

Abstract
The transcription factor family, nuclear factor of activated T cells (NFAT), regulates immune cell phenotype. Four different calcium/calmodulin-regulated isoforms have been identified in the periphery, but isoform expression in microglia, the resident immune cells of the CNS, has not been fully defined. In this study microglial NFAT isoform expression and involvement in regulating inflammatory responses in murine primary microglia culture was examined. Western blot analysis demonstrated robust detection of NFATc1 and c2 isoforms in microglia. Electrophoretic mobility shift assays demonstrated increased NFAT-DNA binding from nuclear extracts of lipopolysaccharide (LPS) stimulated microglia. Moreover, LPS-stimulated microglia behaved similarly to T cell receptor agonist antibody-stimulated Jurkat cells demonstrating a transient increase in NFAT-driven luciferase reporter gene expression. LPS-induced NFAT-luciferase activity in microglia was attenuated by pretreatment with tat-VIVIT, a cell-permeable NFAT inhibitory peptide. Furthermore, LPS-mediated secretion of microglial cytokines, TNF-α and MCP-1, was decreased by treatment with tat-VIVIT but not with tat-VEET, a negative control peptide. These results demonstrate that NFAT plays a role in regulating proinflammatory responses in cultured murine microglia.
Introduction
Modulation of immune responses is one of the major therapeutic objectives in a variety of chronic neurodegenerative diseases. As a therapeutic target for immunomodulation, the nuclear factor of activated T cells (NFAT) has received considerable attention. It was first described as a part of the protein complex which altered transcription of the interleukin-2 (IL-2) gene after antigen receptor activation of T lymphocytes (Shaw et al., 1988). NFAT is now well recognized as a member of the REL family of transcription factors crucially involved in regulating transcription of multiple proinflammatory genes, such as IL-2 and tumor necrosis factor-α (TNF-α) (for a comprehensive review, see Rao et al., 1997). Two groups of differentially regulated NFAT transcription factor isoforms have been identified: (1) Ca2+/calcineurin-activated isoforms, NFATc1 (also known as NFAT2 or NFATc), NFATc2 (NFAT1 or NFATp), NFATc3 (NFAT4 or NFATx) and NFATc4 (NFAT3); and (2) a tonicity-controlled isoform, NFAT5 (NFATz, NFATL1, TonEBP) (Lopez-Rodríguez et al., 1999; Miyakawa et al., 1999). Although NFAT isoforms are expressed in various types of immune and nonimmune cells, the Ca2+/calcineurin-activated NFAT isoforms are critically involved in regulating immune cell phenotypes (Masuda et al., 1998; Macian, 2005). Stimulations leading to calcium-mediated signaling cascades can increase activity of calcineurin, a Ca2+/calmodulin-regulated phosphatase, which then dephosphorylates inactive cytosolic NFAT allowing it to translocate to the nucleus (Shaw et al., 1995). Nuclear NFAT works cooperatively with a number of additional transcription factors including AP-1, NFκB, MEF-2, and PPARγ to regulate transcription (Boise et al., 1993; Jain et al., 1993; Yang et al., 2000; Fisher et al., 2006; Bao et al., 2008; Putt et al., 2009).
NFAT expression has been reported in neurons and astrocytes in the CNS (Graef et al., 2003; Benedito et al., 2005; Luoma and Zirpel, 2008; Pérez-Ortiz et al., 2008; Sama et al., 2008). Neuronal NFAT isoforms have a role in regulating axonal growth (Graef et al., 2003), neuronal survival (Benedito et al., 2005) and apoptosis (Luoma and Zirpel, 2008) during development. Astrocytic NFAT is involved in initiation and maintenance of injury, disease, or aging-mediated neuroinflammatory processes (Pérez-Ortiz et al., 2008; Sama et al., 2008; Abdul et al., 2009). However, relatively few studies have documented expression and function of NFAT in microglia, the resident immune cell of the brain (Ferrari et al., 1999; Kataoka et al., 2009). This study has identified NFAT isoform expression in primary murine microglia cultures and verified that it is involved in regulating proinflammatory gene expression similar to its role in other immune cells.
Materials and Methods
Materials.
The rabbit polyclonal anti-human NFATc1 (NFAT2), NFATc4 (NFAT3), monoclonal NFATc3 (NFAT4), and polyclonal anti-α-tubulin antibodies were purchased from Santa Cruz Biotechnology. The rabbit anti-human NFATc2 (NFAT1) antibody was obtained from Abcam. The CD3 agonist antibody was from eBioscience. Lipopolysaccharide (LPS) and other reagents were obtained from Sigma. The inhibitory peptide, tat-VIVIT (H-YGRKKRRQRRR-AA-MAGPHPVIVITGPHEE-NH2) and control peptide, tat-VEET (H-YGRKKRRQRRR-AA-MAGPPHIVEETGPHVI-NH2) were from the Molecular Biotechnology Core Laboratory at the Cleveland Clinic Foundation (Cleveland, OH).
Tissue culture.
Jurkat cells were acquired from the American Type Culture Collection and maintained in RPMI-1640 medium (Invitrogen) supplemented with 10% heat-inactivated FBS (US Biotechnologies Inc.), 5 mm HEPES, and 1.5 μg/ml penicillin/streptomycin/neomycin. Primary microglial cultures were isolated from cerebral cortices of C57BL/6 mouse brains at postnatal day 0–2 as previously described and purified from mixed glial cultures after 14 d in vitro for use (Floden et al., 2005).
Fluorescent labeling of VIVIT peptide.
A quantity of tat-VIVIT peptide was fluorescent-labeled using the Alexa Fluor 488 Microscale Protein Labeling Kit according to the manufacturer's protocol (Invitrogen Corp.). Fluorescent-labeled tat-VIVIT was incubated with Jurkat or primary microglia cultures for 60 min (10 μm) to verify transduction into cells.
Transient transfection of Jurkat and primary microglia cultures.
NFAT promotor activity in vitro was assessed using an NFAT-luciferase reporter construct (pNFAT-luc) which contained three consecutive NFAT-response elements upstream of the Photinus pyralis luciferase reporter gene (Panomics). Transient transfection of Jurkat cells (5 × 106 cells/1.5 μg of pNFAT-luc and 0.5 μg of pmaxGFP) or primary microglia (1 × 106 cells/1.5 μg of pNFAT-luc and 0.5 μg of pmaxGFP) with the reporter construct was performed using an Amaxa Nucleofector I device with Cell Line Nucleofector Kit V (VCA-1003) or Mouse Macrophage Nucleofector Kit (VPA-1009) according to the manufacturer's protocols. A green fluorescent protein (GFP) expression vector, pmaxGFP, which expressed GFP under the control of the CMV promotor, was also cotransfected with pNFAT-luc to determine the transfection efficiency (Amaxa). Transfected cells were incubated for 24 h in serum-free medium before stimulation.
Stimulation and tat-VIVIT peptide treatment of transiently transfected Jurkat and primary microglia cells.
Transiently transfected Jurkat cells were stimulated in serum-free medium with/without 2 μg/ml anti-CD3 antibody for 4 h at 37°C. Transiently transfected microglia were stimulated with either TNF-α (50 ng/ml) or lipopolysaccharide (LPS) (25 ng/ml) in serum-free DMEM/F-12 medium for 4 h. Designated groups of cells were incubated in the presence of 10 μm tat-VIVIT or tat-VEET peptide (negative control), or 1 μm FK506 (positive control) before (10 min) and during stimulations.
Cell lysate preparation and luciferase assays.
Luciferase activity was measured using the commercial Luciferase Assay System (Promega Corp.). Stimulated cells were harvested in Reporter Lysis Buffer (Promega) and luciferase activity was quantified from supernatants using a TD-20/20 Luminometer (Promega). Values were normalized against fluorescence from the cotransfected pmaxGFP construct using an FLx800 Microplate Fluorescence Reader (BioTek Instruments).
Western blot analysis.
Jurkat or primary microglia cells with or without stimulation were harvested in PBS and resuspended in ice-cold RIPA buffer (1% Triton, 0.1% SDS, 0.5% deoxycholate, 20 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 10 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.2 mmol/L PMSF). Samples were sonicated, centrifuged at 4°C and supernatants quantitated by the method of Bradford (1976). Proteins were resolved using 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes as described previously for chemiluminescent Western blotting (Sondag and Combs, 2004). Signals were captured by a UVP Biospectrum Imaging System (UVP, LLC).
Cytokine array.
Jurkat cells were stimulated in serum-free RPMI medium with/without 1 μg/ml anti-CD3 antibody in the absence or presence of 1 μm tat-VIVIT or tat-VEET peptide for 24 h. The medium from each condition was collected and used to probe a human inflammatory antibody array for relative changes across the treatment paradigms (RayBiotech).
ELISA.
The levels of select proinflammatory cytokine/chemokine were quantified using ELISA. Primary microglial cultures were stimulated in serum-free DMEM/F-12 medium with LPS in the absence or presence of tat-VIVIT or tat-VEET peptide as described above. The medium from each treatment condition was collected and used to quantify concentrations of TNF-α, KC, and MCP-1 using commercially available ELISA reagents (R&D Systems).
Preparation of microglial nuclear extracts and electrophoretic mobility shift assays.
To qualitatively analyze NFAT activation via its ability to form DNA binding complexes, microglia cultures were treated with control, IgG (1 μg/ml) or LPS (25 ng/ml) in serum-free DMEM/F12 medium for 30 min and nuclear extracts were prepared (Panomics). Extracts were incubated with a biotinylated NFAT oligonucleotide probe (NFAT1 probe: AY1027P, Panomics, Inc) using a protocol previously described (Best et al., 1995; Nagamoto-Combs et al., 1997) and modified according to the probe manufacturer's suggestions (Panomics). Reaction mixtures were resolved via nondenaturing 6% polyacrylamide gel, transferred onto Pall Biodyne B nylon membrane, and detected via a Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher Scientific).
Statistics.
Data are presented as mean values ± SDs. Statistical differences were tested using unpaired one-way ANOVA with Tukey-Kramer's post hoc comparison, and considered significant when p < 0.05.
Results
Primary murine microglia expressed NFAT isoforms
To identify whether NFAT isoform(s) were uniquely expressed in microglia we examined primary murine microglia cultures via Western blot analysis for expression of the four Ca2+/calcineurin-regulated NFAT isoforms, NFATc1, c2, c3 and c4, compared with the human Jurkat T cell line (Fig. 1). Although Jurkat cells had detectable levels of all four isoforms as expected, microglia cultures demonstrated immunoreactive bands, in particular, with NFATc1 and c2 antibodies likely representing multiple rodent isoform expression for both subtypes as has been reported (Vihma et al., 2008). This indicated that microglia may exhibit somewhat isoform selective expression compared with Jurkat T cells. Prior work has demonstrated increased NFAT isoform transcription in T cells upon T cell receptor stimulation or activation with phorbol ester (Sareneva et al., 1998). Therefore microglia were stimulated with lipopolysaccharide (LPS) overnight to generate a more reactive phenotype to assess changes in protein levels of NFAT. However, LPS stimulation was insufficient to alter detectable protein levels of NFAT isoforms in microglia (Fig. 1). These data demonstrated that microglia exhibit detectable levels of particularly NFATc1 and c2 isoforms in both basal and proinflammatory stimulated microglia cultures.
Figure 1.

Primary murine microglia expressed NFAT. Microglia were isolated from postnatal day 0 mouse brains and grown as mixed cultures until day 14 then separated for stimulation. Purified microglia were stimulated 24 h with (s, stimulated) or without (u, unstimulated) 25 ng/ml LPS and lysed for comparison to Jurkat T cell lysates. Cell lysates were separated by 10% SDS-PAGE and Western blotted using anti-NFATc1, c2, c3, c4, and α-tubulin (loading control) antibodies and visualized by enhanced chemiluminescence. Blots from three representative experiments are shown.
The tat-VIVIT fusion peptide effectively transduced into murine microglia and human Jurkat cells
Although microglia expressed NFAT isoforms, it was possible that microglial NFAT functioned uniquely from other immune cells, such as the Jurkat T cells. To compare microglial and T cell NFAT functions it was necessary to first verify an NFAT inhibitory reagent. The VIVIT peptide sequence, MAGPHPVIVITGPHEE, interacts with NFAT at its calcineurin binding site and inhibits its activation (Aramburu et al., 1999). Studies using cultured cells and/or in vivo animal models have successfully prevented NFAT activation by the use of a cell-permeable VIVIT peptide fused with poly-arginine (11R-VIVIT) (Noguchi et al., 2004; Kuriyama et al., 2006). Another cell-permeable variation of VIVIT peptide, tat-VIVIT, fused with a widely used HIV-1 TAT protein cationic transduction domain (Brooks et al., 2005; Vives, 2005), was generated to examine the potential of this novel peptide construct for NFAT inhibition. The ability of the peptide to transduce into cells was determined by labeling it with a fluorophore, Alexa 488, and assessing its import into cells. Incubation of both Jurkat cells and microglia for 1 h with 10 μm Alexa 488-labeled tat-VIVIT was sufficient to detect peptide transduction (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) verifying the cell-permeability of this reagent and concentration for further use.
The tat-VIVIT fusion peptide attenuated T-cell receptor-stimulated NFAT activation in vitro
Because this particular tat-VIVIT fusion peptide variant has not been previously characterized for its ability to inhibit NFAT activity, it was necessary to validate its inhibitory action. For this purpose, Jurkat cells were transiently cotransfected with a luciferase construct driven by three copies of an NFAT response element (pNFAT-luc) and a green fluorescent protein construct constitutively driven by the cytomegalovirus promoter (pmaxGFP). Stimulation of T cell receptors by anti-CD3 agonist antibody for 4 h significantly increased NFAT-driven luciferase activity in the transfected Jurkat cells (Fig. 2A). As expected, treatment of the cells with tat-VIVIT peptide decreased luciferase activity significantly greater than that induced by negative control tat-VEET, a peptide with an arbitrary sequence that does not interfere with NFAT (Noguchi et al., 2004) (Fig. 2A). However, tat-VIVIT was unable to decrease luciferase activity to the level achieved by treating cells with the calcineurin inhibitor, FK506 (Fig. 2A).
Figure 2.
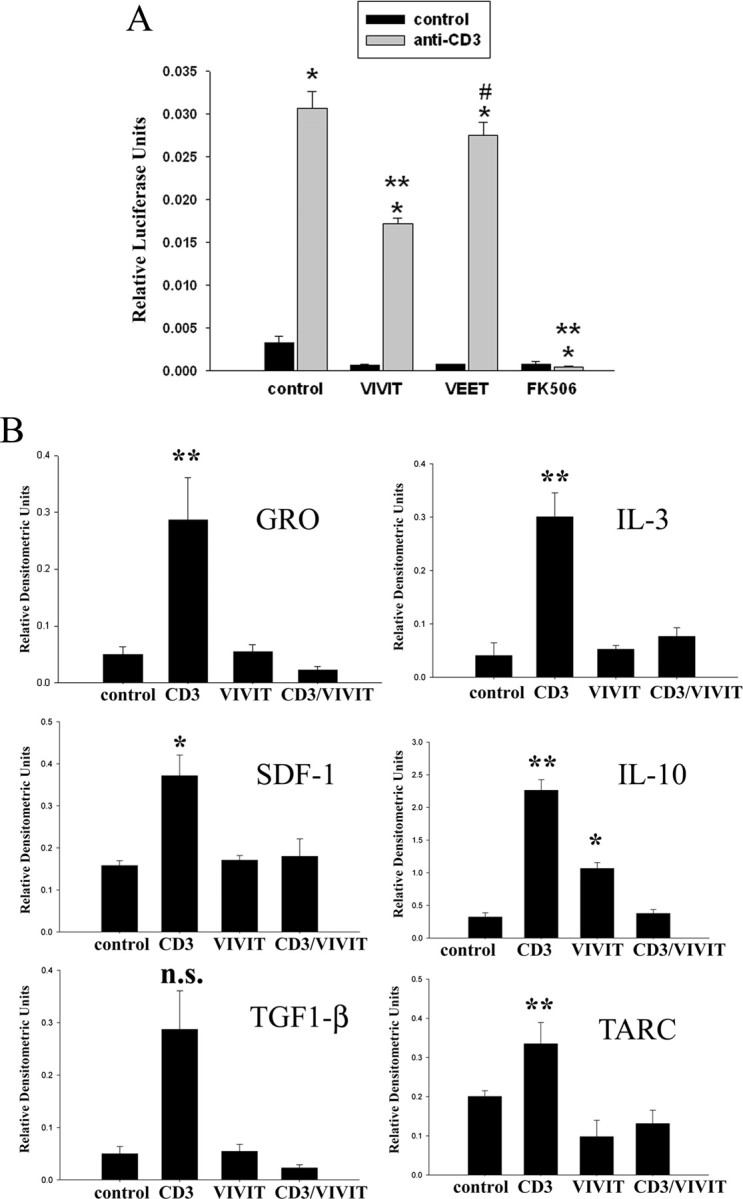
Tat-VIVIT peptide attenuated NFAT-dependent protein expression in immune cells. A, Jurkat cells were transiently cotransfected with an NFAT response element-luciferase construct and a green fluorescent protein construct. After 24 h cells were stimulated with or without 2 μg/ml anti-CD3 agonist antibody to activate T cell receptors. Stimulated cells were preincubated 30 min with or without 10 μm VIVIT peptide or 10 μm VEET peptide (negative control) and during 24 h stimulation. Cells were lysed and luciferase activity was normalized against green fluorescence (transfection control) to express as relative luciferase units. Luciferase units were averaged ± SD and graphed. Representative data from three independent experiments are shown. (*p < 0.001 from control, **p < 0.001 from CD3, #p < 0.05 from CD3). B, Jurkat cells were stimulated with or without 2 μg/ml anti-CD3 antibody in serum-free RMPI for 24 h. Stimulated cells were preincubated 30 min with 10 μm VIVIT peptide and during the 24 h stimulation with the agonist antibody. Media aliquots were analyzed via commercial antibody array to determine changes in select cytokine/chemokine secretion (GRO, SDF-1, IL-3, IL-10, TARC). Densitometric analysis from two independent experiments performed in duplicate were averaged for each condition (±SD) and graphed (**p < 0.001 from control, *p < 0.01 from control).
Based upon the ability of tat-VIVIT to attenuate NFAT-driven luciferase expression, it was next determined whether the peptide exerted the expected attenuation of cytokine secretion. Again Jurkat cells were stimulated with CD3 agonist antibody in the absence or presence of tat-VIVIT peptide. Cytokine array analysis from collected media verified that tat-VIVIT treatment selectively attenuated secretion of a number of CD3-stimulated cytokines (Fig. 2B). In particular, quantitative assessment revealed changes in secreted levels of interleukin-3 (IL-3), interleukin-10 (IL-10), stromal cell-derived factor-1 (SDF-1), growth-related oncogene (GRO), and thymus activation-regulated cytokine (TARC) by tat-VIVIT treatment. Interestingly, VIVIT treatment alone increased IL-10 secretion from the Jurkat cells. These observations indicated that the tat-VIVIT fusion peptide is capable of regulating diverse NFAT-driven gene expression and cytokine secretion validating it as an NFAT inhibitory reagent.
NFAT was activated in primary murine microglia by select proinflammatory stimuli and responsible for regulating cytokine secretion
Once the utility of the tat-VIVIT peptide was confirmed in a classic T cell-dependent proinflammatory response, it was used to directly assess its anti-inflammatory actions on primary microglia. Electrophoretic mobility shift assays verified that typical proinflammatory stimuli for microglial cultures such as LPS and IgG were sufficient to increase NFAT-DNA binding ability (Fig. 3A). NFAT-driven luciferase expression following transient transfection into the primary microglia was also significantly stimulated by LPS but not TNFα suggesting some specificity of activation (Fig. 3B). More importantly, tat-VIVIT was sufficient to attenuate LPS-mediated, NFAT-dependent luciferase expression (Fig. 3C). This result correlated precisely with the ability of tat-VIVIT but not tat-VEET to decrease cytokine secretions. Based upon prior assessment of LPS-stimulated cytokine secretion profiles from microglia cultures (Sondag et al., 2009), increases of highly secreted cytokines were compared with cultures coincubated with LPS and tat-VIVIT. LPS-stimulated secretions of TNFα and MCP-1 were effectively inhibited by tat-VIVIT but not tat-VEET peptide (Fig. 3D,F). However, LPS-stimulated KC secretion was not attenuated by tat-VIVIT, again demonstrating specificity of the NFAT-dependent phenotype for a particular secretory profile (Fig. 3E).
Figure 3.

NFAT activity-regulated proinflammatory secretion and was attenuated by tat-VIVIT peptide in murine microglia cultures. Primary mouse microglia were isolated for 14 d in vitro mixed cultures derived from postnatal day 0 brains for stimulation. A, Microglia were stimulated 30 min with or without 1 μg/ml IgG or 25 ng/ml LPS and NFAT binding ability (arrowhead) to DNA response element oligonucleotide probe was assessed by electromobility shift assay. Blots from three representative experiments are shown. B, C, Mouse microglia cultures were transiently cotransfected with an NFAT response element-luciferase construct and a green fluorescent protein construct. After 24 h, cells were preincubated 30 min with or without 10 μm VIVIT peptide or 10 μm VEET peptide (negative control) and during a 6 h stimulation with TNFα (50 ng/ml) or LPS (25 ng/ml). Cells were lysed and luciferase activity was normalized against green fluorescence (transfection control) to express as relative luciferase units. Luciferase units were averaged ± SD and graphed. Representative data from three independent experiments are shown (*p < 0.01 from control, **p < 0.001 from LPS). D–F, Primary murine microglia cultures were stimulated serum free for 24 h with our without 25 ng/ml LPS, 10 μm VIVIT, or 10 μm VEET (negative control). Media aliquots were taken from the cells and commercial ELISA assays performed to quantify secretion of TNFα, KC and MCP-1 into the media. Mean values ± SD were determined and data shown are representative from 3 independent experiments (*p < 0.05 from control, **p < 0.001 from control, #p < 0.001 from LPS).
Discussion
Although microglia are considered to be the resident immune cells of the CNS, the expression, regulation and function of NFAT in these cells are poorly understood. In this study, microglia cultured from postnatal mouse brain abundantly expressed NFATc1 and c2 isoforms in contrast to the Jurkat T cell line which expressed comparatively high levels of NFATc1-c4 (Fig. 1). However, it is possible that microglia express low, undetectable levels of other NFAT isoforms even though LPS-dependent activation of the cells did not facilitate their detection. This somewhat selective expression suggests that microglial NFAT may function uniquely from NFAT in other immune cells since microglia do not appear to have evolutionarily conserved the multiple isoform expression pattern.
Because of this expression profile, it was necessary to determine whether microglial NFAT behaved similar to peripheral immune cell NFAT by assessing its ability to regulate acquisition of reactive phenotypes. As one of the tools to achieve this goal, the VIVIT peptide, a peptide sequence reported to inhibit NFAT activation, was used (Noguchi et al., 2004). We chose to fuse the VIVIT peptide sequence with an HIV-1 tat protein transduction sequence, instead of the prior reported poly-arginine version described by Noguchi et al. (2004), due to the ability of the tat sequence to deliver functional proteins across the blood–brain barrier (Schwarze et al., 1999) and its precedent in vivo use with brain-derived cells (Vieira et al., 2009; Wang et al., 2009). Interestingly, the transduction efficacy of the tat-VIVT peptide into the Jurkat and microglia differed for reasons that are unclear. The more efficient microglial transduction of peptide (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) correlated with a greater degree of VIVIT-dependent decrease in stimulated NFAT-luciferase expression/activity in microglia versus Jurkat cells (Fig. 3C vs 2A). It is plausible that the larger and flatter morphology of cultured microglia may have been a contributing factor. The incomplete inhibition of NFAT-dependent luciferase activity in Jurkat cells versus microglia could also be related to expression of other NFAT isoforms in the T cells that effectively competed with the tat-VIVIT peptide at the concentrations employed. Use of higher concentrations of tat-VIVIT to compensate for this possibility in either cell type was not tested since the peptide was toxic at higher concentrations as assessed by LDH release assay (data not shown). In contrast to tat-VIVIT, FK506, the calcineurin inhibitor, achieved near complete inhibition of both basal and anti-CD3-stimulated NFAT-luciferase activity in Jurkat cells suggesting that the calcineurin inhibitor prevented activation of all NFAT isoforms and/or attenuated critical calcineurin-dependent signaling events required for proinflammatory phenotype change.
Although LPS proved to be a significant stimulus for increased NFAT-luciferase activity and NFAT-DNA complex formation from microglial extracts, other typical activating stimuli such as TNFα had little effect on luciferase expression. This again suggests that the role of microglial NFAT is highly specialized and only involved in regulating phenotype in response to select stimulations. Indeed, the fact that NFAT inhibition with tat-VIVIT peptide attenuated only LPS-dependent TNFα and MCP-1 secretion but not KC further illustrates the highly complex and unclear role this transcription factor has in coupling-specific stimulations to select proinflammatory expression changes. For example, the increase in IL-10 secretion from VIVIT-treated Jurkat cells was a puzzling finding given the prior data in T cells demonstrating that NFAT activity is required for increasing IL-10 secretion (Lee et al., 2009). Indeed, inhibition of NFAT with VIVIT did attenuate the CD3 stimulated increase in IL-10 secretion from the Jurkat cells (Fig. 2B). One possibility is that in the absence of a stimulatory signal, basal NFAT activity is responsible for generation of some protein that normally suppresses IL-10 production or secretion. Therefore absence of NFAT activity with VIVIT treatment releases this negative control to produce the increase in IL-10 secretion observed (Fig. 2B). The full potential and limitation of NFAT-dependent acquisition of reactive microglial phenotype changes is yet to be described. For example it is not clear whether the NFAT-dependent changes in TNFα and MCP-1 secretions were due to changes in NFAT-dependent transcription of these genes. The precise site of NFAT action affecting the increased secretions of these factors, whether it is at the transcriptional or posttranscriptional level, remains to be elucidated.
The finding that microglia express NFAT presents a possible approach to controlling microglia-mediated neuroinflammation. Although neurons and astrocytes express NFAT isoforms (Canellada et al., 2008; Sama et al., 2008; Abdul et al., 2009; Vashishta et al., 2009) there exists the possibility that isoform subtype selective inhibitors can be designed to preferentially attenuate microglial NFAT activity versus that in other brain cells or immune cells in general.
Footnotes
This work was supported by National Institutes of Health (NIH)–National Center for Research Resources Grant 1P20 RR17699 and NIH/National Institute on Aging Grant 1R01AG026330. This investigation was supported in part by a grant from the National Multiple Sclerosis Society. We thank Angie Floden, Gunjan Dhawan, and Keiko Murase for their technical assistance in microglial culture preparations. We also acknowledge Dr. Satya Yadav for expert help with peptide construction.
References
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu J, Yaffe MB, López-Rodríguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- Bao Y, Li R, Jiang J, Cai B, Gao J, Le K, Zhang F, Chen S, Liu P. Activation of peroxisome proliferator-activated receptor gamma inhibits endothelin-1-induced cardiac hypertrophy via the calcineurin/NFAT signaling pathway. Mol Cell Biochem. 2008;317:189–196. doi: 10.1007/s11010-008-9848-8. [DOI] [PubMed] [Google Scholar]
- Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, Bonni A. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem. 2005;280:2818–2825. doi: 10.1074/jbc.M408741200. [DOI] [PubMed] [Google Scholar]
- Best JA, Chen Y, Piech KM, Tank AW. The response of the tyrosine hydroxylase gene to cyclic AMP is mediated by two cyclic AMP-response elements. J Neurochem. 1995;65:1934–1943. doi: 10.1046/j.1471-4159.1995.65051934.x. [DOI] [PubMed] [Google Scholar]
- Boise LH, Petryniak B, Mao X, June CH, Wang CY, Lindsten T, Bravo R, Kovary K, Leiden JM, Thompson CB. The NFAT-1 DNA binding complex in activated T cells contains Fra-1 and JunB. Mol Cell Biol. 1993;13:1911–1919. doi: 10.1128/mcb.13.3.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brooks H, Lebleu B, Vivès E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev. 2005;57:559–577. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Canellada A, Ramirez BG, Minami T, Redondo JM, Cano E. Calcium/calcineurin signaling in primary cortical astrocyte cultures: Rcan1–4 and cyclooxygenase-2 as NFAT target genes. Glia. 2008;56:709–722. doi: 10.1002/glia.20647. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Stroh C, Schulze-Osthoff K. P2X7/P2Z Purinoreceptor-mediated activation of transcription factor NFAT in microglial cells. J Biol Chem. 1999;274:13205–13210. doi: 10.1074/jbc.274.19.13205. [DOI] [PubMed] [Google Scholar]
- Fisher WG, Yang PC, Medikonduri RK, Jafri MS. NFAT and NFkappaB activation in T lymphocytes: a model of differential activation of gene expression. Ann Biomed Eng. 2006;34:1712–1728. doi: 10.1007/s10439-006-9179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floden AM, Li S, Combs CK. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci. 2005;25:2566–2575. doi: 10.1523/JNEUROSCI.4998-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–670. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature. 1993;365:352–355. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- Kataoka A, Tozaki-Saitoh H, Koga Y, Tsuda M, Inoue K. Activation of P2X<sub>7</sub> receptors induces CCL3 production in microglial cells through transcription factor NFAT. J Neurochem. 2009;108:115–125. doi: 10.1111/j.1471-4159.2008.05744.x. [DOI] [PubMed] [Google Scholar]
- Kuriyama M, Matsushita M, Tateishi A, Moriwaki A, Tomizawa K, Ishino K, Sano S, Matsui H. A cell-permeable NFAT inhibitor peptide prevents pressure-overload cardiac hypertrophy. Chem Biol Drug Des. 2006;67:238–243. doi: 10.1111/j.1747-0285.2006.00360.x. [DOI] [PubMed] [Google Scholar]
- Lee CG, Kang KH, So JS, Kwon HK, Son JS, Song MK, Sahoo A, Yi HJ, Hwang KC, Matsuyama T, Yui K, Im SH. A distal cis-regulatory element, CNS-9, controls NFAT1 and IRF4-mediated IL-10 gene activation in T helper cells. Mol Immunol. 2009;46:613–621. doi: 10.1016/j.molimm.2008.07.037. [DOI] [PubMed] [Google Scholar]
- Lopez-Rodríguez C, Aramburu J, Rakeman AS, Rao A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc Natl Acad Sci U S A. 1999;96:7214–7219. doi: 10.1073/pnas.96.13.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma JI, Zirpel L. Deafferentation-induced activation of NFAT (nuclear factor of activated T-cells) in cochlear nucleus neurons during a developmental critical period: a role for NFATc4-dependent apoptosis in the CNS. J Neurosci. 2008;28:3159–3169. doi: 10.1523/JNEUROSCI.5227-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Masuda ES, Imamura R, Amasaki Y, Arai K, Arai N. Signalling into the T-cell nucleus: NFAT regulation. Cell Signal. 1998;10:599–611. doi: 10.1016/s0898-6568(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. Tonicity-responsive enhancer binding protein, a Rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci U S A. 1999;96:2538–2542. doi: 10.1073/pnas.96.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamoto-Combs K, Piech KM, Best JA, Sun B, Tank AW. Tyrosine hydroxylase gene promoter activity is regulated by both cyclic AMP-responsive element and AP1 sites following calcium influx. Evidence for cyclic amp-responsive element binding protein-independent regulation. J Biol Chem. 1997;272:6051–6058. doi: 10.1074/jbc.272.9.6051. [DOI] [PubMed] [Google Scholar]
- Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, Li ST, Kobayashi N, Matsumoto S, Tanaka K, Tanaka N, Matsui H. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat Med. 2004;10:305–309. doi: 10.1038/nm994. [DOI] [PubMed] [Google Scholar]
- Pérez-Ortiz JM, Serrano-Pérez MC, Pastor MD, Martín ED, Calvo S, Rincón M, Tranque P. Mechanical lesion activates newly identified NFATc1 in primary astrocytes: implication of ATP and purinergic receptors. Eur J Neurosci. 2008;27:2453–2465. doi: 10.1111/j.1460-9568.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- Putt ME, Hannenhalli S, Lu Y, Haines P, Chandrupatla HR, Morrisey EE, Margulies KB, Cappola TP. Evidence for coregulation of myocardial gene expression by MEF2 and NFAT in human heart failure. Circ Cardiovasc Genet. 2009;2:212–219. doi: 10.1161/CIRCGENETICS.108.816686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareneva T, Matikainen S, Vanhatalo J, Melén K, Pelkonen J, Julkunen I. Kinetics of cytokine and NFAT gene expression in human interleukin-2-dependent T lymphoblasts stimulated via T-cell receptor. Immunology. 1998;93:350–357. doi: 10.1046/j.1365-2567.1998.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science. 1988;241:202–205. [PubMed] [Google Scholar]
- Shaw KT, Ho AM, Raghavan A, Kim J, Jain J, Park J, Sharma S, Rao A, Hogan PG. Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor NFAT1 in stimulated immune cells. Proc Natl Acad Sci U S A. 1995;92:11205–11209. doi: 10.1073/pnas.92.24.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondag CM, Combs CK. Amyloid precursor protein mediates proinflammatory activation of monocytic lineage cells. J Biol Chem. 2004;279:14456–14463. doi: 10.1074/jbc.M313747200. [DOI] [PubMed] [Google Scholar]
- Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. 2009;6:1. doi: 10.1186/1742-2094-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashishta A, Habas A, Pruunsild P, Zheng JJ, Timmusk T, Hetman M. Nuclear factor of activated T-cells isoform c4 (NFATc4/NFAT3) as a mediator of antiapoptotic transcription in NMDA receptor-stimulated cortical neurons. J Neurosci. 2009;29:15331–15340. doi: 10.1523/JNEUROSCI.4873-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AS, Rezende AC, Grigoletto J, Rogério F, Velloso LA, Skaper SD, Negro A, Langone F. Ciliary neurotrophic factor infused intracerebroventricularly shows reduced catabolic effects when linked to the TAT protein transduction domain. J Neurochem. 2009;110:1557–1566. doi: 10.1111/j.1471-4159.2009.06259.x. [DOI] [PubMed] [Google Scholar]
- Vihma H, Pruunsild P, Timmusk T. Alternative splicing and expression of human and mouse NFAT genes. Genomics. 2008;92:279–291. doi: 10.1016/j.ygeno.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives E. Present and future of cell-penetrating peptide mediated delivery systems: “Is the Trojan horse too wild to go only to Troy?”. J Control Release. 2005;109:77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Wang Q, Gou X, Jin W, Xiong L, Hou L, Chen S, Zhang H, Zhu X, Xu L. TAT-mediated protein transduction of Nogo extracellular peptide 1–40 and its biological activity. Cell Mol Neurobiol. 2009;29:97–108. doi: 10.1007/s10571-008-9301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XY, Wang LH, Chen T, Hodge DR, Resau JH, DaSilva L, Farrar WL. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J Biol Chem. 2000;275:4541–4544. doi: 10.1074/jbc.275.7.4541. [DOI] [PubMed] [Google Scholar]