

Abstract
Head and neck squamous cell carcinoma is a common human neoplasia with poor prognosis and survival that frequently display Akt overactivation. Here we show that mice displaying constitutive Akt activity (myrAkt) in combination with Trp53 loss in stratified epithelia develop oral cavity tumors that phenocopy human HNSCC. The myrAkt mice develop oral lesions making it a possible model of human oral dysplasia. The malignant conversion of these lesions, which is hampered due to the induction of premature senescence, is achieved by the subsequent ablation of Trp53 gene in the same cells in vivo. Importantly mouse oral tumors can be followed by in vivo imaging, show metastatic spreading to regional lymph nodes and display activation of NFκB and Stat3 pathways and decreased TGFβRII expression, thus resembling human counterparts. In addition, malignant conversion is associated with increased number of putative tumor stem cells. These data identify activation of Akt and p53 loss as a major mechanism of oral tumorigenesis in vivo and suggest that blocking these signaling pathways could have therapeutic implications for the management of HNSCC.
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common type of cancer worldwide. Although new therapeutic approaches, including fractionated radiotherapy, targeted chemotherapy and concurrent radiotherapy and chemotherapy (1–4), have been recently evaluated, the improvement in overall survival in patients with HNSCC is still low. The term HNSCC comprises epithelial tumors that arise in the oral cavity, pharynx, larynx and nasal cavity, with the main risk factors being alcohol and/or tobacco abuse (5). HNSCC results from the accumulation of numerous genetic and epigenetic alterations that occur in a multistep process. The molecular alterations displayed by human HNSCC affect several pathways that influence cell proliferation, apoptosis, differentiation, angiogenesis, inflammation, immune surveillance, and metastasis. The major pathways involved in HNSCC development include the pRb and p53-dependent pathways, EGFR, Stat3, NFκB and TGFβ (reviewed in (6, 7). Moreover, the initiation, growth, recurrence and metastasis of HNSCC, as in many other solid epithelial cancers, have been related to the behavior of a small subpopulation of ‘tumor-initiating’ or cancer stem cells (8, 9). In spite of the fact that the molecular mechanisms of HNSCC are not completely understood, several candidate genes of potential therapeutic relevance are now being validated through in vitro analyses (6, 10, 11); however, these studies cannot recapitulate the complex nature of HNSCC tumors in vivo. Thus, animal models of HNSCC will become essential tools, providing relevant insights of the molecular perturbations of these tumors. Nonetheless, there are few suitable genetically defined mouse models in which to study the progression of this type of tumor under preclinical settings (6), and that fully recapitulate the molecular characteristics of human HNSCC. Here we present a new HNSCC transgenic mouse model based on the expression of constitutively active Akt kinase combined with the ablation of Tp53 gene in stratified epithelia, which phenocopies the molecular alterations previously found in human HNSCC. The characteristics described here make this model an excellent and unique preclinical tool for the therapeutic management of HNSCC at different steps.
MATERIALS AND METHODS
Mice and Histological procedures
The generation of Bk5myrAkt and Trp53F/F;K14cre mice and the protocols for genotyping have been previously described (12–15). These mice were in an immunocompetent mixed C57/Bl6xDBA/2JxFVB/n background. All the animal experiments were approved by the Animal Ethical Committee (CEEA) and conducted in compliance with Centro de Investigaciones Energéticas, Medioambientales y Tecnologicas (CIEMAT) Guidelines. For histological analysis, oral samples were fixed in formalin and embedded in paraffin prior to sectioning, or snap frozen in liquid nitrogen and embedded in OCT for cryosectionning. Sections were stained and processed as described (12, 13). Antibodies were used as follows: 1/50 dilution of anti-Ser473 phosphorylated Akt (Cell Signaling), anti-BrdU (Roche), 1/100 dilution of anti-K15 (NeoMarkers), anti-CD31 (Pharmingen) and anti-CD34 (eBioscience); 1/50 dilution of anti-CycD1 (NeoMarkers), 1/200 dilution of p63 (Santa Cruz Biotechnology and Abcam) and 1/50 dilution of anti-Thr58/Ser42 phosphorylated c-Myc (Cell Signaling); 1/250 dilution of anti-Foxo3a (Upstate Biotechnology), 1/50 dilution of anti-CD44 (Pharmingen) and anti-K13 (Abcam); and 1/500 dilution of anti-HA tag (Covance), 1/500 dilution of anti-K5 (Covance) and anti-p53 (NovoCastra). Biotin-conjugated secondary antibodies for IHC and FITC- or TexasRed-conjugated secondary antibodies for IF were purchased from Jackson ImmunoResearch and used at 1/1000, 1/50 and 1/500, respectively. For IHC, signal was amplified using avidin-peroxidase (ABC elite kit Vector) and peroxidase was visualized using diaminobenzidine as a substrate (DAB kit Vector). Control slides were obtained by replacing primary antibodies with PBS or preimmune sera (data not shown).
Proliferation measurements in the sections were performed by double immunofluorescence using anti K5 (to detect epithelial cells) and anti BrdU mAb essentially as described (12, 15–18) For the quantification of BrdU, at least 5 different samples of each type (normal oral epithelium, dysplasia and SCC) from at least five different mice of each genotype (only non-lesional oral epithelium was observed in control mice) were scored and represented as mean ± SD.
The analysis of blood vessels and microvessel counting was performed as described (15, 19). Briefly, frozen tumor samples embedded in OCT (Tissue Tek, Sakura) and sectioned (6μm thick) were fixed in acetone at −20°C for 5 min and the blood vessels visualized using rat anti-mouse CD31 (Pharmingen, diluted 1:30). Double immunofluorescence was performed using anti K5 to detect the epithelial component as above. Microvessel counting was performed in the five areas of greatest vascularization (double blind analysis) in each sample and expressed as an average. For each type of sample (normal oral epithelium, dysplasia and SCC), the means of 4–6 individual experiments from at least three mice of each genotype (only non-lesional oral epithelium was observed in control mice) were scored.
Cell culture
Oral tissues (oral mucosa, tongue and palate) from adult mice of the different genotypes were digested with 1U/mL dispase (Roche) for 3h at 37°C. Oral epithelium was separated and digested with trypsin (5min, 37°C) and subsequently inactivated and centrifuged. Cell pellet was resuspended in high-calcium medium and left overnight at 37°C, when CNT-24 culture medium (Genicell) was added. When cells reached 80% confluence, they were trypsinized and passaged. Experiments with these cells were done when culture had already stabilized and cells were passaged every 3–4 days. Proliferation curves were performed by seeding 104 cells and counting cell number every 24 hours for five consecutive days. The experiment was performed in triplicate, and mean values and standard deviation were calculated for each time point. Luciferase assays were performed upon transient transfection of the indicated plasmids using Superfect (Qiagen) according to the manufacturer’s recomendations, and cells were harvested 48h after transfection using Promega Dual-Luciferase Assay Kit. Firefly luciferase values were standardized to Renilla luciferase values to account for differences in transfection efficiency. Transfections were performed in triplicate, normalized to the control non transgenic samples and the mean and standard deviation were calculated for each condition.
Western blot analysis
Western blot analyses of oral keratinocytes were performed as previously described (12, 13).
Senescence assay
For senescence assessment, Senescence Associated βgalactosidase Staining kit (Cell Signaling) was used following the manufacturer’s protocol. Briefly, cryosections were fixed in fixing solution for 15min, washed with PBS and then incubated overnight with a staining solution including x-gal. Sections were counterstained with diluted eosin. In the case of the cultured keratinocytes, cells were let to grow until they were sub-confluent and were then fixed and stained as described above. Cells positive for the marker per field (10× magnification) were counted, and at least ten fields per genotype were scored. The mean and standard deviation were calculated for each genotype.
In vivo Imaging (PET)
2-[F-18]fluoro-2-deoxy-D-glucose (FDG), 300 μCi-11.1 MBq- in 0.2mL was i.v. injected after isoflurane anesthesia (2 min, 3% in 100% oxygen) in mice fasting 3–4 h before. Mice were kept awake in cage on a heating pad 30 min before FDG injection and during the uptake period before image acquisition. microPET imaging scans were obtained by using a microPET scanner (ARGUS RC equipment; SUINSA, Spain), and started 30 min after FDG injection, with mice anesthetized by isoflurane inhalation (1,5% in 100% oxygen; Fluovac device -Harvard Apparatus; Holliston, Massachussets, USA-). Imaging adquisition was whole body static during a period of 30 min. Images were reconstructed by 3D-OSEM analysis with the rPET_CT 4.7 Build 441 software (SUINSA and HGUGM biomedical research Foundation, Madrid, Spain).
Fluorescence-activated Cell Sorting analysis
For FACS analysis, oral keratinocytes from the different genotypes were incubated in PBS-EDTA 2mM for 30 minutes at 4°C, then treated with trypsin-EDTA for 5 minutes at 37°C. Cells were then washed in FACS buffer (PBS-FBS 1%, 0′09 azide) and incubated for 20 minutes with the indicated primary antibodies or the appropriate control antibodies. When fluorochrome-conjugated antibodies were used, cells were then washed and fixed in 2% paraformaldehyde. Otherwise, cells were washed, incubated with the appropriate fluorochrome-conjugated secondary antibody and then fixed. Cells were subsequently analyzed in an EPICS XL flow cytometer (Coulter Electronics). Primary antibodies used were rat mAb anti CD34,, and CD44 (Pharmingen), and rabbit polyclonals anti CD133 (Abcam) and ΔNp63 (Abcam).
RESULTS
The Akt activation has been previously involved in the development and progression of HNSCC (20–23). To further confirm this, the expression of phosphorylated Akt was studied in tissue microarrays containing 84 HNSCC and 59 oral dysplasias from human patients (not shown, examples are provided in Supp Fig 1). We found a significant number of dysplasias (42/59) and tumors (41/84) showing the increased Akt activity, suggesting that Akt activation is an early event during human HNSCC development.
To analyze the functions of deregulated Akt activity in vivo, we have recently generated transgenic mice expressing myrAkt in the basal layer of stratified epithelia (12, 13). Several founders and all mice corresponding to one of the established myrAkt lines, all of them characterized by displaying the highest levels of Akt activity (13), developed oral cavity lesions that were easily detectable as oral leukoplakia and erythroplakia in the palate, cheeks and lips (arrows in Fig 1A). Microscopic analyses showed hyperplasia of the tongue and palate glands with signs of adenocarcinomatous conversion (Fig 1A, compare with control in Fig 1A), in situ carcinomas of the oral mucosa (Fig 1A) and lip trichoepithelioma (Fig 1A). We confirmed the expression of the transgene and phosphorylated Akt, indicative of increased Akt activity, in the basal layer of the non lesional oral epithelia of myrAkt mice (Fig 1B), which remains in oral dysplasias (Fig 1B), trichoepithelioma (Fig 1B) and in oral tumors (Fig 1B). BrdU incorporation revealed a mild increase in cell proliferation of myrAkt non tumoral oral epithelia compared with non transgenic mice (Fig 2A and B), but we did not find further increase in dysplasias and tumor samples from myrAkt compared to non-tumoral tissue (Fig 2A and B). With respect to the process of epithelial differentiation, which is affected by deregulated Akt activity (12, 13, 22, 24), we detected an altered expression of keratins, with expansion of K5-expressing cells from the basal location into suprabasal compartment and the suprabasal coexpression of K5 and K13 in myrAkt oral epithelia compared to controls (Fig 2C). Overall, all myrAkt mice develop pretumoral lesions in the oral cavity with age.
Fig 1. Deregulated Akt activity produces preneoplastic lesions in the oral cavity of transgenic mice.

A) Examples of the gross appearance of leukoplakia (left panel) and erythroplakia (right panel) in myrAkt transgenic mice. Histological analysis of these lesions demonstrate the presence of tongue and palate gland hyperplasia compared to control (inset), in situ squamous carcinoma of the oral epithelia and trychoepithelioma of the lips. Bars = 200μm. B) The expression of the transgene and Akt activation can be determined by double immunofluorescence using antibodies against HA epitope and phosphorylated Akt (Ser473) in oral mucosa, oral dysplasia, trychoepithelioma and in situ squamous cell carcinoma in myrAkt transgenic mice.
Fig 2. Altered Proliferation and Differentiation in oral tissues of myrAkt mice.

A) BrdU incorporation demonstrates that the expression of myrAkt promotes increased proliferation, compared with control in non lesional oral mucosa, dysplasia and SCC of the transgenic mice. B) Quantitative analysis of BrdU incorporation. C) Keratin K5 and K13 expression demonstrate that the expression of myrAkt promotes altered differentiation, compared with control in non lesional oral mucosa of the myrAkt transgenic mice. Bars = 150 μm
Notably, although all the myrAkt mice develop oral lesions, few of them progress into aggressive squamous cell carcinoma (9/33). Since deregulated Akt activity may lead to premature senescence (25, 26), we analyzed whether such mechanism might explain the lack of malignant conversion in myrAkt mice. We observed that the areas of malignant tissue display a strong senescence associated β–galactosidase activity (SA-βgal) (denoted by “T” in Fig 3A, n=10), suggestive that in these regions premature senescence occurred, whereas the hyperplastic and dysplastic areas were predominantly SA-βgal negative (denoted by “hyp/dysp” in Fig 3A). In primary oral keratinocyte cultures a significant number of enlarged (Fig 3B) and SA-βgal positive cells (Fig 3B) were detected in myrAkt keratinocytes compared to control (Fig 3B), and myrAkt cultures also display a reduced growth rate (Fig 3C). Akt-induced senescence has been previously associated with the activation of p53-dependent mechanisms (26, 27); in agreement, western blot and luciferase assays (Fig 3D) revealed the increased expression of p53 and p53-dependent genes in primary oral myrAkt keratinocytes in parallel with the phosphorylation of Akt and Foxo3a and increased expression of βcatenin and ΔNp63 (Fig 3D). The mechanisms leading to premature senescence and thus p53 activation may involve overexpression of βcatenin (28), the induction of Pml (29–31) and/or the production of reactive oxygen species due to reduced expression and/or inhibition of Foxo3a (25, 32). Analysis of reactive oxygen species by FACS revealed no major differences between control and myrAkt oral keratinocytes (not shown). On the other hand, western blot analysis showed that the increased Akt activity of myrAkt cells is associated with the upregulation of Pml and βcatenin expression (Fig 3D), thus suggesting that, in this system, Akt can mediate a premature senescence that is associated with the increased expression of these two proteins.
Fig 3. Premature senescence induction by deregulated Akt activity.
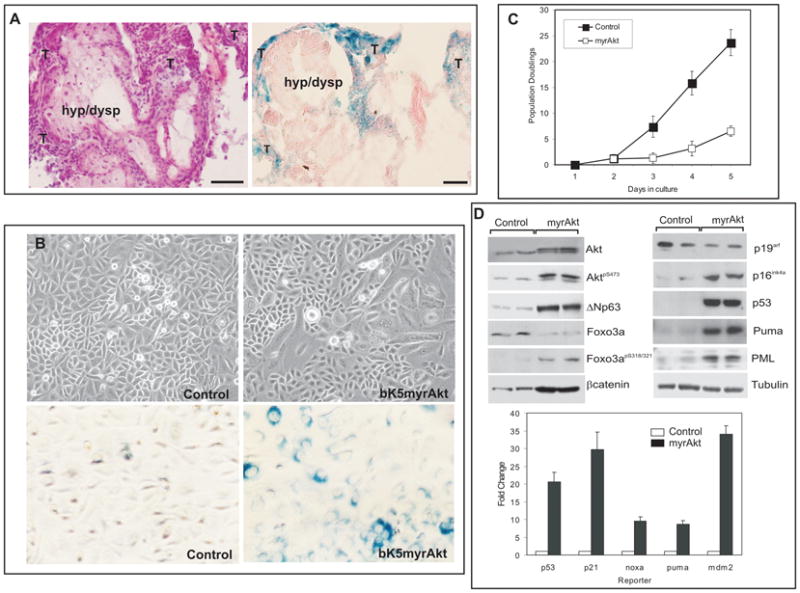
A) Example of Hematoxilyn-eosin stained and Senescence-associated βGal (SA-βGal) staining of oral SCC in myrAkt transgenic mice demonstrating the presence of SA-βGal positive areas in tumoral (denoted by T) but not in hyperplasic or dysplasic (denoted by hyp/dysp) parts. Bars =100 μm. B) Primary oral keratinocyte cultures from control and myrAkt transgenic mice show the presence of enlarged flattened cells in transgenic cultures. SA-βGal staining shows increased number of senescent cells in transgenic compared with control cultures. C) Doubling population of primary keratinocyte cultures derived from control or myrAkt transgenic mice. D) Western blot analysis of primary keratinocytes showing the increased expression of βcatenin, ΔNp63, p53, Puma, Pml, p16ink4a, phosphorylated Foxo3a and reduced total Foxo3a in parallel with increased phosphorylation of Akt. Of note, no changes were observed in p19arf; Tubulin was used as a loading control. Luciferase experiments in oral primary keratinocytes demonstrating increased transcription from p21, Noxa, Puma and mdm2 promoters together with increased activity of p53-responding element.
To functionally demonstrate that p53-mediated senescence was responsible for the reduced malignant conversion of oral lesions in myrAkt mice, we bred them with a mouse line bearing p53 loss in stratified epithelial tissues (Trp53F/F; K14Cre) (14, 15). While somatic ablation of the Trp53 gene in stratified epithelia leads to spontaneous tumor development (15) affecting primarily the epidermis and the oral cavity to a much lesser extent (14/57), the Trp53F/F;K14Cre;myrAkt mice displayed overt malignant tumors in the oral cavity (Fig 4A), which also affected the palate (Fig 4A) ventral side of the tongue (Fig 4A) and the external and internal side of the lips (Fig 4A). Tumor incidence studied in a large cohort of Trp53F/F;myrAkt (n=33), Trp53F/+;K14Cre;myrAkt (n=20) and Trp53F/FK14Cre;myrAkt mice (n=29) revealed that the complete absence of Trp53 leads also to earlier tumor appearance (Fig 4B).
Fig 4. Trp53F/F;K14Cre myrAkt mice develop malignant oral cavity tumors.

A) Gross appearance of oral cavity aggressive SCC in Trp53F/F;K14Cre myrAkt mice and examples of the histological appearance of aggressive tumors in the oral mucosa, gland and epithelia of the palate, ventral side of the tongue and internal side of the lips. B) Kaplan Meyer curves showing the percentage of tumor free mice as a function of the genotype and age showing the accelerated and complete penetrance of HNSCC in Trp53F/F;K14Cre myrAkt mice (p denotes the statistical significance by long rank test between Trp53F/F; myrAkt and Trp53F/F;K14Cre;myrAkt mice). C) Examples of PET imaging after FDG administration of Trp53F/F;K14Cre;myrAkt mice showing coronal (left), sagittal (mid) and axial (right) sections of a tumor bearing mouse at day 0, day 21 and day 42 after initial detection of the tumor (actual mouse age at day 0 was 6 weeks). Open arrows denotes the primary tumor, solid white arrows denote a distal secondary tumor and yellow arrows denote local secondary tumors. Dashed lines in coronal sections denote the corresponding position at which sagittal and coronal sections were obtained. Hg denotes harderian glands and b denotes brain. Bars in C= 1cm.
The use of mouse models as preclinical tools to asses the efficiency of targeted therapies requires that the tumors generated can be followed by in vivo imaging in individualized manner. This is of a particular relevance in the case of tumors arising in places where they can only be detected after necropsy, such as the internal parts of the oral cavity. Moreover this may allow a more defined quantification of the tumor growth properties and the presence of secondary tumors and/or metastasis. We thus studied whether the Trp53F/FK14Cre;myrAkt mouse tumors can be monitored by positron emission tomography (PET) imaging. Upon FDG administration, tumors can be easily detected in oral area (arrow in Fig 4C) and, importantly, the tumor progression at different time points can also be followed in vivo in the same animals (Fig 4C). In addition, in vivo PET imaging studies also revealed the development of secondary proximal (denoted by yellow arrows in Fig 4C) and distant tumors (denoted by white arrow in Fig 4C) in mice with time. Histopathological examination revealed that these distant tumors corresponded to lymph node metastases (Supplementary Fig 2). These metastases can be detected only when the primary tumors display undifferentiated highly aggressive characteristics (Supp Fig 2a) at advanced stages. The metastatic cells, detected by the expression of keratin K5, affected primarily the proximal lymph nodes located at the submandibular region (Supp. Fig 2b′-d). In addition, K5-positive cells were also detected infiltrating distal mesenteric lymph nodes (Supp Fig 2e) and forming micrometastatic nodules in the lungs (Supp Fig 2f, f′).
We have also generated two other transgenic mouse models. The first one expresses myrAkt and is deficient for pRb in stratified epithelial tissues (RbF/F;K14Cre) (18). The somatic loss of Rb1 in stratified epithelia does not lead to spontaneous tumor formation (18, 33). Neither the pathological characteristics of the oral lesions that developed in myrAkt mice nor the tumor development rate were affected in the absence of pRb in myrAkt mice (Supp Fig 3). The second model bears the simultaneous ablation of Pten (34) and Trp53 suppressor genes in stratified epithelia (PtenF/F; Trp53F/FK14Cre). In contrast to the corresponding parentals, mice lacking both Pten and p53 displayed a progressive weakening and died of yet unknown causes or had to be sacrificed due to ethical reasons (Supp Fig 4a). However, all these animals developed tumors in the oral cavity or lips with complete penetrance by 18 weeks of age (Supp Fig 4b). In some instances, only sporadic tumors, primarily localized in the mouth margins were observed in mice lacking Pten or p53 (Supp Fig 4c, d). Overall, the gross appearance (Supp Fig 4e), localization and the histological characteristics of the tumors (Supp Fig 4f–h) in double deficient mice were indistinguishable from those observed in Trp53F/FK14Cre;myrAkt mice. These results demonstrate that the activation of Akt, either by myrAkt expression or by Pten ablation, in conjunction with Trp53 (but not Rb1) loss leads to the development of malignant carcinomas in the oral region in mice.
We next studied whether p53 loss can overcome the myrAkt induced premature senescence. The expression of SA-βgal was almost negligible in tumors from Trp53F/FK14Cre;myrAkt mice (n=15; Fig 5A) compared with the expression of this senescence marker in myrAkt tumors (see Fig 3). Moreover, the proliferative arrest observed in myrAkt keratinocytes did not take place in Trp53F/FK14Cre;myrAkt cells (Supp Fig 5). In agreement proliferation was increased in Trp53F/FK14Cre;myrAkt compared with myrAkt oral tumors (Fig 5A and Supp Fig 6), and Trp53F/FK14Cre;myrAkt primary oral keratinocytes displayed no significant SAβgal staining (Fig 5B) nor reduced growth rate (not shown). Luciferase experiments also showed that, as expected, the induction of p53-dependent targets was negligible in Trp53F/FK14Cre;myrAkt keratinocytes (Supp Fig 7). These data also reinforce the suggestion that deregulated Akt contributes to a premature senescence that prevents further malignant conversion and increased proliferation, and that this process can be evaded by the loss of p53.
Fig 5. Trp53F/F;K14Cre myrAkt tumors bypass senescence and phenocopy human HNSCC alterations.

A) Example of Hematoxilyn-Eosin stained and senescence-associated βGal (SA-βgal) staining of oral SCC in Trp53F/F;K14Cre;myrAkt mouse showing the almost complete absence of SA-βGal staining in tumoral (T) and hyperplasic-dysplasic (hyp/dysp) areas. Bars in =100 μm. Lower panel shows the summary of proliferation analysis measured in non lesional, dysplasic and tumoral areas of the oral epithelia form control (black bars), myrAkt (blue bars) and Trp53F/F;K14Cre myrAkt (red bars) mice (statistical significance *, p≤0.01; **, p ≤ 0.005). B) Quantitative analysis of SA-βGal positive cells in primary oral cultures derived from mice of the quoted genotype. C) Western blot analysis of primary oral keratinocytes showing the expression of the quoted proteins. Lower panel shows the luciferase activity showing the increased activation of NFκB and Stat3 reporters in Trp53F/F;K14Cre myrAkt oral keratinocytes. D) Immunohistochemistry analysis of TGFβRII expression in myrAkt and Trp53F/F;K14Cre;myrAkt oral tumors showing the reduced expression in Trp53F/F;K14Cre;myrAkt sample. Squared areas show higher magnification or tumoral areas. (Bar in D=150μm)
Because deregulated Akt activity in keratinocytes can also modulate angiogenesis resulting in increased epithelial tumorigenicity (19), we have also analyzed this process in normal oral mucosa, dysplasia and tumor samples in myrAkt and Trp53F/FK14Cre;myrAkt mice. CD31 staining of normal oral epithelia of control and myrAkt mice revealed increased formation of blood vessels by the expression of constitutively active Akt (Supp Fig 8 and data not shown). CD31 staining also revealed that the number blood vessel was enhanced in dysplasias and tumors compared with normal oral epithelia in myrAkt mice (Supp Fig 8 and data not shown). However, we did not detect significant differences in the number of blood vessels in Trp53F/FK14Cre;myrAkt compared to myrAkt samples (Supp Fig 8 and data not shown). Similarly, the altered differentiation of oral epithelia promoted by myrAkt expression (Fig 2C) was not further aggravated by the subsequent p53 loss (Supp Fig 9). These results suggest that the augmented tumor susceptibility and malignancy mediated by ablation of the Trp53 gene in myrAkt mice is not primarily mediated by altered differentiation nor augmented angiogenesis.
Among the most common alterations found in human HNSCC are the activation of EGFR and the subsequent activation of NFκB and Stat3 pathways (35–37). We found by western blot analysis (Fig 5C) a mild increase in EGFR expression in myrAkt and Trp53F/FK14Cre;myrAkt keratinocytes compared with control cells. On the other hand, activation of EGFR (as observed by Tyr-phosphorylated EGFR; Fig 5C) was only observed in transgenic samples; although no significant differences were found between myrAkt and Trp53F/FK14Cre;myrAkt cells. Regarding the NFκB pathway, we observed decreased expression of IκBα and increased expression of IKKγ, IKKα, p50 and p65/RelA, suggesting increased NfκB activity. This was further corroborated by luciferase experiments (Fig 5C). With respect to Stat3 activation, no significant changes were observed in Tyr-phosphorylated, active Stat3 in myrAkt oral keratinocytes compared with controls or in luciferase experiments (Fig 5C). In contrast, Trp53F/FK14Cre;myrAkt keratinocytes showed a remarkable increase in both NFκB and Stat3 pathways (Fig 5C). Indeed, the expression of IκBα was further decreased, whereas the expression of p50, p65/relA, phosphorylated p65 and Tyr-phosphorylated Stat3 were increased in Trp53F/FK14Cre;myrAkt compared with myrAkt cells (Fig 5C). As above, these findings were further corroborated by luciferase experiments in primary oral keratinocytes using the same Stat3 and NFκB responding elements (Fig 5C). Together, these data indicated that activation of NfκB and Stat3 takes place in the Trp53F/FK14Cre;myrAkt cells independently from EGFR activation, which showed similar levels in Trp53F/FK14Cre;myrAkt and Trp53F/F;myrAkt keratinocytes.
Another common alteration of human HNSCC is the reduced expression of TGFβRII (38). We also observed by western blot a reduction in TGFβRII expression in myrAkt and in Trp53F/FK14Cre;myrAkt oral keratinocytes (Fig 5C). However, when tumors were studied by immunohistochemistry, we observed that the expression of TGFβRII was reduced in Trp53F/FK14Cre;myrAkt compared with myrAkt oral tumors (Fig 5D). Finally, we have previously shown that human HNSCC displaying increased Akt activation are characterized by several alterations such as increased expression of CycD1, c-myc, βcatenin and ΔNp63 and reduced Foxo3a expression (22). Western blot analysis of primary keratinocytes (Fig 5C), luciferase experiments and immunohistochemistry of tumor samples (Supp Fig 10) confirmed these alterations in Trp53F/FK14Cre;myrAkt tumors and keratinocytes, although no major differences were found between Trp53F/FK14Cre;myrAkt and myrAkt samples. Collectively, these data demonstrate that mouse Trp53F/FK14Cre;myrAkt tumors and primary cells recapitulate and phenocopy most of the molecular alterations found in human HNSCCs.
The initiation, growth, recurrence and metastasis of solid tumors, including HNSCC, have been related to the behavior of a small subpopulation of ‘tumor-initiating’ or cancer stem cells. In human HNSCC, these cells are characterized by the expression of specific markers such as CD44 and CD133 (8, 9). On the other hand, we have recently shown that myrAkt expression leads to an increased number of adult epidermal stem cells of the hair bulge characterized by K15 and CD34 expression (13). Consequently, we monitored whether the expression of myrAkt may affect stem cell behavior in the tumors and how this may be modulated by subsequent p53 loss. We observed increased expression of K15 and CD34 in parallel with Akt activity (Fig 6A) in tumors derived from Trp53F/F;K14Cre;myrAkt mice, compared with myrAkt samples (Fig 6A). We also noticed that the population expressing ΔNp63 in myrAkt tumors (Fig 6A) is increased due to the expansion into the suprabasal-like compartments in Trp53F/F;K14Cre;myrAkt samples (Fig 6A). Finally, we also observed CD44 and CD133 expression in myrAkt and in Trp53F/F;K14Cre;myrAkt tumors (Fig 6A). As in the case of ΔNp63, the number of cells expressing these cancer stem cell markers is increased in Trp53F/F;K14Cre;myrAkt samples compared to myrAkt tumors due to the expansion of positive population into suprabasal-like compartments (see insets in Fig 6A). To further corroborate these observations, we also carried out FACS analysis of primary oral keratinocytes. These demonstrated an increase in the population of cells expressing CD34, ΔNp63 and CD44 (Fig 6B) in myrAkt cultures, which in the case of CD34 is strikingly increased in Trp53F/F;K14Cre;myrAkt cells (Fig 6B). Nonetheless, no significant changes were observed in the population of cells expressing CD44 o ΔNp63 between myrAkt and Trp53F/F;K14Cre;myrAkt keratinocyte cultures (Fig 6B), indicating that, in both cases, the observed increase in tumors (Fig 6B) is limited to the in vivo situation. With respect to CD133, we observed, in agreement with previous data of human HNSCC lines (8, 9), that the number of cells expressing this cancer stem cell marker is very low, although it is augmented in myrAkt (4× fold) and further increased (10x fold) in Trp53F/F;K14Cre;myrAkt keratinocytes (Fig 6B). Collectively these data indicate that in our myrAkt mouse models there is an increase in the population of cells bearing characteristics of epithelial and tumor stem cells, specially in the case of Trp53F/F;K14Cre;myrAkt mouse.
Fig 6. Increased population of adult and putative cancer stem cells in Trp53F/F;K14Cre myrAkt mice.

A) Examples of double immunofluorescence images of myrAkt and Trp53F/F;K14Cre myrAkt tumors showing increased expression of K15, CD34, ΔNp63, CD44 and CD133. K5 was used to detect the epithelial component of the tumors. Right panel of A denotes the same area as in left and mid panels showing the expression of the transgene and activity of Akt analyzed by double immunofluorescence against HA epitope and phosphorylated Akt (Ser473). B) FACS analysis showing the expression of CD34,ΔNp63, CD44 and CD133 in primary oral keratinocyte cultures from mice of the quoted genotypes. Bars = 100 μm.
DISCUSSION
The recent improvements of therapies, using fractionated radiotherapy, targeted chemotherapy and concurrent radiotherapy and chemotherapy (1–4), have led only to a moderate increase in the overall survival in HNSCC patients. Molecularly targeted therapies are promising in HNSCC management and several candidate molecules of potential therapeutic relevance are now being validated through in vitro analyses (6, 10, 11). Therefore, in vivo systems aimed to the analysis of these therapies are necessary. However, these approaches have been hindered by a lack of appropriate animal models mimicking these tumors at both the pathological and molecular levels. In recent years several possible mouse models for HNSCC have been described. Nevertheless, the molecular characteristics of these tumors were not systematically compared with human counterparts. The first report of a genetically engineered mouse model was the targeted expression of cyclin D1 transgene to the oral-esophageal epithelium (L2-cyclinD1 transgenic mice) (39). These mice exhibited dysplasia in the tongue, esophagus, and forestomach by 16 months of age but did not develop tumors (39) unless they were crossbred with p53 heterozygous mice, which resulted in invasive oroesophageal cancer development in L2-cyclinD1/p53+/− mice (40). More recently, using inducible systems, it has been reported that the activation of an oncogenic K-rasG12D allele in the oral cavity of the mouse induces oral tumor formation in mice, but these lesions were classified as benign squamous papillomas (41). Finally, the TGFβRII deletion in combination with activation of either K-ras or H-ras in mouse head-and-neck epithelia caused HNSCC (38), in contrast with the targeted ablation of this gene in all stratified epithelia, which results in anal and genital SCCs (42), indicating that progression to cancers occurs rapidly when the TGFβRII null epithelial tissues are exposed to activated oncogenes and/or loss of additional tumor suppressors.
Here we present a novel and plausible mouse model of human HNSCC to study the consecutive steps involved in initiation and progression that will allow us to address questions like the cell of origin and the role of cancer stem cells in the maintenance of these tumors. In this mouse model, constitutively active Akt leads to dysplasia in oral tissues, demonstrating that the activation of Akt is sufficient to initiate oral carcinogenesis. In agreement, we found Akt activation in a vast majority of human oral dysplasias. Consequently the myrAkt mice could also be used in preclinical intervention and prevention studies. This is also of a particular relevance for studying radiotherapy, one of the current therapies for HNSCC, as the activation of Akt is associated with intrinsic radioresistance, tumor-cell proliferation, and hypoxia, the three major radioresistance mechanisms (reviewed in 43).
Importantly, in myrAkt transgenic mice, aggressive HNSCC are developed only sporadically due to the induction of p53-dependent premature senescence. Although the mechanism by which Akt activity induces premature senescence in oral lesions is unknown at present, the overactivation of βcatenin (28) and/or the induction of Pml (29–31) are likely candidates. Of note the expression of Pml is sustained in Trp53F/F;K14Cre;myrAkt cells (Fig 4D) whereas the transcriptional activity of βcatenin is slightly reduced in Trp53F/F;K14Cre;myrAkt keratinocytes (Supp Fig 8). On the other hand, this observation suggests that those therapies targeted to sustain p53 activation may help to prevent the development of aggressive SCCs. We also demonstrated that the loss of p53, but not of pRb, in conjunction with constitutive Akt activity abrogated premature senescence, and that myrAkt expression or Pten ablation, in mice bearing the p53 loss in epithelia allows the development of aggressive HNSCC. Similar processes of escape to premature senescence have been previously described for PTEN-deficient prostate tumor formation (26). We also demonstrated that several changes taking place in human HNSCC, and affecting EGFR, NFκB, Stat3 and TGFβRII (6, 7), also occur in the mouse tumors. These observations, together with the fact that these tumors can be followed by in vivo imaging using PET, and display some of the pathological characteristics presented by human lesions, strongly support that this represents a suitable preclinical model for intervention studies in which questions with respect to therapy response and resistance can be addressed (discussed in (43)).
Our observations, besides reinforcing the current criteria of premature senescence as a tumor suppression mechanism, also suggest the relevance of putative stem cells in the development of tumors. In agreement with our previous findings (13, 22), we observed that the constitutive Akt activity promotes increased expression of putative adult and cancer stem cell markers, which in particular cases are further increased due to Trp53 ablation. In conclusion, we have developed a mouse model of primary HNSCC that will be useful for future studies with translational applications including the molecular and genetic analysis of HNSCC, molecular imaging and testing new therapeutic agents.
Supplementary Material
Acknowledgments
We want to express our gratitude to Dr. A. Berns and Dr. P. Krimpenfort (Netherlands Cancer Institute, NKI) for providing different mutant mice, to Jesus Martinez and the personnel of the animal facility of CIEMAT for the excellent care of the animals, and to Pilar Hernandez (CIEMAT) for the histological preparations. This work is partially supported by Grants: SAF2005-00033 (MEC), Oncocycle (S2006/BIO-0232) from CAM, PS-090100-2006-3 form MICINN and ISCIII-RETIC RD06/0020 (MSC) to JMP and by NIH grant CA37111, NIEHS Center grant ES00784 and Cancer Center Support Grant CA16672 to JD.
Contributor Information
Marta Moral, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Carmen Segrelles, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Ana Belen Martinez-Cruz, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Corina Lorz, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Mirentxu Santos, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Ramon Garcia-Escudero, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Jerry Lu, Department of Carcinogenesis, Science Park-Research Division, University of Texas M.D. Anderson Cancer Center, Smithville, TX 78957, USA.
Kaoru Kiguchi, Department of Carcinogenesis, Science Park-Research Division, University of Texas M.D. Anderson Cancer Center, Smithville, TX 78957, USA.
Agueda Buitrago, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Clotilde Costa, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Cristina Saiz, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Jose L Rodriguez-Peralto, Pathology Department, Hospital Universitario 12 de Octubre, Crta. Andalucía, 5,4 28041 Madrid. Spain.
Francisco J Martinez-Tello, Pathology Department, Hospital Universitario 12 de Octubre, Crta. Andalucía, 5,4 28041 Madrid. Spain.
Maria Rodriguez-Pinilla, Centro Nacional de Investigaciones Oncológicas (CNIO), Melchor Fernandez Almagro, 3. 28029 Madrid, Spain.
Montserrat Sanchez-Cespedes, Centro Nacional de Investigaciones Oncológicas (CNIO), Melchor Fernandez Almagro, 3. 28029 Madrid, Spain.
Marina Garin, Division of Hematopoiesis and Gene Therapy, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Teresa Grande, Unit of Medical Applications, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
Ana Bravo, Department of Veterinary Clinical Sciences, Veterinary Pathology Unit, Veterinary Faculty, University of Santiago de Compostela, E-27002 Lugo, Spain.
John DiGiovanni, Department of Carcinogenesis, Science Park-Research Division, University of Texas M.D. Anderson Cancer Center, Smithville, TX 78957, USA.
Jesus M. Paramio, Molecular Oncology Unit. Division of Biomedicine, CIEMAT. Ave. Complutense 22, E-28040 Madrid, Spain.
References
- 1.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006 Feb 9;354(6):567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 2.Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003 Nov 27;349(22):2091–8. doi: 10.1056/NEJMoa031317. [DOI] [PubMed] [Google Scholar]
- 3.Bourhis J, Overgaard J, Audry H, et al. Hyperfractionated or accelerated radiotherapy in head and neck cancer: a meta-analysis. Lancet. 2006 Sep 2;368(9538):843–54. doi: 10.1016/S0140-6736(06)69121-6. [DOI] [PubMed] [Google Scholar]
- 4.Baselga J, Trigo JM, Bourhis J, et al. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol. 2005 Aug 20;23(24):5568–77. doi: 10.1200/JCO.2005.07.119. [DOI] [PubMed] [Google Scholar]
- 5.Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin. 2002 Jul–Aug;52(4):195–215. doi: 10.3322/canjclin.52.4.195. [DOI] [PubMed] [Google Scholar]
- 6.Lu SL, Herrington H, Wang XJ. Mouse models for human head and neck squamous cell carcinomas. Head Neck. 2006 Oct;28(10):945–54. doi: 10.1002/hed.20397. [DOI] [PubMed] [Google Scholar]
- 7.Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004 Apr;5(4):311–6. doi: 10.1016/s1535-6108(04)00090-x. [DOI] [PubMed] [Google Scholar]
- 8.Harper LJ, Piper K, Common J, Fortune F, Mackenzie IC. Stem cell patterns in cell lines derived from head and neck squamous cell carcinoma. J Oral Pathol Med. 2007 Nov;36(10):594–603. doi: 10.1111/j.1600-0714.2007.00617.x. [DOI] [PubMed] [Google Scholar]
- 9.Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007 Jan 16;104(3):973–8. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Ordonez B, Beauchemin M, Jordan RC. Molecular biology of squamous cell carcinoma of the head and neck. J Clin Pathol. 2006 May;59(5):445–53. doi: 10.1136/jcp.2003.007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jimeno A, Kulesza P, Wheelhouse J, et al. Dual EGFR and mTOR targeting in squamous cell carcinoma models, and development of early markers of efficacy. Br J Cancer. 2007 Mar 26;96(6):952–9. doi: 10.1038/sj.bjc.6603656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Segrelles C, Lu J, Hammann B, et al. Deregulated Activity of Akt in Epithelial Basal Cells Induces Spontaneous Tumors and Heightened Sensitivity to Skin Carcinogenesis. Cancer Res. 2007 November 15;67(22):10879–88. doi: 10.1158/0008-5472.CAN-07-2564. [DOI] [PubMed] [Google Scholar]
- 13.Segrelles C, Moral M, Lorz C, et al. Constitutively Active Akt Induces Ectodermal Defects and Impaired Bone Morphogenetic Protein Signaling. Mol Biol Cell. 2008 Jan;19(1):137–49. doi: 10.1091/mbc.E07-08-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29(4):418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Cruz AB, Santos M, Lara MF, et al. Spontaneous squamous cell carcinoma induced by the somatic inactivation of retinoblastoma and Trp53 tumor suppressors. Cancer Res. 2008 Feb 1;68(3):683–92. doi: 10.1158/0008-5472.CAN-07-3049. [DOI] [PubMed] [Google Scholar]
- 16.Lara MF, Garcia-Escudero R, Ruiz S, et al. Gene profiling approaches help to define the specific functions of retinoblastoma family in epidermis. Mol Carcinog. 2008 Mar;47(3):209–21. doi: 10.1002/mc.20376. [DOI] [PubMed] [Google Scholar]
- 17.Lara MF, Santos M, Ruiz S, et al. p107 acts as a tumor suppressor in pRb-deficient epidermis. Mol Carcinog. 2008 Feb;47(2):105–13. doi: 10.1002/mc.20367. [DOI] [PubMed] [Google Scholar]
- 18.Ruiz S, Santos M, Segrelles C, et al. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development. 2004 Jun;131(11):2737–48. doi: 10.1242/dev.01148. [DOI] [PubMed] [Google Scholar]
- 19.Segrelles C, Ruiz S, Santos M, Martinez-Palacio J, Lara MF, Paramio JM. Akt mediates an angiogenic switch in transformed keratinocytes. Carcinogenesis. 2004 Jul;25(7):1137–47. doi: 10.1093/carcin/bgh132. [DOI] [PubMed] [Google Scholar]
- 20.Amornphimoltham P, Sriuranpong V, Patel V, et al. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res. 2004 Jun 15;10(12 Pt 1):4029–37. doi: 10.1158/1078-0432.CCR-03-0249. [DOI] [PubMed] [Google Scholar]
- 21.Molinolo AA, Hewitt SM, Amornphimoltham P, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. 2007 Sep 1;13(17):4964–73. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- 22.Segrelles C, Moral M, Lara MF, et al. Molecular determinants of Akt-induced keratinocyte transformation. Oncogene. 2006 Feb 23;25(8):1174–85. doi: 10.1038/sj.onc.1209155. [DOI] [PubMed] [Google Scholar]
- 23.Pedrero JM, Carracedo DG, Pinto CM, et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005 Mar 20;114(2):242–8. doi: 10.1002/ijc.20711. [DOI] [PubMed] [Google Scholar]
- 24.Segrelles C, Ruiz S, Perez P, et al. Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene. 2002;21(1):53–64. doi: 10.1038/sj.onc.1205032. [DOI] [PubMed] [Google Scholar]
- 25.Minamino T, Miyauchi H, Tateno K, Kunieda T, Komuro I. Akt-induced cellular senescence: implication for human disease. Cell Cycle. 2004 Apr;3(4):449–51. [PubMed] [Google Scholar]
- 26.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005 Aug 4;436(7051):725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007 Jan;27(2):662–77. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saegusa M, Hashimura M, Kuwata T, Hamano M, Okayasu I. Beta-catenin simultaneously induces activation of the p53-p21WAF1 pathway and overexpression of cyclin D1 during squamous differentiation of endometrial carcinoma cells. Am J Pathol. 2004 May;164(5):1739–49. doi: 10.1016/s0002-9440(10)63732-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Stanchina E, Querido E, Narita M, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell. 2004 Feb 27;13(4):523–35. doi: 10.1016/s1097-2765(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 30.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002 Jan 25;108(2):165–70. doi: 10.1016/s0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 31.Pearson M, Carbone R, Sebastiani C, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000 Jul 13;406(6792):207–10. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 32.Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. Embo J. 2004 Jan 14;23(1):212–20. doi: 10.1038/sj.emboj.7600045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz S, Santos M, Lara MF, Segrelles C, Ballestin C, Paramio JM. Unexpected roles for pRb in mouse skin carcinogenesis. Cancer Res. 2005 Nov 1;65(21):9678–86. doi: 10.1158/0008-5472.CAN-05-1853. [DOI] [PubMed] [Google Scholar]
- 34.Marino S, Krimpenfort P, Leung C, et al. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development. 2002;129(14):3513–22. doi: 10.1242/dev.129.14.3513. [DOI] [PubMed] [Google Scholar]
- 35.Allen CT, Ricker JL, Chen Z, Van Waes C. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 2007 Oct;29(10):959–71. doi: 10.1002/hed.20615. [DOI] [PubMed] [Google Scholar]
- 36.Leeman RJ, Lui VW, Grandis JR. STAT3 as a therapeutic target in head and neck cancer. Expert Opin Biol Ther. 2006 Mar;6(3):231–41. doi: 10.1517/14712598.6.3.231. [DOI] [PubMed] [Google Scholar]
- 37.Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63(11):2948–56. [PubMed] [Google Scholar]
- 38.Lu SL, Herrington H, Reh D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006 May 15;20(10):1331–42. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakagawa H, Wang TC, Zukerberg L, et al. The targeting of the cyclin D1 oncogene by an Epstein-Barr virus promoter in transgenic mice causes dysplasia in the tongue, esophagus and forestomach. Oncogene. 1997 Mar 13;14(10):1185–90. doi: 10.1038/sj.onc.1200937. [DOI] [PubMed] [Google Scholar]
- 40.Opitz OG, Harada H, Suliman Y, et al. A mouse model of human oral-esophageal cancer. J Clin Invest. 2002 Sep;110(6):761–9. doi: 10.1172/JCI15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caulin C, Nguyen T, Longley MA, Zhou Z, Wang XJ, Roop DR. Inducible activation of oncogenic K-ras results in tumor formation in the oral cavity. Cancer Res. 2004 Aug 1;64(15):5054–8. doi: 10.1158/0008-5472.CAN-04-1488. [DOI] [PubMed] [Google Scholar]
- 42.Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007 Oct;12(4):313–27. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bussink J, van der Kogel AJ, Kaanders JH. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. The lancet oncology. 2008 Mar;9(3):288–96. doi: 10.1016/S1470-2045(08)70073-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.