.200501056. [DOI] [PubMed] [Google Scholar]; (c) Gawley RE, Narayan S. Chem Comm. 2005;40:5109–5111. doi: 10.1039/b509958g. [DOI] [PubMed] [Google Scholar]
- 25.For a recent improvement and extension of this method to a broad substrate scope, see: Xu Q, Appella DH. Org Lett. 2008;10:1497–1500. doi: 10.1021/ol800288b.
- 26.Chakraborty TK, Ghosh A, Raju TV. Chem Lett. 2003;32:82–83. [Google Scholar]
- 27.For a review, see: Hu XE. Tetrahedron. 2004;60:2701–2743.
- 28.Dess DB, Martin JC. J Am Chem Soc. 1991;113:7277–7287. [Google Scholar]
- 29.Surendra K, Krishnaveni NS, Reddy MA, Nageswar YVD, Rao KR. J Org Chem. 2003;68:9119–9121. doi: 10.1021/jo034079c. [DOI] [PubMed] [Google Scholar]
- 30.(a) Khuddus MA, Swern D. J Am Chem Soc. 1973;95:8393–8402. [Google Scholar]; (b) Santosusso TM, Swern D. J Org Chem. 1975;40:2764–2769. [Google Scholar]; (c) Cohen T, Tsuji T. J Org Chem. 1961;26:1681–1681. [Google Scholar]; (d) Trost BM, Fray MJ. Tetrahedron Lett. 1988;29:2163–2166. [Google Scholar]
- 31.For N-aroylaziridines: Heine HW, Newton T. Tetrahedron Lett. 1967;8:1859–1860.For N-alkoxycarbonylaziridines: Fujita S, Hiyama T, Nozaki H. Tetrahedron Lett. 1969;10:1677–1678.Fujita S, Hiyama T, Nozaki H. Tetrahedron. 1970;26:4347–4352.We thank J. Du Bois and K. Guthikonda for drawing our attention to application of this thermal method for opening trichloroethoxysulfamoylaziridines: Guthikonda K, Wehn PM, Caliando BJ, Du Bois J. Tetrahedron. 2006;62:11331–11342.
- 32.The amount of In(OT)3 was not optimized.
- 33.Yadav JS, Subba Reddy VS, Mahesh Kumar G, Murthy ChVSR. Syn Comm. 2002;32:1797–1802. and earlier references therein. [Google Scholar]
- 34.Cervello J, Sastre T. Synthesis. 1990:221–222. [Google Scholar]
- 35.Nadir UK, Krishna RV. J Heterocyclic Chem. 2004;41:737–739. [Google Scholar]
- 36.Carato P, Yous S, Sellier D, Poupaert JH, Lebegue N, Berthelot P. Tetrahedron. 2004;60:10321–10324. [Google Scholar]
- 37.(a) Badr MZA, Aly MM, Fahmy AM. J Org Chem. 1981;46:4784–4787. [Google Scholar]; (b) Pete JP, Portella C. J Chem Res Synopses. 1979;1:20–21. [Google Scholar]
- 38.Vedejs E, Lin S. J Org Chem. 1994;59:1602–1603. [Google Scholar]
- 39.Di-anion and in situ silyl protection strategies have been tried but failed.
- 40.Anderson GT, Chase CE, Koh YH, Stien D, Weinreb SM, Shang M. J Org Chem. 1998;63:7594–7595. [Google Scholar]
- 41.(a) Sharpless KB, Hori T. J Org Chem. 1976;41:176–177. [Google Scholar]; (b) Bussas R, Kresze G. Liebigs Ann Chem. 1980:629–649. [Google Scholar]