

Abstract
Regio- and diastereoselective reactions of a homoproline enolate enable the synthesis of novel extended dipeptide surrogates. Bicyclic carbamate 9 and fused β-lactam scaffold 11 were prepared from L-pyroglutamic acid via substrate-controlled electrophilic azidation. Synthesis of orthogonally-protected hexahydropyrrolizine, hexahydropyrrolizinone, and hexahydropyrroloazepinone dipeptide surrogates relied on allylation of proline derivative 5, followed by Curtius rearrangement to introduce the N-terminal carbamate group. A total of six azabicycloalkane derivatives were evaluated for conformational mimicry of extended dipeptides by a combination of x-ray diffraction and molecular modeling. Analysis of putative backbone dihedral angles and N- to C-terminal dipeptide distances indicate that compounds (α′S)-14b and 21 approximate the conformation of dipeptides found in β-sheets, while tripeptide mimic 28 is also highly extended in the solid state. Structural data suggest that ring size and relative stereochemistry have a profound effect on the ability of these scaffolds to act as β-strand mimetics and should inform the design of related conformational probes.
Keywords: Beta-strands, peptidomimetics, proline, extended peptides, dipeptide surrogates, azabicycloalkanes, constrained amino acids
Introduction
The β-strand consists of a highly extended or “sawtooth” amino acid arrangement and is the simplest peptide secondary structure motif. β-strands lack intramolecular hydrogen bonds between backbone residues and typically interact with complimentary peptide chains to form β-sheets. These super-secondary structures are key recognition elements in protein-protein and protein-DNA interactions relevant to cell proliferation, infectious diseases and neurological disorders.1 The biological relevance of such interactions has prompted the design and synthesis of various peptidomimetic β-strand inducers. Nucleation of β-strand conformations in model systems has generally relied on β-hairpin templates that facilitate intramolecular backbone contacts between peptide appendages and extended peptide surrogates that replace short sections of the peptide backbone.2
While conformationally extended peptidomimetics have often been employed to study the dynamics of β-sheet formation,3 a number of important enzymatic and protein surface binding events involve interactions with the side-chain pharmacophores of isolated β-strand substrates.2,4 For example, farnesyl transferases and Akt (PKB), both of which are implicated in oncogenesis, are notable examples of proteins that recognize single β-strand peptides5 as well as constrained isosteres.6 Previously reported scaffolds often feature substituted aromatic motifs to impart backbone rigidity (Figure 1).2b,3g,6a,7 Artificial β-strands comprised entirely of non-peptidic subunits have also been developed as potential disruptors of protein-protein interactions.8
Figure 1.

Selected examples of β-strand peptidomimetics.
In efforts toward β-strand peptidomimetics targeting cell signaling pathways, our laboratory is pursuing the synthesis of constrained scaffolds to mimic the conformational and electronic properties of extended peptides. Although rigid dipeptide mimics have been extensively studied with respect to turn nucleation, extended dipeptide surrogates are less common.9 Peptidomimetics based on azabicycloalkane scaffolds, for example, have been widely employed as peptide turn inducers and their synthesis has been the subject of comprehensive reviews.10 Conceptually, these scaffolds arise from a 3-amino (Freidinger-type) lactam constraint,11 followed by additional covalent tethering to afford structures of type A (Figure 2). We envisioned that a 4-, 5-, or 6-amino lactam constraint (transposition of the carbonyl group) could be introduced in conjunction with a second backbone tether to provide scaffolds of type B. The unique azabicycloalkane substitution pattern and highly constrained ψ, φ, and ω dihedral angles are designed to maintain a sawtooth peptide backbone arrangement.
Figure 2.

Design of azabicycloalkane-based dipeptide surrogates.
Here, we report our efforts toward novel dipeptide surrogates based on structure B. The synthesis of these scaffolds relies on regio- and diastereoselective reactions of a homoproline enolate and subsequent elaboration into bicyclic core structures. The current work highlights the synthetic versatility of key chimeric proline intermediates and explores the ability of our scaffolds to mimic the conformation of extended dipeptides. Although structures such as B are devoid of amino acid side chain functionality, the established role of the extended peptide conformation in molecular recognition suggests that these surrogates could serve as useful β-strand inducers and conformational probes.
Results and Discussion
Synthesis
We recently reported the diastereoselective electrophilic azidation of a homoproline en route to analogs of the Pseudomonas siderophore pyochelin (Scheme 1).12 In our studies, it became apparent that the ability to react enolates of 2 with alkylating reagents could lead to synthetically useful and diversely substituted proline derivatives. Although similar alkylations of urethane-protected homoprolines have been reported to proceed uneventfully,13 we found that the enolates formed from 2 failed to give satisfactory yields of α′-substituted products. In most cases, we recovered unreacted starting material along with ring-opened enoates resulting from reverse-Michael addition. In contrast, when the urethane protecting group was replaced with a methyl substituent (3), azidation in the presence of LiHMDS and 2,4,6-triisopropylbenezensulfonyl azide afforded 4 in 75% isolated yield. Analysis of the crude product mixture by 1H NMR revealed the presence of only one diastereomer, later identified as the anti isomer by x-ray diffraction of an advanced intermediate. The stereochemical outcome can be rationalized by minimization of 1,3-allylic strain and reaction with the electrophile on the less hindered face of the enolate.14
Scheme 1.

Diastereoselective azidation of homoproline derivative 3.
While derivative 4 served as a useful precursor to the carbapyochelins (which also harbor an N-Me group), failed attempts at demethylation15 severely limited the synthetic utility of the azidation product. We then evaluated the N-benzyl group as a more convenient protecting group alternative. As shown in Scheme 2, acidolysis of the Boc group of 2a was followed by benzylation to give derivative 5 in 67% yield. Electrophilic azidation of 5 under the same conditions used for 312 resulted in low conversion, indicating that the benzyl group has a deleterious effect on the reaction. After some optimizitaion it was found that the addition of HMPA in the presence of 2.2 equivalents of KHMDS and 2.2 equivalents of 2,4,6-triisopropylbenezensulfonyl azide gave 84% yield of the desired product (6a) after acetic acid quenching. We later confirmed that HMPA was essential to ensure good conversions in the reactions of 5 with other electrophiles. Similar conditions in the presence of methylbromoacetate afforded 45% isolated yield of 6b, while the use of allyl bromide resulted in 95% yield of 6c. As with proline 3, only one diastereomer was observed in reactions with the enolate derived from 5. Single-crystal x-ray diffraction carried out on 6a confirmed the expected stereochemistry at the newly formed α′ chiral center.
Scheme 2.

Synthesis of scaffolds 9 and 11.
Intermediate 6a was elaborated into novel bicyclic scaffolds as depicted in Scheme 2. Azide reduction and Boc protection gave rise to the orthogonally protected bis-amino acid 7. Ethyl ester reduction with lithium borohydride then afforded amino alcohol 8 in high yield. Finally, carbamate scaffold 9 was obtained after hydrogenolysis and treatment of the crude amine with 1,1′-carbonyldiimidazole. A 1-azabicyclo[3.2.0]heptan-7-one scaffold (11) was also efficiently prepared from intermediate 7 by way of protecting group removal and treatment with Mukaiyama’s condensation reagent. Although β-lactam 11 shares its core structure with the carbapencillins and is reminiscent of β-turn-inducing azabicycloalkanes, its evaluation in the context of dipeptide mimicry has not been previously investigated. Unfortunately, while 11 was stable to flash chromatography over silica gel, we found that an unsoluble gel formed during attempted acidolysis of the N-Boc group (TFA/DCM), and even upon prolonged exposure to CHCl3.
We next turned our attention to the synthesis of larger bicyclic lactams starting from 6b and 6c (Scheme 3). Although we previously observed modest conversion of 5 into triester 6b, the introduction of a methoxycarbonylmethyl group allowed rapid access to a hexahydropyrrolizinone scaffold. Hydrogenolysis of the N-benzyl group, followed by heating in toluene, resulted in selective lactamization to afford 12 in 89% yield. Somewhat surprisingly, we found that treatment of diester 12 with 1M aq. LiOH in MeOH resulted in isolation of the diacid as the major product. The unusual lability of the tert-butyl ester required the use of 1.5 equivalents of LiOH and close monitoring to effect selective saponification.16 Although near-quantitave yield of 13 was obtained, the 1H and 13C NMR specta revealed significant epimerization despite the mild hydrolysis conditions. The inseparable acids were then subjected to Curtius rearrangement in the presence of various alcohols to give carbamates 14a–c as diastereomeric mixtures.
Scheme 3.

Epimerization en route to bicyclic lactams 14 and 17.
In order to prepare a homologous hexahydropyrroloazepinone core, allyl derivative 6c was subjected to cross-metathesis with benzyl acrylate to give 15. Removal of both benzyl groups and concomitant reduction of the alkene was followed by lactamization in the presence of HBTU (O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate) to give 16. As with 12, alkaline hydrolysis of the ethyl ester resulted in two diastereomeric lactam products. However, in this case the tert-butyl ester group remained intact in the presence of 1M aq. NaOH. The mixture of diastereomers were then subjected to Curtius rearrangement to give compounds 17, which were separable by careful column chromatography over silica gel. 1D nOe studies revealed a correlation between the carbamate N-H proton and Hδ in only one of the two diastereomers of 17. These results confirmed that epimerization occurs at the α′ rather than α center, and allowed stereochemical assignment of each product.
To circumvent the configurational lability of 12 and 16, we opted to change the order of operations in our synthetic strategy. As shown in Scheme 4, the ethyl ester of 6c was efficiently hydrolyzed without epimerization in the presence of 2M aq. NaOH and catalytic tetrabutylammonium hydroxide at 50°C.17 Curtius rearrangement in the presence of 2,2,2-trichloroethanol then afforded orthogonally-protected diamino acid 19 in good yield. Hexahydropyrrolizinone 21 was formed via oxidative olefin cleavage, followed by aldehyde oxidation, hydrogenolysis, and lactamization in the presence of HBTU. To access the hexahydropyrroloazepinone variant (23), intermediate 19 was subjected to cross metathesis prior to hydrogenation and condensation. We found the hydrogenation steps particularly challenging due to the lability of the chlorine atoms of the trichloroethoxycarbonyl (Troc) protecting groups. The use of Pearlman’s catalyst was required for efficient removal of the N-benzyl group, but the des-chloro ethyl carbamate analogs of 21 and 23 were also isolated as side products. Optimized hydrogenation conditions and careful monitoring of reaction progress did, however, provide 21 and 23 as single diastereomers suitable for incorporation into peptide host sequences.
Scheme 4.
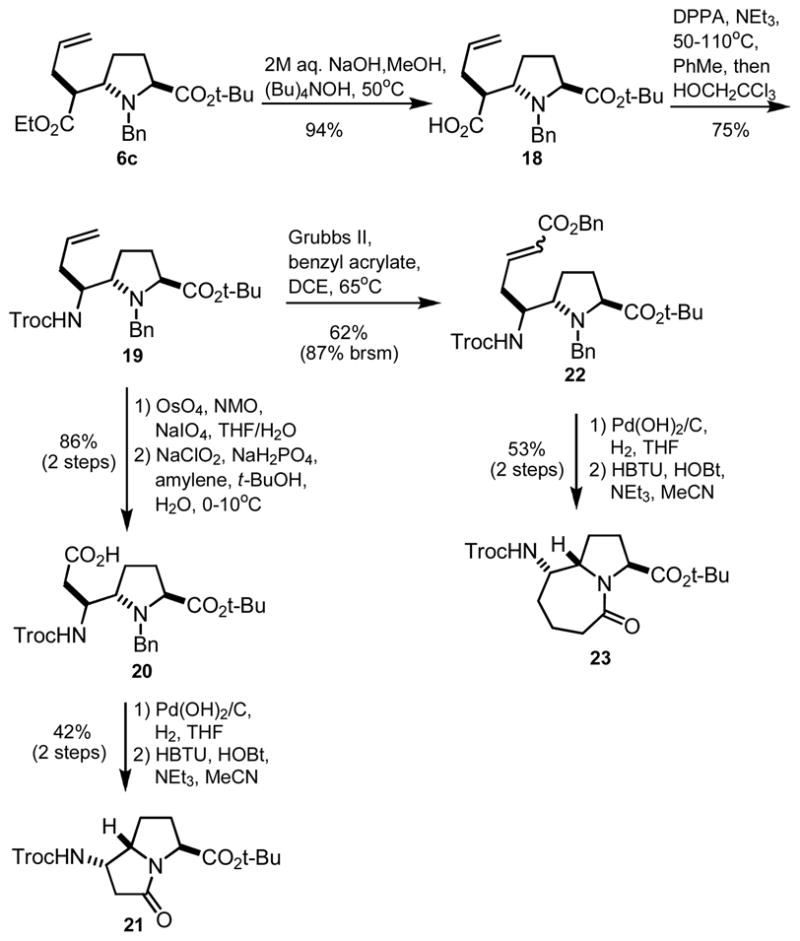
Diastereoselective synthesis of 21 and 23.
The synthesis of a [5,7]-fused carbamate from N-trimethylsilylethoxycarbonyl derivative 24 was also investigated (Scheme 5). In this case, dihydroxylation and cleavage of olefin 24 was followed by aldehyde reduction and debenzylation. Treatment of the resulting amine with 1,1′-carbonyldiimidazole (CDI) resulted in the formation of hexahydropyrrolizine 26 instead of the desired 7-membered cyclic carbamate, likely through 5-exo-tet displacement.18 Attempts to carry out the same transformation with triphosgene and nitrophenylchloroformate also failed to provide carbamate 27.
Scheme 5.

Unexpected formation of hexahydropyrrolizine 26.
We next carried out the coupling of our scaffolds in order to demonstrate their suitability for incorporation into peptides (Scheme 6). Various attempts to selectively remove the N-Boc group in 9 under mildly acidic conditions resulted in concomitant tert-butyl ester cleavage. The previously observed sensitivity of the tert-butyl ester in compound 12 toward hydrolysis prompted us to investigate aqueous base as a selective deprotection alternative. We found that 2M aq. NaOH at room temperature was effective for providing the carboxylic acid in high yield. Coupling to phenylalanine methyl ester proceeded uneventfully to give tripeptide mimic 28 in diastereomerically pure form, indicating negligible epimerization during hydrolysis and C-terminal condensation. In the case of lactams 21 and 23, tert-butyl ester cleavage with TFA was followed by condensation with phenylalanine tert-butyl ester to give 29 and 30, which were also diastereomerically pure by RP-HPLC and NMR.
Scheme 6.
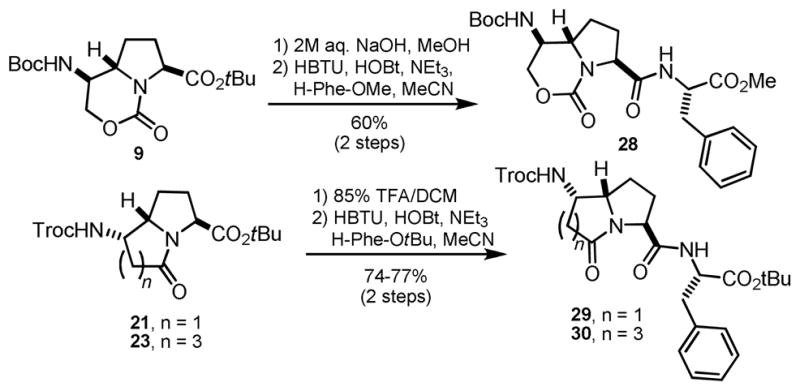
C-terminal coupling of scaffolds 9, 21, and 23.
Finally, we sought to demonstrate the ability to introduce lactam constraints following incorporation into a short peptide (Scheme 7). We utilized the configurationally stable N-benzyl derivative 19 as a starting material for dipetide formation. Cross-metathesis, hydrogenation, and lactamization, as described above, afforded tripeptide mimic 30 in an slightly higher overall yield relative to the route in Scheme 6. Analysis by RP-HPLC and NMR revealed a single diastereomer. Compound 31 was also transformed into lactam 29 in reasonable yield and high diastereomeric purity. This strategy represents a potentially useful alternative in cases where peptide coupling to pre-formed bicyclic scaffolds may present a challenge.
Scheme 7.

Introduction of lactam constraint after incorporation.
Configurational and Conformational Analysis
The ability of the above described azabicycloalkanes to act as structurally defined dipeptide surrogates was evaluated by a combination of x-ray crystallography and molecular modeling. With respect to β-strand mimicry, we have targeted scaffolds that severely restrict the ψ and φ dihedral angles of sequential amino acid residues (in addition to the ω torsion defined by the central trans amide bond). In β-sheet peptides, ψ and φ torsions are generally of similar magnitude (in the range of 113° to 139°), but of opposite sign.9,19 This alternation is critical for maintaining an extended conformation over longer sections of a peptide. Due to the transposition of the carbonyl group in our scaffolds, we have labeled the dihedral angles in relation to the backbone torsions they are meant to replace, as shown in Figure 3.
Figure 3.

Backbone torsions and N-to-C dipeptide distance for β-strands and synthetic dipeptide surrogates.
The x-ray structure of (α′S)-14b, the major isomer of which crystallized out of EtOAc/hexanes, is shown in Figure 4A. In the solid state, (α′S)-14b exhibits an “up-down” relationship between the N-H and C-terminal carbonyl group reminiscent of a sawtooth extended dipeptide. Moreover, the distance between the terminal nitrogen and carbonyl carbon is 5.7Å, as compared to ~5.9Å typically found across the dipeptide of a β-strand. Examination of the putative ψ1 and φ2 dihedral angles in (α′S)-14b reveals values of −112.8° and +102.4°, respectively. These values correspond well to the torsions found in a typical parallel β-sheet peptide.20 In addition, the ω surrogate dihedral angle is +156.8°, which deviates only slightly from the ideal 180° despite the tetrahedral geometry at C5. Bicyclic lactam 21, which differs only in N-terminal protection, is expected to exhibit the same conformational characteristics as (α′S)-14b. Since 21 is readily accessible in diastereomerically pure form, it should serve as a useful building block for the introduction of a β-strand dipeptide mimic into host structures.
Figure 4.

X-ray structures and calculated torsions, in degrees, for compounds (α′S)-14b, 23, and 28 (most hydrogens omitted for clarity).
Hexahydropyrroloazepinone 23 yielded diffraction quality crystals by slow evaporation from diethyl ether/hexanes (Figure 4B). While the putative φ2 angle in compound 23 is close to that found in hexahydropyrrolizinone (α′S)-14b, the ψ1 torsion deviates significantly as the result of the axial disposition of the Troc-carbamate substituent. Moreover, the putative ω angle is more acute relative to that in the hexahydropyrrolizinone scaffold. Taken together, these constraints result in an N- to C-terminal distance much shorter (4.8 Å) than that expected for an extended dipeptide.
Although we did not obtain diffraction quality crystals of carbamate scaffold 9, we did obtain an x-ray structure of phenylalanyl derivative 28 (Figure 4C). Interestingly, this compound features two intermolecular H-bonds at either end of the bicyclic scaffold and exists as a head-to-tail dimer in the solid state. Since the rigid core is intended to replace a sawtooth dipeptide, the observed conformation for the N-H and carbonyl groups deviates from the expected alternating pattern (both the hydrogen bond donor and acceptor are oriented in the same direction, linking the two molecules in a macrocyclic motif). In addition to crystal packing forces, this conformation is assumed to be largely dependent on the stereochemical relationship between the chiral centers of the bicyclic scaffold. As opposed to compounds (α′S)-14b and 23, the terminal acyloxyamine in 28 resides on the exo face of the ring system. This difference is clearly manifested in the ψ1 dihedral angle of +171.4°,21 which is not only higher magnitude, but of the same sign as the φ1 torsion. As with (α′S)-14b, the distance between the terminal nitrogen and the scaffold carbonyl carbon in 28 was near that observed in an ideal extended dipeptide (5.9 Å for each molecule in the dimer).
Azabicycloalkanes 11, (α′R)-17, and 26 represent additional scaffolds that were synthetically accessible, but not initially targeted as β-strand mimics. Still, we sought to evaluate their conformational properties by molecular modeling after attempts to obtain diffraction quality crystals were unsuccessful. We performed a conformational search using Macromodel with the MM3* force field.22 In each case the 50 lowest energy conformers exhibited only slight differences in the constrained dihedral angles. Calculated torsions and distances from molecular mechanics (given for the lowest energy conformer) are shown in Table 1.
Table 1.
Calculated torsions (in degrees) and distances (in Å) for 11, (α′R)-17, and 26 from MM3* conformational searches.22
| scaffold | Ψ1 | φ2 | ω | N…CO distance |
|---|---|---|---|---|
| 11 | +116.0 | +115.6 | +148.0 | 5.4 |
| (α′R)-17 | +160.3 | +105.0 | +111.6 | 5.9 |
| 26 | −147.3 | +147.1 | +121.2 | 6.0 |
As expected, azabicyclo[3.2.0]heptan-7-one scaffold 11 exhibits ψ1 and φ2 angles that are of the same sign (as in the case of 28), owing to the exo carbamate substituent. The low energy conformer of compound (α′R)-17 features a similar relationship, in addition to a putative ω torsion that deviates significantly from planarity. Energy minimization of hexahydropyrrolizine 26, which lacks a carbonyl group, resulted in an opening of the ψ1 torsion relative to the bicyclic lactam. However, the calculated ω dihedral angle closes as the result of a change in nitrogen bond order. Although scaffold 26 is not isoelectronic with a native peptide backbone, its structure and conformation suggest potential utility as an extended dipeptide scaffold. We further note that compound 26 was also synthesized in a more direct manner from 24 by oxidation followed by hydrogenolysis (75% overall yield). 23
Finally, the x-ray structures in Figure 4 serve to establish the relative configuration of each bicyclic scaffold. The anti relationship resulting from functionalization of 5 is thus confirmed, supporting the proposed A1,3-minimized stereochemical model for the N-benzyl series. The structure of 23 also provides confirmation of the proposed stereochemistry of 21 and 26, both of which are derived from allylated intermediate 6c.
Conclusion
We have prepared a series of novel azabicycloalkanes as potential extended dipeptide surrogates via regio- and stereoselective reactions of a chimeric homoproline enolate. Orthogonally-protected bicyclic scaffolds are obtained in reasonable overall yields and high diastereomeric purities. Conformational analysis by x-ray diffraction and molecular modeling indicate that relative stereochemistry and ring size have a profound effect on key dihedral angles. Hexahydropyrrolizinones (α′S)-14b and 21 share a number of conformational characteristics with extended dipeptides found in parallel β-sheets. Scaffold 9 may also serve as a useful extended dipeptide surrogate based on x-ray structure data obtained for tripeptide mimic 28. The synthetic routes described here highlight the versatility of proline derivative 5 and should allow access to additional probes of local peptide conformation. We are currently pursuing the synthesis of functionalized derivatives to mimic a wider array of extended dipeptides. Studies on the incorporation of selected scaffolds into β-strand peptides involved in oncogenic signaling are also underway in our laboratory.
Experimental Section
General
Unless stated otherwise, reactions were performed in flame-dried glassware under a positive pressure of argon or nitrogen gas using dry solvents. Commercial grade reagents and solvents were used without further purification except where noted. Diethyl ether, toluene, dimethylformamide dichloromethane, and tetrahydrofuran were purified by solvent purification system. Other anhydrous solvents were purchased directly from chemical suppliers. Thin-layer chromatography (TLC) was performed using silica gel 60 F254 pre-coated plates (0.25 mm). Flash chromatography was performed using silica gel (60 μm particle size). The purity of all compounds was judged by TLC analysis (single spot/two solvent systems) using a UV lamp, CAM (ceric ammonium molybdate), ninhydrin, or basic KMnO4 stain(s) for detection purposes. NMR spectra were recorded on a 400 MHz spectrometer. 1H and 13C NMR chemical shifts are reported as δ using residual solvent as an internal standard. Analytical high performance liquid chromatography (HPLC) was performed C18 reverse phase analytical column. HPLC elution was carried out with a 20 min linear gradient of MeCN in water (each containing 0.1% formic acid buffer).
(2S,5S)-tert-butyl 1-benzyl-5-(2-ethoxycarbonylmethyl)pyrrolidine-2-carboxylate (5)
A solution of 2a (9.50 g, 26.6 mmol) in 10% TFA/DCM was stirred at rt for 5 h. The reaction solution was diluted with EtOAc and evaporated under reduced pressure (dilution and evaporation was repeated two times). The resulting sticky foam was dissolved in 150 mL of acetone and treated with benzyl bromide (7.90 mL, 66.5 mmol) and K2CO3 (36.7 g, 266 μmol) and stirred for 1 d. The resulting white suspension was filtered through a celite pad and rinsed with excess acetone. The filtrate was evaporated to afford a white slurry, which was then dissolved in water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification by flash chromatography over silica gel (3% to 20% EtOAc/hexanes as eluent) afforded 5 as a colorless oil (6.15 g, 67% over 2 steps, 72% based on recovered starting material). [α]25D −74.5 (c 3.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.38–7.16 (m, 5H), 4.11 (q, J = 7.1, 2H), 3.94 (d, J = 13.6, 1H), 3.80 (d, J = 13.6, 1H), 3.43 (m, 1H), 3.43 (dd, J = 8.1, 1.3, 1H), 2.58 (dd, J = 14.5, 3.9, 1H), 2.26 (ddd, J = 17.2, 13.4, 8.8, 2H), 2.06 (ddd, J = 18.4, 12.9, 9.7, 1H), 1.75 (m, 2H), 1.44 (s, 9H), 1.24 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 173.9, 172.5, 139.8, 128.8, 128.4, 127.1, 80.7, 63.9, 60.4, 58.9, 52.9, 40.3, 29.7, 28.4, 27.9, 14.5; HRMS (ESI-TOF) (m/z) [MH]+ calcd for C20H29NO4 348.21693, found 348.21646.
(2S,5S)-tert-butyl 5-((S)-1-azido-2-ethoxycarbonylmethyl)-1-benzylpyrrolidine-2-carboxylate (6a)
A solution of 5 (1.00 g, 2.88 mmol) in 20 mL of THF under argon at −78°C was treated with KHMDS (0.5 M in toluene, 12.7 mL, 6.34 mmol) and stirred for 45 min. HMPA (1.10 mL, 6.34 mmol) was added and the reaction was stirred another 15 min at the same temperature. A solution of 2,4,6-triisopropylbenzenesulfonyl azide (1.78 mL, 5.76 mmol) in 2.0 mL of THF was cannulated into the reaction mixture. After 2–3 min the reaction was quenched with glacial acetic acid (830μL, 14.4 mmol) and stirred for 16 h with gradual warming to rt. The reaction solution was evaporated, taken up in 10% aq. NaHCO3, and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification by flash chromatography over silica gel (5% EtOAc/hexanes as eluent) afforded 6a as a white solid (930 mg, 84%). mp 82–84°; [α]25D −80.0 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.35–7.21 (m, 5H), 4.20 (qd, J = 7.2, 2.7, 2H), 4.08 (d, J = 2.6, 1H), 4.03 (d, J=13.3, 1H), 3.94 (d, J = 13.3, 1H), 3.77 (m, 1H), 3.59 (d, J = 7.1, 1H), 2.12 (m, 2H), 1.81 (m, 2H), 1.43 (s, 9H), 1.27 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 173.6, 169.1, 138.9, 129.1, 128.6, 127.5, 81.0, 64.7, 64.6, 64.5, 61.9, 53.6, 28.9, 28.3, 25.5, 14.4; HRMS (ESI-TOF) (m/z) [MH]+ calcd for C20H28N4O4, 389.21833, found 389.22139, [M+Na+] calcd 411.20028, found 411.20342.
(S)-1-ethyl 4-methyl 2-((2S,5S)-1-benzyl-5-(tert-butoxycarbonyl)pyrrolidin-2-yl)succinate (6b)
A solution of 5 (1.00 g, 2.88 mmol) in 12 mL of THF under argon at −78° C was treated dropwise with KHMDS (0.5 M in toluene, 7.49 mL, 3.75 mmol) and stirred for 30 min. HMPA (1.10 mL, 6.34 mmol) was added and stirred another 10 min at the same temperature. Methyl bromoacetate (580 μL, 6.34 mmol) was then added dropwise into the mixture and the reaction stirred for 40 min. The reaction was quenched with sat. aq. NH4Cl and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification by flash chromatography over silica gel (5% to 10% EtOAc/hexanes as eluent) afforded 6b as a colorless oil (540 mg, 45%, 79% borsm). [α]25D −39.9 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.33–7.18 (m, 5H), 4.15 (m, 2H), 3.96 (d, J = 13.3, 1H), 3.80 (m, 2H), 3.67 (s, 3H), 3.44 (d, J = 7.5, 1H), 3.24 (dt, J = 10.4, 3.8, 1H), 2.76 (dd, J = 16.8, 3.6, 1H), 2.67 (dd, J = 16.7, 10.4, 1H), 2.04 (m, 1H), 1.87 (m, 1H), 1.66 (m, 2H), 1.43 (s, 9H), 1.26 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 173.9, 173.8, 173.6, 139.2, 128.9, 128.5, 127.3, 80.9, 63.8, 62.0, 60.9, 52.8, 51.9, 43.7, 29.6, 28.6, 28.4, 25.2, 14.4; HRMS (ESI-TOF) (m/z) [MH]+ calcd for C23H33NO6 420.23806, found 420.23877, [M+Na+] calcd 442.22001, found 442.2197.
(2S,5S)-tert-butyl 1-benzyl-5-((S)-1-ethoxy-1-oxopent-4-en-2-yl)pyrrolidine-2-carboxylate (6c)
Triester 6c was prepared from 5 following the same procedure described for 6b, with methyl bromoacetate in place of allyl bromide. Purification of the crude rmaterial by flash chromatography over silica gel (5% EtOAc/hexanes as eluent) afforded 6c as a colorless oil (3.20 g, 95%). [α]25D −55.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.27 (m, 5H), 5.74 (ddt, J = 17.0, 10.1, 6.8, 1H), 5.04 (ddd, J = 17.1, 3.2, 1.5, 2H), 4.96 (m, 1H), 4.11 (m, 2H), 3.96 (d, J = 13.7, 1H), 3.83 (d, J = 13.6, 1H), 3.64 (dt, J = 9.3, 3.7, 1H), 3.49 (d, J = 7.0, 1H), 2.63 (m, 1H), 2.47 (m, 1H), 2.37 (m, 1H), 1.93 (m, 3H), 1.73 (dd, J = 11.3, 8.6, 1H), 1.44 (s, 9H), 1.24 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 174.4, 173.8, 139.8, 137.0, 128.8, 128.5, 127.1, 116.1, 80.8, 64.1, 63.2, 60.5, 53.1, 48.4, 29.5, 28.8, 28.4, 25.5, 14.5; HRMS (ESI-TOF) (m/z) [MH]+ calcd for C23H33NO4 388.24901, found 388.24665.
(2S,5S)-tert-butyl 1-benzyl-5-((S)-1-(tert-butoxycarbonylamino)-2-ethoxycarbonylmethyl) pyrrolidine-2-carboxylate (7)
A solution of 6a (810 mg, 2.10 mmol) and triphenylphosphine (1.21 g, 4.62 mmol) in 12 mL THF was refluxed for 2 h and 500 μL of water was added. After refluxing another 24 h triethylamine (880 μL, 6.30 mmol) was added followed by di-tert-butyl dicarbonate (590 mg, 2.73 mmol) and the reaction was stirred for an additional 24 h at rt. The reaction was quenched with sat. aq. NH4Cl and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. Purification by flash chromatography over silica gel (5% to 10% EtOAc/hexanes as eluent) afforded 7 as a thick colorless oil (920 mg, 94%). [α]25D −26.9 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24 (m, 5H), 5.16 (m, 1H), 4.49 (m, 1H), 4.20 (q, J = 7.1, 2H), 4.02 (d, J = 12.6, 1H), 3.77 (d, J = 12.8, 2H), 3.40 (d, J = 7.0, 1H), 1.97 (m, 2H), 1.75 (m, 2H), 1.45 (m, 18H), 1.27 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 173.6, 171.9, 156.1, 139.0, 129.2, 128.4, 127.3, 80.9, 79.9, 63.4, 62.5, 61.4, 54.7, 52.3, 28.6, 28.3, 28.1, 25.0, 14.4, 1.3; HRMS (ESI-TOF) (m/z) [MH]+ calcd for C25H38N2O6 463.28026, found 463.28064, [M+Na+] calcd 485.26221, found 485.26586.
(2 S,5S)-tert-butyl 1-benzyl-5-((S)-1-(tert-butoxycarbonylamino)-2-hydroxyethyl)pyrrolidine-2-carboxylate (8)
A solution of 7 (870 mg, 1.88 mmol) in Et2O under argon atmosphere at rt was treated with LiBH4 (2M in THF, 1.60 mL, 3.20 mmol) and stirred for 6.5 h. The reaction was quenched with 1M aq. NaOH (4 mL) and stirred for 10 min. After dilution with water, the mixture was stirred vigorously for 30 m. The aqueous layer was extracted with EtOAc and the combined organic layers were dried over anhydrous Na2SO4 and evaporated. Purification by flash chromatography over silica gel (50% EtOAc/hexanes as eluent) afforded 8 as a white solid (730 mg, 92%). mp 113–115°; [α]25D −47.3. (c 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.27 (m, 5H), 5.43 (bs, 1H), 5.01 (bs, 1H), 3.94 (d, J = 13.2, 1H), 3.78 (m, 2H), 3.60 (m, 3H), 3.45 (d, J = 7.4, 1H), 1.99 (m, 2H), 1.76 (dd, J = 12.4, 9.9, 1H), 1.63 (m, 1H), 1.46 (s, 9H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.3, 158.4, 138.8, 129.1, 128.6, 127.5, 81.2, 80.3, 65.8, 63.4, 62.7, 54.6, 52.8, 28.6, 28.4, 28.2, 24.8; (ESI-TOF) (m/z) [MH]+ calcd for C23H36N2O5 421.26970, found 421.27058, [M+Na+] calcd 443.25164, found 443.25554.
(4S,4aS,7S)-tert-butyl 4-(tert-butoxycarbonylamino)-1-oxohexahydro-1H-pyrrolo[1,2-c][1,3]oxazine-7-carboxylate (9)
A solution of 8 (715 mg, 1.70 mmol) in 7 mL of MeOH was treated with 250 mg of 20% Pd(OH)2/C and stirred under H2 (balloon) at rt for 2.5 h. The reaction solution was filtered through a celite pad and rinsed with excess MeOH. The filtrate was evaporated under reduced pressure to afford a thick colorless oil (562 mg, quantitative yield).
The above amino alcohol (360 mg, 1.09 mmol) was dissolved in 10 mL of THF and treated with triethylamine (230 μL, 1.64 mmol), 4-dimethylaminopyridine (130 mg, 1.09 mmol), and 1,1′-carbonyldiimidazole (880 mg, 5.45 mmol) respectively. After stirring 3.5 h at rt the reaction was evaporated, taken up in EtOAc, and washed with 1M aq. HCl. The aqueous layer was dried over anhydrous Na2SO4, filtered, and evaporated. The crude residue was adsorbed onto silica gel and purified by flash chromatography over silica (60% to 80% EtOAc/hexanes as eluent) to afford 9 as a white solid (357 mg, 92%). mp 208–210°(dec); [α]25D −43.1 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.76 (d, J = 8.8, 1H), 4.37 (t, J = 8.3, 1H), 4.29 (dd, J = 10.5, 4.6, 1H), 3.98 (t, J = 10.7, 1H), 3.79 (m, 1H), 3.52 (td, J = 9.4, 5.0, 1H), 2.36 (m, 1H), 2.24 (m, 1H), 1.71 (m, 2H), 1.46 (s, 9H), 1.42 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.6, 155.1, 151.7, 82.2, 80.8, 68.5, 61.6, 60.8, 48.2, 31.5, 28.5, 28.2, 28.0; (ESI-TOF) (m/z) [MH]+ calcd for C17H28N2O6 357.20201, found 357.20442, [M+Na+] calcd 379.18396, found 379.18658.
(S)-2-((2S,5S)-5-(tert-butoxycarbonyl)pyrrolidin-2-yl)-2-(tert-butoxycarbonylamino)acetic acid (10)
A solution of 7 (280 mg, 605 μmol) in 6 mL of THF:H2O (1:1) at rt was treated with LiOH (78.8 mg, 1.88 mmol) and stirred for 7 h. The reaction solution was evaporated under reduced pressure. The aqueous layer was washed with Et2O, acidified with 1M aq. HCl, and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and evaporated to afford a white solid (250 mg, 96%). [α]25D −15.5 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.28 (m, 5H), 5.45 (bs, 1H), 4.45 (bs, 1H), 4.13 (m, 1H), 3.97 (m, 2H), 3.52 (d, J = 7.6, 1H), 2.18 (m, 1H), 2.03 (m, 1H), 1.83 (m, 2H), 1.45 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 174.3, 172.1, 157.0, 137.1, 129.4, 128.7, 127.9, 81.8, 80.6, 63.9, 63.5, 54.9, 52.8, 28.6, 28.3, 28.2, 25.2.
The above acid (200 mg, 460 μmol) was dissolved in 2 mL of MeOH and treated with 69 mg of 20% Pd(OH)2/C and stirred under H2 (balloon) for 1.25 h. The reaction solution was filtered through a celite pad and rinsed with excess MeOH. The filtrate was evaporated in vacuo to afford 10 as a white solid (160 mg, quantitative yield). mp 163–165° (dec); 1H NMR (400 MHz, CDCl3) δ 7.26 (m, 2H), 5.96 (d, J = 6.4, 1H), 4.23 (m, 1H), 4.15 (t, J = 7.2, 1H), 3.94 (dd, J = 13.3, 6.7, 1H), 2.45 (m, 1H), 2.17 (m, 1H), 2.05 (m, 2H), 1.47 (s, 9H), 1.41 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.1, 168.6, 157.0, 84.0, 80.0, 62.7, 59.5, 54.6, 28.6, 28.5, 28.1, 26.9; (ESI-TOF) (m/z) [MH]+ calcd for C17H28N2O6 345.20201, found 345.20310, [M+Na+] calcd 367.18396, found 367.18288.
(2S,5S,6S)-tert-butyl 6-(tert-butoxycarbonylamino)-7-oxo-1-azabicyclo[3.2.0]heptane-2-carboxylate (11)
A solution of Mukaiyama’s reagent (210 mg, 810 μmol) in 15 mL of acetonitrile was treated with triethylamine (240 μL, 1.70 mmol) and heated to 70°C. A solution of 10 (70.0 mg, 203 μmol) in 15 mL of acetonitrile was cannulated into the mixture and the reaction was allowed to cool to rt gradually and stirred for 2 days. The reaction solution was evaporated, taken up in EtOAc, and washed with water. The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. Purification by flash chromatography over silica gel (30% EtOAc/hexanes as eluent) afforded 11 as a sticky colorless oil (57.0 mg, 86%). [α]25D −58.1 (c 0.4, DMSO); 1H NMR (400 MHz, CDCl3) d 5.36 (d, J = 8.5, 1H), 4.57 (d, J = 8.4, 1H), 4.34 (dd, J = 7.8, 5.8, 1H), 3.78 (t, J = 5.8, 1H), 2.39 (m, 1H), 2.24 (dt, J = 11.4, 6.4, 1H), 2.13 (dt, J = 14.1, 7.4, 1H), 1.70 (m, 1H), 1.42 (d, J = 2.3, 18H). 1H NMR (400 MHz, DMSO-d6) δ 7.81 (d, J = 8.6, 1H), 4.35 (d, J = 8.6, 1H), 4.19 (dd, J = 7.7, 5.7, 1H), 3.68 (m, 1H), 2.33 (m, 1H), 2.03 (m, 2H), 1.71 (m, 1H), 1.39 (s, 9H), 1.37 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 174.7, 170.7, 155.3, 81.7, 79.3, 63.1, 63.0, 59.9, 34.8, 28.8, 28.5, 28.2; (ESI-TOF) (m/z) [MH]+ calcd for C16H26N2O5 327.19217, found 327.19028, [M+Na+] calcd 349.17339, found 349.17259.
(1S,5S,7aS)-5-tert-butyl 1-ethyl 3-oxohexahydro-1H-pyrrolizine-1,5-dicarboxylate (12)
A solution of 6b (295 mg, 703 μmol) in 4 mL of MeOH was treated with 100 mg of 20% Pd/C and stirred for 3h under H2 (balloon) atmosphere. The reaction solution was filtered through a celite pad and rinsed with excess MeOH. The filtrate was evaporated in vacuo to afford a yellowish oil, which was then dissolved in 5 mL of toluene and stirred at 90 °C for 20 h. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography over silica gel (50% EtOAc/hexanes as eluent) to furnish 12 as a pale yellow solid (186 mg, 89% over 2 steps). mp 64–66°; [α]25D −140.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.39 (dd, J = 8.7, 6.6, 1H), 4.29 (m, 1H), 4.17 (m, 2H), 3.45 (m, 1H), 2.82 (dd, J = 7.4, 2.4, 2H), 2.41 (m, 1H), 1.94 (m, 2H), 1.71 (s, 1H), 1.45 (s, 9H), 1.26 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 174.7, 171.8, 171.3, 82.1, 62.6, 61.4, 56.4, 39.7, 35.2, 31.4, 28.2, 27.7, 14.5; (ESI-TOF) (m/z) [MH]+ calcd for C15H23NO5 298.16490, found 298.16433, [M+Na+] calcd 320.14684, found 320.14613.
(5S,7aS)-5-(tert-butoxycarbonyl)-3-oxohexahydro-1H-pyrrolizine-1-carboxylic acid (13)
A solution of 12 (218 mg, 734 μmol) in 5 mL of THF:H2O (1:1) at rt was treated with LiOH (45.0 mg, 1.07 mmol) and stirred for 15 min. The reaction was diluted with 1M aq. HCl and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and evaporated to afford 13 as a ~3:1 mixture of diastereomers (196 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 6.70 (bs, 1H), 4.36 (m, 2H), 3.48 (td, J = 8.0, 6.2, 1H), 2.87 (m, 2H), 2.48 (m, 1.5H), 2.27 (s, 0.5H), 2.04 (m, 2H), 1.46 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 175.7, 175.5, 174.8, 172.7, 171.1, 170.8, 82.5, 82.3, 63.9, 62.7, 56.4, 55.7, 45.8, 39.9, 37.8, 35.7, 32.3, 31.8, 31.6, 28.2, 27.8.; (ESI-TOF) (m/z) [MH]+ calcd for C13H19NO5 270.13360, found 270.13479, [M+Na]+ calcd 292.11554, found 292.11663.
(3S,7aS)-tert-butyl 7-(benzyloxycarbonylamino)-5-oxohexahydro-1H-pyrrolizine-3-carboxylate (14a)
A solution 13 (140 mg, 520 mmol) in toluene at 50 °C was treated with triethylamine (181 μL, 1.30 mmol) followed by diphenylphosphoryl azide (DPPA) (281 mL, 1.30 mmol), dropwise. The reaction was stirred from 50 °C to 110 °C over 1 h and at 110 °C for 4.5 h. Benzyl alcohol (107 μL, 1.04 mmol) was added and the reaction was stirred at 110 °C for 20 h. The reaction solution was evaporated under reduced pressure and adsorbed onto silica gel. Purification by flash chromatography over silica gel (5% to 10% EtOAc/hexanes as eluent) afforded 14a as a ~3:1 mixture of diastereomers (118 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 5H), 5.36 (d, J = 8.0, 1H), 5.11 (m, 2H), 4.41 (m, 1H), 4.27 (dd, J = 13.5, 6.1, 1.5H), 4.09 (m, 0.25H), 3.86 (dt, J = 13.1, 6.5, 0.25H), 3.13 (dd, J = 17.0, 7.2, 1H), 2.81 (dd, J = 16.3, 8.6, 0.25H), 2.64 (dt, J = 8.8, 7.3, 0.25H), 2.39 (m, 1H), 2.24 (d, J = 17.1, 1H), 2.05 (m, 1H), 1.89 (m, 1H), 1.61 (m, 1H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.8, 170.8, 156.2, 136.4, 128.8, 128.8, 128.6, 128.5, 128.4, 128.3, 82.2, 67.2, 66.0, 55.7, 49.4, 42.0, 32.5, 28.2, 24.6; (ESI-TOF) (m/z) [MH]+ calcd for C20H26N2O5 375.19217, found 375.19207, [M+Na]+ calcd 397.17339, found 397.17330.
(3S,7aS)-tert-butyl 7-(tert-butoxycarbonylamino)-5-oxohexahydro-1H-pyrrolizine-3-carboxylate (14b)
N-Boc derivative 14b was prepared from 13 following the same procedure described for 14a, with t-butanol in place of benzyl alcohol. Purification by flash chromatography over silica gel (5% to 10% EtOAc/hexanes as eluent) afforded 14b as a ~3:1 mixture of diastereomers (26%). 1H NMR (400 MHz, CDCl3) δ 4.95 (dd, J = 21.2, 6.8, 1H), 4.29 (m, 3H), 4.00 (m, 0.25 H), 3.82 (m, 0.25), 3.12 (dd, J = 17.0, 7.2, 1H), 2.79 (m, 1H), 2.61 (m, 0.25H), 2.43 (m, 1.25H), 2.22 (d, J = 17.0, 1H), 2.05 (m, 1H), 1.88 (bs, 1H), 1.60 (m, 1H), 1.43 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 171.8, 171.1, 170.9, 155.5, 82.1, 82.0, 80.2, 66.2, 56.0, 55.6, 48.7, 41.9, 32.4, 32.0, 30.9, 28.6, 28.5, 28.2, 24.7; (ESI-TOF) (m/z) [MH]+ calcd for C17H28N2O5 341.20710, found 341.20804, [M+Na]+ calcd 363.18904, found 363.18941.
(3S,7aS)-tert-butyl 5-oxo-7-((2-(trimethylsilyl)ethoxy)carbonylamino)hexahydro-1H-pyrrolizine-3-carboxylate (14c)
N-Teoc derivative 14c was prepared from 13 following the same procedure described for 14a, with 2-trimethylsilylethanol in place of benzyl alcohol. Purification by flash chromatography over silica gel (5% to 10% EtOAc/hexanes as eluent) afforded 14c as a ~3:1 mixture of diastereomers (57%); 1H NMR (400 MHz, CDCl3) δ 5.25 (d, J = 8.1, 1H), 4.38 (m, 1H), 4.24 (m, 2H), 4.11 (m, 2H), 3.12 (dd, J = 17.0, 7.2, 1H), 2.37 (m, 1H), 2.21 (d, J = 17.0, 1H), 2.04 (m, 1H), 1.86 (m, 1H), 1.62 (m, 1H), 1.43 (s, 9H), 0.93 (m, 2H), 0.00 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.2, 172.2, 157.8, 83.4, 67.3, 65.0, 57.2, 56.9, 50.5, 43.3, 33.7, 33.2, 32.1, 29.5, 25.8, 19.2, 0.00. (ESI-TOF) (m/z) [MH]+ calcd for C18H32N2O5Si 385.21533, found 385.21625, [M+Na]+ calcd 407.19727, found 407.19812.
(S)-1-benzyl 6-ethyl 5-((2S,5S)-1-benzyl-5-(tert-butoxycarbonyl)pyrrolidin-2-yl)hex-2-enedioate (15)
A solution of 6c (100 mg, 258 μmol) in 1.70 mL of 1,2-dichloroethane was treated with methyl acrylate (418 μL, 2.58 mmol) and Grubbs’ 2nd generation catalyst (21.0 mg, 24.7 μmol) and stirred for 1d at 65° C. The reaction solution was evaporated under reduced pressure and adsorbed onto silica gel. Purification by flash chromatography over silica gel (5% to 10% EtOAc/hexanes as eluent) afforded 15 as a 14:1 mixture of E:Z isomers (72.0 mg, 53%, 66% based on recovered starting material). Data given for the E isomer: 1H NMR (400 MHz, CDCl3) δ 7.40–7.17 (m, 10H), 6.89 (dt, J = 15.5, 7.1, 1H), 5.84 (d, J = 15.7, 1H), 5.16 (s, 1H), 4.11 (q, J = 7.1, 2H), 3.89 (q, J = 13.5, 2H), 3.70 (dt, J = 9.6, 3.5, 1H), 3.53 (d, J = 7.4, 1H), 2.63 (m, 2H), 2.50 (ddd, J = 14.7, 10.5, 7.5, 1H), 2.05 (ddd, J = 21.2, 10.6, 6.4, 1H), 1.91 (m, 1H), 1.76 (m, 2H), 1.44 (s, 9H), 1.23 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 173.7, 173.7, 166.5, 148.5, 139.5, 136.3, 128.8, 128.8, 128.6, 128.4, 128.4, 127.3, 122.2, 80.9, 66.2, 64.5, 63.2, 60.8, 53.3, 47.2, 28.7, 28.4, 27.7, 25.3, 14.5; (ESI-TOF) (m/z) [MH]+ calcd for C31H39NO6 522.280501, found 522.28167, [M+Na]+ calcd 544.26696, found 544.26345.
(3S,9S,9aS)-3-tert-butyl 9-ethyl 5-oxooctahydro-1H-pyrrolo[1,2-a]azepine-3,9-dicarboxylate (16)
A solution of 15 (165 mg, 317 μmol) in 3 mL of MeOH was treated with 80 mg of Pd(OH)2/C and the reaction was stirred under H2 (balloon) for 1.25 h. The reaction solution was filtered through a celite pad and rinsed with excess MeOH/EtOAc. The filtrate was evaporated under reduced pressure to afford a thick oil, which was then dissolved in DMF and treated with triethylamine (86.0 μL, 620 μmol), HBTU (140 mg, 370 μmol), and hydroxybenzotriazole (HOBt) (8.50 mg, 62.9 μmol). The reaction was stirred for 24 h. The reaction solution was evaporated at 60°C under reduced pressure and the residue was dissolved in EtOAc. The organic layer was washed with 1M aq. HCl and 10% aq. Na2CO3. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. Purification by flash chromatography over silica gel (50% EtOAc/hexanes as eluent) afforded 16 as a colorless oil (75.0 mg, 73%, 2 steps). [α]25D −57.8 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.36 (dd, J = 9.1, 2.3, 1H), 4.23 (dt, J = 9.5, 1.9, 1H), 4.10 (m, 2H), 2.70 (bs, 1H), 2.55 (m, 2H), 2.42 (m, 1H), 2.16 (m, 2H), 1.98 (m, 2H), 1.75 (m, 3H), 1.42 (s, 9H), 1.24 (t, J = 7.1, 3H); 13C NMR (101 MHz, CDCl3) δ 173.9, 172.6, 172.1, 81.3, 61.9, 60.9, 60.0, 47.3, 38.1, 32.7, 31.3, 28.2, 27.4, 19.9, 14.4; (ESI-TOF) (m/z) [MH]+ C17H27NO5 326.19690, found 326.19382.
(3S,9aS)-tert-butyl 9-(benzyloxycarbonylamino)-5-oxooctahydro-1H-pyrrolo[1,2-a]azepine-3-carboxylate (17)
A solution of 16 (41.0 mg, 138 μmol) in 2 mL of THF:MeOH (2:1) was treated with 1 mL of 1M aq. NaOH and stirred at 40 °C for 4.5 h. The reaction was diluted with water and the solution mixture was washed with Et2O. After acidification to pH < 3 with 1M aq. HCl, the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure to afford the carboxylic acid as a ~2:1 mixture of diastereomers. (36.0 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 7.88 (bs, 1H), 4.41 (m, 1H), 4.21 (m, 1H), 2.77 (s, 0.5H), 2.60 (m, 2H), 2.46 (m, 1H), 2.33 (m, 1.5H), 2.11 (m, 2H), 2.01–1.70 (m, 4H), 1.57 (m, 1H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 178.1, 177.1, 174.9, 174.7, 172.0, 171.3, 81.8, 81.6, 62.3, 61.3, 60.1, 59.6, 50.0, 46.8, 37.8, 36.9, 33.2, 32.7, 31.5, 30.5, 28.2, 27.6, 27.3, 21.7, 19.7.
The above distereomeric mixture of carboxylic acids was subjected to the same Curtius rearrangement conditions described for 14a. Purification by flash chromatography over silica gel (50% EtOAc/hexanes as eluent) afforded (α′S)-17 (22% isolated yield, 2 steps) as a colorless oil and (α′R)-17 (38% isolated yield, 2 steps) a a white solid.
Data for (α′S)-17a: [α]25D −60.9 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 5H), 5.09 (s, 2H), 4.97 (d, J = 10.0, 1H), 4.43 (d, J = 8.6, 1H), 4.16 (d, J = 8.6, 1H), 4.05 (m, 1H), 2.59 (dd, J = 14.4, 6.7, 1H), 2.48 (m, 1H), 2.32 (m, 1H), 2.02 (m, 3H), 1.85–1.54 (m, 4H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.9, 171.5, 156.3, 136.4, 128.8, 128.5, 128.4, 81.7, 67.3, 62.1, 61.4, 52.4, 38.1, 35.5, 30.6, 28.2, 27.9, 18.2; (ESI-TOF) (m/z) [MH]+ calcd for C22H30N2O5 403.22347, found 403.22247, [M+Na]+ calcd 435.20469, found 425.20439.
Data for (α′R)-17a: mp 121–123°; [α]25D −57.9 (c 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.32 (m, 5H), 5.09 (d, J = 1.9, 2H), 4.74 (d, J = 9.4, 1H), 4.39 (d, J = 8.1, 1H), 3.82 (t, J = 8.6, 1H), 3.52 (m, 1H), 2.49 (m, 2H), 2.27 (m, 1H), 2.09 (m, 3H), 1.68 (m, 5H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.0, 171.7, 156.2, 136.4, 129.7, 128.8, 128.5, 128.4, 81.5, 67.3, 63.7, 61.4, 52.6, 37.4, 37.0, 28.2, 27.8, 27.1, 22.1; (ESI-TOF) (m/z) [MH]+ calcd for C22H30N2O5 403.22347, found 403.22207, [M+Na]+ calcd 435.20469, found 425.20409.
(S)-2-((2S,5S)-1-benzyl-5-(tert-butoxycarbonyl)pyrrolidin-2-yl)pent-4-enoic acid (18)
A solution of 6c (1.00 g, 2.58 mmol) in 17 mL of MeOH was treated with 13 mL of 2M aq. NaOH and tetrabutylammonium hydroxide (134 μL, 516 μmol) as a phase transfer catalyst and stirred at 50° C for 24 h. The reaction solution was concentrated, acidified to pH = 3 with 1M aq. HCl, and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure to afford 18 as a white solid (870 mg, 94%); mp 76–78°; [α]25D −104.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.31 (m, 5H), 5.80 (ddt, J = 16.9, 10.1, 6.8, 1H), 5.08 (m, 2H), 4.11 (d, J = 13.2, 1H), 3.97 (d, J = 13.2, 1H), 3.75 (dt, J = 10.0, 3.2, 1H), 3.52 (d, J = 7.6, 1H), 2.65 (ddd, J = 8.8, 6.4, 2.7, 1H), 2.53 (m, 2H), 2.28 (m, 1H), 2.03 (m, 1H), 1.78 (m, 2H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.9, 172.4, 137.7, 136.0, 129.1, 128.9, 127.90, 117.3, 81.5, 64.2, 62.5, 53.9, 49.1, 33.2, 28.3, 28.2, 28.1; (ESI-TOF) (m/z) [MH]+ calcd for C21H29NO4 360.21766, found 360.21856.
(2S,5S)-tert-butyl 1-benzyl-5-((S)-1-((2,2,2-trichloroethoxy)carbonylamino)but-3-enyl)pyrrolidine-2-carboxylate (19)
N-Troc derivative 19 was prepared from 18 following the same procedure described for 14a, with 2,2,2-tricloroethanol in place of benzyl alcohol. Purification by flash chromatography over silica gel (50% EtOAc/hexanes as eluent) afforded 19 as a colorless oil (75%); [α]25D −48.7 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.27 (m, 5H), 5.75 (ddt, J = 16.8, 10.2, 6.9, 1H), 5.08 (m, 3H), 4.72 (q, J = 12.1, 2H), 4.04 (d, J = 13.9, 1H), 3.91 (d, J = 13.9, 1H), 3.78 (tdd, J = 9.7, 4.5, 2.9, 1H), 3.56 (m, 2H), 2.56 (dt, J = 14.2, 5.3, 1H), 2.24 (m, 1H), 2.12 (m, 1H), 1.95 (tt, J = 12.0, 8.3, 1H), 1.74 (dd, J = 12.7, 8.3, 1H), 1.62 (m, 1H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.9, 154.7, 139.7, 135.3, 128.7, 128.6, 127.3, 117.6, 96.1, 80.9, 74.6, 65.1, 64.1, 54.7, 54.5, 35.8, 29.1, 28.4, 27.1; (ESI-TOF) (m/z) [MH]+ calcd for C23H31Cl3N2O4 505.14222, found 505.14626, [M+Na]+ calcd 527.12416, found 527.12359.
(S)-3-((2S,5S)-1-benzyl-5-(tert-butoxycarbonyl)pyrrolidin-2-yl)-3-((2,2,2-trichloroethoxy)carbonylamino)propanoic acid (20)
A solution of 19 (100 mg, 197 μmol) in 2.5 mL of THF:H2O (3:1) was treated with 4-methylmorpholine N-oxide (51.0 mg, 435 μmol) and osmium tetroxide (2.5 % solution in 2-methyl-2-propanol, 220 μL, 20.0 μmol). After 2 h stirring at rt, the resulting diol intermediate was treated with sodium periodate (93.0 mg, 435 μmol) and the reaction was stirred for another 15 h. The reaction was quenched with 5 % aq. Na2S2O3 and diluted with brine. The aqueous layer was extracted with EtOAc and the combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated. Purification by flash chromatography over silica gel (50% EtOAc/hexanes as eluent) afforded the aldehyde as a colorless thick oil (95.0 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 9.58 (t, J = 2.5, 1H), 7.28 (ddd, J = 12.0, 10.8, 7.6, 5H), 5.14 (d, J=8.8, 1H) 4.72 (m, 2H), 4.35 (m, 1H), 3.98 (m, 2H), 3.57 (dt, J = 9.8, 2.9, 1H), 3.51 (d, J = 7.5, 1H), 2.72 (ddd, J = 15.6, 6.0, 2.4, 1H), 2.47 (ddd, J = 15.6, 8.1, 2.7, 1H), 2.23 (m, 1H), 1.91 (tt, J = 12.0, 8.4, 1H), 1.77 (dd, J = 12.9, 8.4, 1H), 1.61 (m, 2H), 1.42 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 199.7, 173.2, 154.3, 138.8, 128.9, 128.7, 127.6, 95.7, 81.2, 74.8, 64.9, 64.5, 54.4, 49.3, 44.9, 28.8, 28.3, 25.7; (ESI-TOF) (m/z) [MH]+ calcd for C22H29Cl3N2O5 507.12220, found 507.11920.
A solution of the above aldehyde (65.0 mg, 128 μmol) in 0.7 mL of t-BuOH was treated with amylene (160 μL, 1.52 mmol) and cooled to 0° C. A solution of NaH2PO4 (158 mg, 1.15 mmol) and NaClO2 (87.0 μL, 767 μmol) was added dropwise and the reaction was stirred from 0° C to 10° C over 1 h. The reaction was quenched with 5% aq. Na2S2O3 and diluted with brine. The pH of the aqueous layer was adjusted to 3with 1M aq. HCl and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated. Purification by flash chromatography over silica gel (from 70% to 100% EtOAc/hexanes as eluent) afforded carboxylic acid 20 as a thick colorless oil (67.0 mg, 90%). [α]25D −43.6 (c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.29 (m, 5H), 5.45 (d, J = 8.7, 1H), 4.77 (m, 2H), 4.36 (m, 1H), 4.11 (dd, J = 31.3, 10.1, 1H), 3.96 (m, 1H), 3.67 (d, J = 9.8, 1H), 3.49 (dd, J = 14.8, 7.3, 1H), 2.86 (dd, J = 15.9, 6.6, 1H), 2.51 (ddd, J = 23.8, 15.5, 7.4, 1H), 2.24 (m, 1H), 1.95 (m, 1H), 1.77 (m, 2H), 1.43 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 175.8, 172.6, 154.3, 137.7, 129.2, 128.8, 127.8, 95.8, 81.5, 74.8, 64.4, 64.1, 54.1, 50.3, 36.1, 28.9, 28.3, 25.6; (ESI-TOF) (m/z) [M-H]-calcd for C22H29Cl3N2O6 521.10184, found 521.10280.
(3S,7S,7aS)-tert-butyl 5-oxo-7-((2,2,2-trichloroethoxy)carbonylamino)hexahydro-1H-pyrrolizine-3-carboxylate (21)
A solution of 20 (60.0 mg, 116 μmol) in 2 mL of THF was treated with 25 mg of Pd(OH)2/C was purged with H2 and stirred under H2 (balloon) for 7.5 h. The reaction solution was filtered through a celite pad and rinsed with excess MeOH/EtOAc. The filtrate was evaporated under reduced pressure to afford a thick oil, which was then dissolved in 3 mL of acetonitrile and treated with triethylamine (53.0 μL, 378 μmol), HBTU (62.0 mg, 164 μmol), and HOBt (3.00 mg, 25.0 μmol) and stirred at rt for 20 h. The reaction mixture was evaporated under reduced pressure. The residue was dissolved in EtOAc and washed with 1M aq. HCl followed by 10% aq. Na2CO3. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated. Purification by flash chromatography over silica gel (50% to 100 % EtOAc/hexanes as eluent) afforded 21 as a white solid (20.0 mg, 42%, 2 steps) in addition to 10 mg of the corresponding ethyl carbamate. Data for 21; mp 147–149°; [α]25D −120.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.93 (d, J = 8.3, 1H), 4.78 (d, J = 12.0, 1H), 4.68 (d, J = 12.0, 1H), 4.49 (dd, J = 12.9, 7.4, 1H), 4.32 (dd, J = 13.5, 6.4, 2H), 3.19 (dd, J = 16.9, 7.1, 1H), 2.45 (dtd, J = 12.7, 8.3, 4.2, 1H), 2.30 (d, J = 17.0, 1H), 2.09 (m, 1H), 1.92 (m, 1H), 1.71 (m, 2H), 1.46 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.7, 170.7, 154.5, 95.7, 82.3, 74.7, 65.9, 55.7, 49.8, 42.1, 32.5, 28.2, 24.5; (ESI-TOF) (m/z) [MH]+ calcd for C15H21Cl3N2O5 415.05888, found 415.05815, [M+Na]+ calcd 437.04083, found 437.03907.
(2S,5S)-tert-butyl 1-benzyl-5-((S)-5-(benzyloxy)-5-oxo-1-((2,2,2-trichloroethoxy)carbonylamino)pent-3-enyl)pyrrolidine-2-carboxylate (22)
A solution of 19 (265 mg, 520 μmol) in 750 μL of dichloroethane was treated with benzyl acrylate (850 mg, 5.23 mmol) and Grubbs’ 2nd generation catalyst (40.0 mg, 47.1 μmol) and stirred at 65° C for 1 d (catalyst was added in 3 portions). The reaction solution was evaporated and adsorbed onto silica gel. Purification by flash chromatography over silica gel (0% to 20 % EtOAc/hexanes as eluent) afforded 22 as a 7.4:1 mixture of E:Z isomers (210 mg, 62%, 87%, borsm). Data given for the E isomer: [α]25D −26.4 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.30 (m, 10H), 6.92 (dt, J = 14.6, 7.1, 1H), 5.87 (d, J = 15.6, 1H), 5.16 (d, J = 5.3, 1H), 5.12 (m, 2H), 4.73 (m, 2H), 3.90 (m, 3H), 3.55 (m, 2H), 2.69 (dt, J = 13.4, 4.8, 1H), 2.24 (m, 2H), 1.94 (tt, J = 12.0, 8.4, 1H), 1.77 (dd, J = 12.7, 8.5, 1H), 1.65 (m, 1H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.5, 166.1, 154.5, 146.2, 139.5, 136.2, 128.8, 128.7, 128.6, 128.4, 127.5, 123.4, 96.0, 81.1, 74.6, 66.4, 65.3, 64.4, 54.5, 53.7, 33.7, 29.0, 28.4, 26.6; (ESI-TOF) (m/z) [MH]+ ]+ calcd for C31H37Cl3N2O6 639.17900, found 639.17605.
(3S,9S,9aS)-tert-butyl 5-oxo-9-((2,2,2-trichloroethoxy)carbonylamino)octahydro-1H-pyrrolo[1,2-a]azepine-3-carboxylate (23)
A solution of 22 (82.0 mg, 128 μmol) in 2.5 mL of THF was treated with 30.0 mg of 20% Pd(OH)2/C and stirred for 6 h at rt under H2 (balloon) atmosphere. The reaction was filtered through a celite pad, rinsed with EtOAc, and evaporated under reduced pressure. The resulting amino acid was dissolved in acetonitrile and treated with triethylamine (54.0 μL, 384 μmol), HBTU (63.0 mg, 166 μmol), and HOBt (3.50 mg, 25.9 μmol). After 24 h stirring at rt the reaction was evaporated and dissolved in EtOAc. The organic layer was washed with 1M aq. HCl and 10 % aq. Na2CO3, dried over anhydrous Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography over silica gel (60% to 80% EtOAc/hexanes as eluent) to afford 23 as a white solid (30.0 mg, 53%, over 2 steps) along with 10 mg of the corresponsing ethyl carbamate. Data for 23: mp 198–200°; [α]25D −51.8 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.21 (d, J = 10.2, 1H), 4.83 (d, J = 12.1, 1H), 4.64 (d, J = 12.0, 1H), 4.51 (dd, J = 8.7, 1.5, 1H), 4.19 (d, J = 8.1, 1H), 4.07 (dt, J = 9.7, 3.3, 1H), 2.63 (dd, J = 14.5, 6.9, 1H), 2.50 (m, 1H), 2.36 (tt, J = 12.5, 8.3, 1H), 2.15 (ddd, J = 12.6, 10.6, 6.3, 1H), 2.05 (m, 1H), 186 (m, 4H), 1.63 (m, 1H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 175.0, 171.5, 154.6, 121.6, 81.8, 74.7, 62.2, 61.2, 52.9, 38.1, 35.4, 30.6, 28.2, 27.9, 18.2; (ESI-TOF) (m/z) [MH]+ calcd for C17H25Cl3N2O5 443.09090, found 443.09069, [M+Na]+ 465.07212, found 465.07230.
(2S,5S)-tert-butyl 1-benzyl-5-((S)-1-((2-(trimethylsilyl)ethoxy)carbonylamino)but-3-enyl)pyrrolidine-2-carboxylate (24)
N-Teoc derivative 24 was prepared from 18 following the same procedure described for 14a, with 2-trimethylsilylethanol in place of benzyl alcohol. Purification by flash chromatography over silica gel (5% to 10% EtOAc/hexanes as eluent) afforded 24 as a colorless oil (58%). [α]25D −58.9 (c 2.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24 (m, 5H), 5.72 (m, 1H), 5.01 (m, 2H), 4.75 (d, J = 9.0, 1H), 4.10 (m, 2H), 4.02 (d, J = 14.1, 1H), 3.86 (d, J = 14.0, 1H), 3.72 (m, 1H), 3.48 (dd, J = 12.8, 4.8, 2H), 2.49 (m, 1H), 2.18 (m, 1H), 2.05 (m, 1H), 1.90 (tt, J = 12.5, 8.6, 1H), 1.69 (d, J = 12.7, 8.4, 1H), 1.58 (m, 1H), 1.40 (s, 9H), 0.95 (dd, J = 16.9, 8.5, 2H), 0.00 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.9, 158.0, 141.1, 136.9, 129.9, 129.8, 128.4, 118.5, 82.1, 66.3, 65.3, 64.3, 55.6, 55.3, 37.2, 30.4, 29.6, 28.2, 19.2, 0.00; (ESI-TOF) (m/z) [MH]+ calcd for C26H42N2O4Si 475.29866, found 475.29934.
(2S,5S)-tert-butyl 1-benzyl-5-((S)-3-hydroxy-1-((2-(trimethylsilyl)ethoxy)carbonylamino)propyl)pyrrolidine-2-carboxylate (25)
A solution of 24 (90.0 mg, 190 μmol) in 2.5 mL of THF:H2O (3:1) was treated with 4-methyl morpholine N-oxide (54.0 mg, 464 μmol) and osmium tetroxide (2.5% solution in 2-methyl 2-propanol, 231 μL, 22.7 μmol). After stirring at rt for 2 h, the resulting diol intermediate was treated with sodium periodate (99.0 mg, 464 μmol) and stirred for another 15 h. The reaction was quenched with 5% aq. Na2S2O3 and diluted with brine. The aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered, and evaporated. Purification by flash chromatography over silica gel (50% EtOAc/hexanes as eluent) afforded the desired aldehyde as a colorless thick oil (60.0 mg, 66%). [α]25D −55.3 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 1H), 7.24 (m, 5H), 4.69 (d, J = 8.3, 1H), 4.32 (m, 1H), 4.10 (m, 2H), 3.94 (dd, J = 41.0, 13.5, 2H), 3.50 (dt, J = 9.9, 3.0, 1H), 3.45 (d, J = 7.5, 1H), 2.63 (ddd, J = 15.4, 6.1, 2.7, 1H), 2.37 (dd, J = 13.8, 9.1, 1H), 2.17 (tt, J = 12.0, 9.6, 1H), 1.86 (tt, J = 12.0, 8.4, 1H), 1.71 (dd, J = 12.8, 8.5, 1H), 1.56 (m, 1H), 1.39 (s, 9H), 0.93 (dd, J = 11.8, 5.3, 2H), 0.00 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 201.5, 174.5, 157.6, 140.2, 130.2, 129.9, 128.7, 82.3, 66.0, 65.9, 64.9, 55.6, 50.1, 46.6, 30.1, 29.6, 26.9, 19.2, 0.0.
A solution of above aldehyde (50.0 mg, 105 μmol) in 2 mL of THF was treated with NaBH4 (6.00 mg, 157 μmol) and stirred for 30 min at rt. The reaction was quenched with sat. aq. NH4Cl and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure to afford 25 as a sticky foam (50.0 mg, quantitative yield). 1H NMR (400 MHz, CDCl3) δ 7.23 (m, 5H), 5.02 (d, J = 9.0, 1H), 4.12 (m, 2H), 3.99 (d, J = 13.7, 1H), 3.84 (d, J = 13.6, 2H), 3.59 (m, 3H), 3.46 (m, 2H), 2.24 (m, 1H), 1.89 (m, 3H), 1.69 (dd, J = 12.8, 8.4, 1H), 1.58 (m, 1H), 1.47 (m, 1H), 1.39 (s, 9H), 0.95 (m, 2H), 0.00 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.7, 159.2, 140.4, 130.0, 129.9, 128.6, 82.2, 66.2, 66.1, 64.9, 60.7, 55.9, 52.8, 37.2, 30.2, 29.6, 29.1, 19.2, 0.0; (ESI-TOF) (m/z) [MH]+ calcd for C25H42N2O5Si 479.29358, found 479.29459, [M+Na]+ 501.27552, found 501.27650.
(3S,7S,7aS)-tert-butyl 7-((2-(trimethylsilyl)ethoxy)carbonylamino)hexahydro-1H-pyrrolizine-3-carboxylate (26)
Method A: A solution of 25 (48.0 mg, 100 μmol) in 2 mL of MeOH was treated with 20 mg of 20% Pd(OH)2/C and stirred for 1.5 h at rt under H2 (balloon) atmosphere. The reaction was filtered through a celite pad, rinsed with MeOH/EtOAc and evaporated under reduced pressure. The resulting colorless oil was dissolved in 2 mL of THF and treated with triethylamine (41.8 μl, 300 μmol), 4-dimethylaminopyridine (12.2 mg, 100 μmol), and 1,1′-carbonyldiimidazole (81.0 mg, 500 μmol). The reaction was stirred under argon atmosphere at rt for 20 h. The mixture was concentrated under reduced pressure, diluted with EtOAc, and washed with sat. aq. NH4Cl. The combined organic layers were dried over anhydrous Na2SO4 and evaporated. Purification by flash chromatography over silica gel (70% to 100% EtOAc/hexanes as eluent) afforded 26 as a colorless oil (30.0 mg, 82%).
Method B: Bicyclic amine 26 was also prepared from 24 via alkene oxidation as described above, followed by treatment of the intermediate aldehyde (40.0 mg, 84.0 μmol) with 20.0 mg of 20% Pd(OH)2/C in 2 mL of MeOH. After stirring for 20 h at rt under H2 (balloon) atmosphere, the reaction was filtered through a celite pad, rinsed with MeOH/EtOAc, and evaporated under reduced pressure. Purification by flash chromatography over silica gel (70% to 100% EtOAc/hexanes and then 5% MeOH/EtOAc as eluents) afforded 26 as a colorless oil (29.0 mg, 93% for the last step). [α]25D −30.0 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.44 (d, J = 7.5, 1H), 4.13 (m, 3H), 3.82 (dd, J = 14.1, 7.0, 1H), 3.20 (m, 2H), 2.58 (dt, J = 11.0, 7.1, 1H), 2.16 (m, 2H), 1.99 (dt, J = 21.9, 9.3, 1H), 1.83 (m, 1H), 1.70 (m, 1H), 1.51 (m, 1H), 1.44 (s, 9H), 0.96 (m, 2H), 0.00 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.4, 157.8, 82.2, 71.4, 69.1, 64.7, 54.4, 53.5, 34.3, 33.3, 29.6, 27.1, 19.2, 0.0; (ESI-TOF) (m/z) [MH]+ calcd for C18H34N2O4Si 371.23606, found 371.23664.
Boc-[5,6-carbamate]-Phe-OMe (28)
A solution of 9 (164 mg, 440 μmol) in 4 mL of MeOH was treated with 5 mL of 2M aq. NaOH at rt and stirred for 24 h. The reaction was evaporated under reduced pressure and the aqueous layer was washed with Et2O. The pH of the aqueous layer was adjusted to 3, and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure to afford the desired carboxylic acid as a white solid (120 mg, 87%).
The above carboxylic acid (24.0 mg, 79.9 μmol) was dissolved in 2 mL of MeCN and treated with triethylamine (33.5 μL, 240 μmol), HBTU (40.0 mg, 105 μmol), and HOBt (2.00 mg, 16.0 μmol) and stirred for 5 min before adding phenylalanine methyl ester (20.0 mg, 100 μmol). After stirring at rt for 20 h, the reaction was evaporated under reduced pressure and diluted with EtOAc. The organic layer was washed with 1M aq. HCl followed by 10% aq. Na2CO3, dried over anhydrous Na2SO4, and evaporated. Purification by flash chromatography over silica gel (70% to 80% EtOAc/hexanes as eluent) afforded 28 as a white solid (23.0 mg, 60%, 2 steps). mp 140–142°; [α]25D −26.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25 (m, 5H), 7.05 (d, J = 8.2, 1H), 4.85 (td, J = 8.0, 5.5, 1H), 4.63 (d, J = 8.7, 1H), 4.38 (t, J = 8.1, 1H), 4.26 (dd, J = 10.4, 4.4, 1H), 3.90 (dd, J = 13.4, 7.5, 1H), 3.72 (m, 3H), 3.29 (td, J = 9.8, 5.7, 1H), 3.20 (dd, J = 13.9, 5.4, 1H), 3.04 (dd, J = 13.9, 7.9, 1H), 2.17 (m, 1H), 2.10 (ddd, J = 11.2, 6.6, 4.1, 1H), 1.98 (m, 1H), 1.60 (m, 1H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.1, 170.8, 155.0, 153.3, 136.3, 129.6, 129.4, 128.7, 127.1, 68.2, 61.7, 61.6, 53.5, 52.7, 48.1, 37.8, 31.3, 28.6, 28.5, 26.1; (ESI-TOF) (m/z) [MH]+ calcd for C23H31N3O7 462.22420, found 462.22620, [M+Na]+ 484.20542, found 484.20800.
Troc-[5,5-lactam]-Phe-OtBu (29)
A solution of 21 (15.0 mg, 36.0 μmol) in 1.5 mL of 75% THF/DCM was stirred at rt for 6.5 h. The reaction was diluted with EtOAc and evaporated under reduced pressure (dilution and evaporation was repeated three more times). The resulting colorless oil was dissolved in 1.5 mL of acetonitrile and treated with triethylamine (30.0 μL, 216 μmol), HBTU (17.7 mg, 46.8 μmol), and HOBt (972 μg, 7.20 μmol) and stirred for 5 min before adding phenylalanine tert-butyl ester (12.0 mg, 47.0 μmol). After stirring at rt for 20 h, the reaction was evaporated under reduced pressure and diluted with EtOAc. The organic layer was washed with 1M aq. HCl followed by 10% aq. Na2CO3, dried over anhydrous Na2SO4, and evaporated. Purification by flash chromatography over silica gel (60% EtOAc/hexanes as eluent) afforded 29 as a thick oil (15.0 mg, 74%, 2 steps). [α]25D −83.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.7, 1H), 7.23 (m, 3H), 7.12 (dd, J = 11.4, 4.9, 2H), 5.97 (d, J = 8.4, 1H), 4.69 (m, 3H), 4.45 (dd, J = 13.3, 7.5, 1H), 4.31 (t, J = 7.9, 1H), 4.04 (dt, J = 9.1, 6.0, 1H), 3.12 (ddd, J = 24.4, 15.6, 6.8, 2H), 2.98 (dd, J = 13.9, 6.9, 1H), 2.34 (m, 3H), 1.83 (dtd, J = 9.0, 6.7, 2.6, 1H), 1.62 (m, 1H), 1.41 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.3, 170.6, 170.0, 154.4, 136.6, 129.7, 128.4, 127.1, 95.6, 82.5, 74.7, 66.4, 56.9, 54.1, 49.4, 42.1, 38.1, 30.5, 28.2, 24.6; (ESI-TOF) (m/z) [MH]+ calcd for C24H30Cl3N3O6 562.12730, found 562.12662, [M+Na]+ 584.10924, found 584.10870.
Compound 29 was also obtained from 33 as follows: A solution of 33 (50.0 mg, 74.5 μmol) in 1.5 mL of MeOH at rt was treated with 18 mg of 20% Pd(OH)2/C and stirred for 2 h under H2 (balloon). The reaction was filtered through a celite pad, rinsed with MeOH/EtOAc, and evaporated under reduced pressure to afford a white solid, which was then dissolved in 2 mL of DMF and treated with triethylamine (20.8 μL, 149 μmol), HBTU (37.0 mg, 96.8 μmol), and HOBt (2.00 mg, 14.9 μmol) respectively. After 20 h stirring at rt, the reaction was evaporated under reduced pressure and diluted with EtOAc. The organic layer was washed with 1M aq. HCl followed by 10% aq. Na2CO3, dried over anhydrous Na2SO4, and evaporated. Purification by flash chromatography over silica gel (60% EtOAc/hexanes as eluent) afforded 29 as a colorless oil (23.0 mg, 54%, 2 steps)
Troc-[5,7-lactam]-Phe-OtBu (30)
Tripeptide mimic 30 was prepared from 23 using the same two-step procedure described for 29. Purification of the crude material by flash chromatography over silica gel (60% EtOAc/hexanes as eluent) afforded 30 as a white solid (42.0 mg, 77%, 2 steps). mp 97–99°; [α]25D −9.62 (c 1.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24 (m, 5H), 6.62 (d, J = 7.7, 1H), 5.36 (d, J = 10.1, 1H), 4.81 (d, J = 12.1, 1H), 4.66 (m, 3H), 4.08 (m, 2H), 3.09 (dd, J = 5.9, 3.6, 2H), 2.60 (dd, J = 14.1, 6.8, 1H), 2.40 (m, 2H), 2.03 (m, 3H), 1.82 (m, 3H), 1.61 (m, 1H), 1.40 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 175.3, 171.1, 170.7, 154.6, 136.4, 129.9, 128.5, 127.1, 95.7, 82.6, 74.7, 62.3, 61.5, 53.8, 53.1, 38.3, 38.0, 35.4, 31.3, 28.2, 27.5, 18.0; (ESI-TOF) (m/z) [MH]+ calcd for C26H34Cl3N3O6 590.15860, found 590.15828, [M+Na]+ 612.14054, found 612.14093.
Compound 30 was also obtained from 32 following the same procedure use to convert 33 to 29. Purification by flash chromatography over silica gel (60% to 100% EtOAc/hexanes as eluent) afforded 30 as a colorless oil (32.0 mg, 53% 2 steps).
Dipeptide 31
Compound 31 was prepared from 19 using the same two-step procedure described for 29. Purification by flash chromatography over silica gel (30% EtOAc/hexanes as eluent) afforded 31 as a white solid (100 mg, 77% 2 steps). mp 116–118°; [α]25D −21.1 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24 (m, 8H), 7.09 (dd, J = 7.5, 1.5, 2H), 6.35 (d, J = 8.0, 1H), 5.73 (ddt, J = 14.0, 10.2, 7.0, 1H), 5.08 (dd, J = 13.3, 5.4, 2H), 4.94 (d, J = 9.3, 1H), 4.71 (m, 3H), 3.84 (m, 3H), 3.45 (dd, J = 8.1, 1.5, 1H), 3.26 (m, 1H), 3.05 (m, 2H), 2.46 (dt, J = 14.3, 5.2, 1H), 2.08 (m, 3H), 1.82 (m, 1H), 1.61 (m, 1H), 1.36 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.5, 170.7, 154.5, 139.3, 136.5, 134.5, 129.6, 128.7, 128.7, 128.6, 128.5, 127.3, 127.2, 118.0, 96.0, 82.5, 74.7, 65.4, 64.6, 53.5, 53.4, 52.9, 38.3, 36.7, 28.6, 28.2, 28.1, 27.2; (ESI-TOF) (m/z) [MH]+ calcd for C32H40Cl3N3O5 652.21064, found 652.21560, [M+Na]+ 674.19258, found 674.20121.
Dipeptide 32
A solution of 31 (105 mg, 160 μmol) in 1 mL of 1,2-dichloroethane was treated with Grubbs’ 2nd generation catalyst (13.0 mg, 18.0 μmol) and benzyl acrylate (296 mg, 1.83 mmol) and stirred for 24 h at 65° C. The reaction was evaporated, adsorbed onto silica gel, and purified by flash chromatography over silica gel (20% to 40% EtOAc/hexanes as eluent) to afford 32 as a thick colorless oil (102 mg, 80%, 97% borsm). Data given for the E isomer: [α]25D −15.8 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.30 (m, 13H), 7.07 (dd, J = 7.3, 1.8, 2H), 6.92 (m, 1H), 6.15 (d, J = 8.1, 1H), 5.88 (d, J = 15.6, 1H), 5.16 (s, 2H), 4.96 (dd, J = 16.6, 9.5, 1H), 4.70 (m, 3H), 3.90 (m, 3H), 3.46 (d, J = 6.9, 1H), 3.35 (dt, J = 8.9, 4.4, 1H), 3.05 (m, 2H), 2.61 (m, 1H), 2.22 (m, 2H), 2.01 (m, 1H), 1.85 (m, 1H), 1.62 (m, 2H), 1.37 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.3, 170.8, 166.0, 154.4, 145.5, 139.2, 136.4, 136.2, 129.6, 128.8, 128.8, 128.7, 128.5, 128.4, 128.4, 127.4, 127.3, 123.7, 95.8, 82.6, 74.6, 66.4, 65.4, 64.7, 53.7, 53.1, 52.9, 38.3, 34.8, 28.8, 28.1, 27.1; (ESI-TOF) (m/z) [MH]+ calcd for C40H46Cl3N3O7 786.24741, found 786.25238, [M+Na]+ 808.22936, found 808.23375.
Dipeptide 33
A solution of 31 (180 mg, 276 μmol) in 6 mL of THF:H2O (4:2) at rt was treated with 4-methylmopholine N-oxide (71.1 mg, 607 μmol) and osmium tetroxide (2.5% solution in 2-methyl2-propanol, 308 μL, 30.0 μmol) and stirred for 4.5 h. Sodium periodate (130 mg, 607 μmol) was then added into the diol intermediate and the reaction was stirred for 14 h. The reaction was quenched with 5% aq. Na2S2O3 and diluted with brine. The aqueous layer was extracted with EtOAc and the combined organic layers were dried over anhydrous Na2SO4, and evaporated. Purification by flash chromatography over silica gel (20% to 40% EtOAc/hexanes as eluent) afforded the desired aldehyde as a colorless oil (110 mg, 61%). [α]25D −39.7 (c 2.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.62 (t, J = 2.5, 1H), 7.24 (m, 8H), 7.00 (dd, J = 6.5, 2.8, 2H), 5.87 (d, J = 8.1, 1H), 5.13 (d, J = 8.7, 1H), 4.72 (m, 3H), 4.35 (m, 1H), 3.88 (m, 2H), 3.54 (m, 1H), 3.42 (d, J = 7.6, 1H), 3.05 (dd, J = 14.0, 6.0, 1H), 2.94 (dd, J = 14.0, 6.3, 1H), 2.67 (ddd, J = 15.6, 6.2, 2.6, 1H), 2.49 (ddd, J = 15.7, 7.7, 2.5, 1H), 2.27 (m, 1H), 1.92 (m, 1H), 1.81 (m, 1H), 1.61 (ddt, J = 12.3, 6.6, 3.6, 1H), 1.36 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 199.5, 172.9, 170.7, 154.2, 138.8, 136.2, 129.5, 128.8, 128.7, 128.6, 127.5, 127.3, 95.6, 82.6, 74.8, 65.0, 64.7, 53.9, 52.9, 49.1, 45.4, 38.4, 29.0, 28.1, 26.0; (ESI-TOF) (m/z) [MH]+ calcd for C31H38Cl3N3O6 654.18990, found 654.18966, [M+Na]+ 676.17184, found 676.17397.
A solution of above aldehyde (56.0 mg, 85.5 μmol) in 500 μL of t-BuOH was treated with amylene (108 μL, 1.02 mmol) and cooled to 0° C. A solution of NaH2PO4 (105 mg, 765 μmol) and NaClO2 (58.0 μL, 510 μmol) in water (1.00 mL) was added dropwise and the reaction was stirred from 0° C to 10° C over 1 h. The reaction was quenched with 5% aq. Na2S2O3 and diluted with brine. The pH of the aqueous layer was adjusted to 5 and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, and evaporated. Purification by flash chromatography over silica gel (60% to 100 % EtOAc/hexanes and 10% MeOH/EtOAc as eluent) afforded 33 as a thick colorless oil (51.0 mg, 89%). 1H NMR (400 MHz, CDCl3) δ 7.25 (m, 8H), 7.07 (d, J = 6.3, 2H), 6.45 (m, 1H), 5.53 (bs, 1H), 4.71 (dt, J = 20.3, 8.3, 3H), 4.28 (m, 1H), 4.00 (m, 2H), 3.79–3.46 (m, 2H), 3.02 (m, 2H), 2.75 (dd, J = 16.3, 7.2, 1H), 2.55 (dd, J = 16.2, 6.1, 1H), 2.29 (m, 1H), 2.05–1.82 (m, 2H), 1.72 (m, 1H), 1.36 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 175.4, 171.8, 170.6, 154.3, 136.4, 129.5, 129.4, 129.1, 128.9, 128.7, 128.1, 127.3, 95.7, 82.6, 74.8, 65.3, 64.5, 53.8, 53.4, 49.7, 38.1, 36.9, 29.0, 28.1, 25.8; (ESI-TOF) (m/z) [MH]+ calcd for C31H38Cl3N3O7 670.18481, found 670.18508, [M+Na]+ 692.16675, found 692.16718.
Supplementary Material
Acknowledgments
This research was supported by the Moffitt Cancer Center and a grant from National Institutes of Health (U54 CA132383). We thank Dr. Eileen Duesler (University of New Mexico) for carrying out x-ray diffraction studies and Drs. Wayne Guida and Kenyon Daniel (Moffitt Cancer Center) for assistance with molecular modeling.
Footnotes
Supporting Information Available: NMR spectra for all new compounds and crystal structure data (CIF files) for compounds 6a, (α′S)-14, 23, and 28. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Somers WS, Phillips SEV. Nature. 1992;359:387–393. doi: 10.1038/359387a0. [DOI] [PubMed] [Google Scholar]; (b) Puglisi JD, Chen L, Blanchard S, Frankel AD. Science. 1995;270:1200–1203. doi: 10.1126/science.270.5239.1200. [DOI] [PubMed] [Google Scholar]; (c) Derrick JP, Wigley DB. Nature. 1992;359:752–754. doi: 10.1038/359752a0. [DOI] [PubMed] [Google Scholar]; (d) Colon W, Kelly JW. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 2.(a) Glenn MP, Fairlie DP. Mini Reviews in Medicinal Chemistry. 2002;2:433–445. doi: 10.2174/1389557023405747. [DOI] [PubMed] [Google Scholar]; (b) Loughlin WA, Tyndall JDA, Glenn MP, Fairlie DP. Chem Rev. 2004;104:6085–6118. doi: 10.1021/cr040648k. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kemp DS, Bowen BR, Muendel CC. J Org Chem. 1990;55:4650–4657. [Google Scholar]; (b) Khakshoor O, Nowick JS. Curr Opin Chem Biol. 2008;12:722–729. doi: 10.1016/j.cbpa.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Levin S, Nowick JS. J Am Chem Soc. 2007;129:13043–13048. doi: 10.1021/ja073391r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nowick JS. Acc Chem Res. 1999;32:287–296. [Google Scholar]; (e) Nowick JS, Smith EM, Noronha G. J Org Chem. 1995;60:7386–7387. [Google Scholar]; (f) Nowick JS, Cary JM, Tsai JH. J Am Chem Soc. 2001;123:5176–5180. doi: 10.1021/ja010220s. [DOI] [PubMed] [Google Scholar]; (g) Phillips ST, Rezac M, Abel U, Kossenjans M, Bartlett PA. J Am Chem Soc. 2002;124:58–66. doi: 10.1021/ja0168460. [DOI] [PubMed] [Google Scholar]; (h) Zeng H, Yang X, Flowers RA, Gong B. J Am Chem Soc. 2002;124:2903–2910. doi: 10.1021/ja010701b. [DOI] [PubMed] [Google Scholar]
- 4.(a) Smith CK, Regan L. Acc Chem Res. 1997;30:153–161. [Google Scholar]; (b) Fairlie DP, Tyndall JDA, Reid RC, Wong AK, Abbenante G, Scanlon MJ, March DR, Bergman DA, Chai CLL, Burkett BA. J Med Chem. 2000;43:1271–1281. doi: 10.1021/jm990315t. [DOI] [PubMed] [Google Scholar]; (c) Tyndall JDA, Fairlie DP. J Mol Recog. 1999;12:363–370. doi: 10.1002/(SICI)1099-1352(199911/12)12:6<363::AID-JMR478>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]; (d) Wolfram B, Robert H.Eur J Biochem 1992204433–451.1541261 [Google Scholar]; (e) Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. Nature. 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]; (f) Sawyer TK, Bohacek RS, Dalgarno DC, Eyermann CJ, Kawahata N, Metcalf CA, Shakespeare WC, Sundaramoorthi R, Wang Y, Yang MG. Mini-Reviews in Medicinal Chemistry. 2002;2:475–488. doi: 10.2174/1389557023405765. [DOI] [PubMed] [Google Scholar]
- 5.(a) Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS, Weber PC. Biochemistry. 1998;37:16601–16611. doi: 10.1021/bi981197z. [DOI] [PubMed] [Google Scholar]; (b) Long SB, Casey PJ, Beese LS. Structure. 2000;8:209–222. doi: 10.1016/s0969-2126(00)00096-4. [DOI] [PubMed] [Google Scholar]; (c) Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. Nat Struct Mol Biol. 2002;9:940–944. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- 6.(a) Qian Y, Blaskovich MA, Saleem M, Seong CM, Wathen SP, Hamilton AD, Sebti SM. J Biol Chem. 1994;269:12410–3. [PubMed] [Google Scholar]; (b) Clerc FF, Guitton JD, Fromage N, Lelièvre Y, Duchesne M, Tocqué B, James-Surcouf E, Commerçon A, Becquart J. Bioorg Med Chem Lett. 1995;5:1779–1784. [Google Scholar]; (c) Qian Y, Marugan JJ, Fossum RD, Vogt A, Sebti SM, Hamilton AD. Bioorg Med Chem. 1999;7:3011–24. doi: 10.1016/s0968-0896(99)00252-7. [DOI] [PubMed] [Google Scholar]; (d) Kayser KJ, Glenn MP, Sebti SM, Cheng JQ, Hamilton AD. Bioorg Med Chem Lett. 2007;17:2068–73. doi: 10.1016/j.bmcl.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 7.(a) Nowick JS, Pairish M, Lee IQ, Holmes DL, Ziller JW. J Am Chem Soc. 1997;119:5413–5424. [Google Scholar]; (b) Blomberg D, Brickmann K, Kihlberg J. Tetrahedron. 2006;62:10937–10944. [Google Scholar]; (c) Martin SF, Austin RE, Oalmann CJ, Baker WR, Condon SL, DeLara E, Rosenberg SH, Spina KP, Stein HH. J Med Chem. 1992;35:1710–1721. doi: 10.1021/jm00088a005. [DOI] [PubMed] [Google Scholar]; (d) Hagihara M, Anthony NJ, Stout TJ, Clardy J, Schreiber SL. J Am Chem Soc. 1992;114:6568–6570. [Google Scholar]; (e) Burns CJ, Guitton JD, Baudoin B, Lelievre Y, Duchesne M, Parker F, Fromage N, Commercon A. J Med Chem. 1997;40:1763–1767. doi: 10.1021/jm9701177. [DOI] [PubMed] [Google Scholar]; (f) Chandrasekhar S, Sudhakar A, Kiran MU, Babu BN, Jagadeesh B. Tetrahedron Lett. 2008;49:7368–7371. [Google Scholar]
- 8.(a) Smith AB, Guzman MC, Sprengeler PA, Keenan TP, Holcomb RC, Wood JL, Carroll PJ, Hirschmann R. J Am Chem Soc. 1994;116:9947–9962. [Google Scholar]; (b) Angelo NG, Arora PS. J Am Chem Soc. 2005;127:17134–17135. doi: 10.1021/ja056406z. [DOI] [PubMed] [Google Scholar]; (c) Wyrembak PN, Hamilton AD. J Am Chem Soc. 2009;131:4566–4567. doi: 10.1021/ja809245t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillespie P, Cicariello J, Olson GL. Peptide Science. 1997;43:191–217. [Google Scholar]
- 10.(a) Hanessian S, McNaughtonSmith G, Lombart HG, Lubell WD. Tetrahedron. 1997;53:12789–12854. [Google Scholar]; (b) Cluzeau J, Lubell WD. Peptide Science. 2005;80:98–150. doi: 10.1002/bip.20213. [DOI] [PubMed] [Google Scholar]; (c) Halab L, Gosselin F, Lubell WD. Peptide Science. 2000;55:101–122. doi: 10.1002/1097-0282(2000)55:2<101::AID-BIP20>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 11.Freidinger RM, Perlow DS, Veber DF. J Org Chem. 1982;47:104–109. [Google Scholar]
- 12.Liyanage W, Weerasinghe L, Strong RK, Del Valle JR. J Org Chem. 2008;73:7420–7423. doi: 10.1021/jo801294p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Knight DW, Share AC, Gallagher PT. J Chem Soc Perkin Trans 1. 1997:2089–2098. [Google Scholar]; (b) Hanessian S, Sharma R. Heterocycles. 2000;52:1231–1239. [Google Scholar]; (c) Yi JJ, Hua ZM, Rong TG. Synth Comm. 2003;33:3913–3917. [Google Scholar]
- 14.Similar stereochemical outcomes were observed in the examples from refs. 43–45.
- 15.(a) Olofson RA, Martz JT, Senet JP, Piteau M, Malfroot T. J Org Chem. 1984;49:2081–2082. [Google Scholar]; (b) Olofson RA, Schnur RC. Tetrahedron Lett. 1977;18:1571–1574. [Google Scholar]
- 16.Use of excess trimethyltin hydroxide as a mild ester deprotection reagent gave only trace amounts of the desired acid. See: Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. Angew Chem Int Ed. 2005;44:1378–1382. doi: 10.1002/anie.200462207.
- 17.The configurational instability of 12 and 16 versus 6c is likely due to steric interactions of the endo α′ substituent in the convex bicyclic frameworks. Based on the x-ray structure of 23, the ethylcarboxy substituent in 16 presumably also occupies an axial position.
- 18.For a study on similar reaction pathways with 1,1′-carbonyldiimidazole, see: de Figueiredo RM, Fröhlich R, Christmann M. J Org Chem. 2006;71:4147–4154. doi: 10.1021/jo060130b.
- 19.Venkatraman J, Shankaramma SC, Balaram P. Chem Rev. 2001;101:3131–3152. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- 20.It should be noted that these values are of roughly equal and opposite sign, which is the principle requirement for our surrogates. However, mimicry of a natural L,L-dipeptide strand would require the synthesis of the enantiomer scaffold starting from D-pyroglutamic acid.
- 21.Measured torsions and distances for 28 are given as the average between the two molecules in the dimer.
- 22.See Supporting Information for details.
- 23.See Experimental Section for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.