

Abstract
IgE production is inversely regulated by circulating and B cell surface levels of the low affinity IgE receptor, CD23. To begin to understand physiologic determinants of CD23 expression, we analyzed effects of BCR and TLR stimulation on CD23 levels. BCR and TLR 2, 3, 4, 6, and 9 agonists induced CD23 down-modulation from the cell surface. However, among the ligands only TLR4 agonists induced transcriptional activation of CD23 and generation of significant soluble CD23. These responses were induced by LPS both in vitro and in vivo, and were seen in both murine and human B cells. LPS also induced expression of matrix metalloprotease 9 (MMP9) and failed to induce CD23 cleaving activity in MMP9−/− cells, thus implicating MMP9 in the LPS-induced release of CD23 from the cell surface. Finally, type 1 transitional B cells uniquely produce MMP9 in response to LPS, suggesting a mechanism wherein endotoxin induces T1 cell expression of MMP9, which mediates cleavage of CD23 on distinct, mature B cells.
Activation of mast cells and basophils by IgE binding to its high affinity receptor, FcεRI, is a central feature of allergic inflammation, inducing de novo production and release of mediators such as histamine, leukotrienes, prostaglandins, and cytokines (1, 2). Interaction of IgE with its low affinity receptor (CD23 or FcεRII), which is expressed on B cells and found in soluble form, regulates IgE production. IgE production appears to be differentially regulated by membrane-bound (m)4 and soluble (s) forms of CD23. In humans, sCD23 enhances IgE production via an interaction with the membrane-bound B cell coreceptor CD21 (3). However, murine CD21 does not contain the inverse RGD motif and seven amino acid tail required for CD23 binding (2). In mice, reduction of membrane CD23 by treatment of animals with an anti-CD23 mAb (19G5), which renders membrane-bound CD23 highly susceptible to cleavage, results in a significant increase in total and DNP-KLH specific IgE relative to controls. However, Ag-specific IgE is equally increased in 19G5 treated wild-type and CD21-deficient mice (4), demonstrating that in mice the regulatory activity of CD23 is not mediated by CD21.
The function of human sCD23 in enhancing IgE production is consistent with observations that high serum levels of the protein are directly correlated with severity of allergy and asthma. In humans, sCD23 is strikingly increased in the blood of patients with allergic disorders (5). Allergic patients who undergo successful immunotherapy show a significant decrease in the amount of sCD23 in their blood (5-7). Preincubation of ex vivo human cells with Lumiliximab, an anti-CD23 mAb, before allergen stimulation, significantly decreases the production of cytokines associated with allergic responses (8). In preliminary clinical trials, patients who received Lumiliximab had a 40% decrease in serum IgE levels (9). The exact mechanism of anti-CD23-induced reductions in cytokine and IgE production remains unknown. In view of the demonstrated role of sCD23 in enhancing IgE-mediated allergic responses, understanding the mechanisms that regulate CD23 expression and release from the cell surface may provide important insight regarding improved therapy for patients with allergy and asthma.
Soluble CD23 is generated from the cleavage of mCD23 from B lymphocytes. Der p1, a major allergen of house dust mite Dermatophagoides pteronyssinus, has been shown to induce cleavage of mCD23 from human B lymphocytes resulting in accumulation of sCD23 in culture supernatants (10, 11). Patients with atopic dermatitis (12, 13) or allergies to grass pollen (6), birch pollen (14), parietaria pollen (15), ragweed pollen (16), or cow’s milk (17) exhibit increased sCD23 in peripheral blood suggesting that there must be alternative mechanisms for induction of sCD23 generation. Interestingly, it has been shown that the severity of asthma induced by exposure to the house dust mite is not solely a function of the concentration of the mite in dust, rather the amount of endotoxin in the dust (18). In addition, LPS, a component of endotoxin, significantly enhances IgE-mediated histamine release from basophils in patients allergic to grass pollen and dog dander (19).
As many naturally occurring allergens are found associated with endotoxin, we hypothesized that Ag and/or innate immune system signals may trigger synthesis, membrane expression, and/or cleavage of CD23 from the B cell surface, and this may explain accumulation of sCD23 in peripheral blood of allergic patients. We tested the ability of a panel of Ag receptor and TLR agonists to induce modulation of CD23 from the surface of ex vivo B cells. All of the stimuli we tested induced down modulation of surface CD23 on B cells. However, only the TLR4 agonist, LPS, induced increased accumulation of sCD23 in culture supernatants and peripheral blood. This effect was dependent on the TLR4 signaling adaptor molecule MyD88 and matrix metalloprotease (MMP) 9 expression. Interestingly, only type 1 transitional B cells produced MMP9 in response to LPS stimulation. Our results suggest that MyD88-dependent TLR4 agonists may enhance allergic responses by inducing the production of both CD23 and MMP9, resulting in increased cleavage of mCD23 and sCD23 accumulation. Taken together, these data provide a mechanism by which endotoxin containing allergens may promote IgE production.
Materials and Methods
Mice
All experiments with mice were performed in accordance with the regulations and with approval of The National Jewish Health (Denver, CO) and Institutional Animal Care and Use Committee. C57BL/6 mice were purchased from The Jackson Laboratory and the colony maintained in our facilities. MyD88-deficient C57BL/6 mice and littermate controls were provided by Phillipa Marrack (National Jewish Health). MMP9-deficient mice were purchased from The Jackson Laboratory. All mice were maintained in a pathogen-free facility of National Jewish Health.
Medium
All cells were cultured in complete medium: 500 ml of HyQ IMDM from HyClone, 10% heat inactivated FBS (BioSource), 1 mM sodium pyruvate (Life Technologies), 50 μg/ml gentamicin (Life Technologies), 100 U/ml Pen-Strep, 2 mM l-glutamine (Life Technologies), 5 × 10−5 M 2-ME.
Splenocyte culture
Splenectomy was performed on the mice indicated. Whole spleens were homogenized in 15 ml of complete medium in a dounce homogenizer and passed through a 70-μM filter (BD Falcon). The splenic cellular suspension was centrifuged at 1200 × g for 5 min. The supernatant was removed and the cellular pellet was resuspended in 1 ml of ice-cold ACK buffer (0.155 M ammonium chloride, 1 M disodium EDTA, 0.01 M potassium bicarbonate) for 1 min to deplete the mixture of RBC. The ACK buffer was diluted by the addition of 15 ml complete medium and centrifuged at 1200 × g for 5 min. The supernatant was removed and the cellular pellet was resuspended in 5 ml of complete medium. The cellular suspension was counted using a hemocytometer and suspended at 10 × 106/ml in complete medium. One × 106 splenocytes were cultured in 100 μl of complete medium in a 96-well plate for each experiment noted.
Processing and stimulation of human peripheral blood cells
With National Jewish Health institutional review board approval, human blood was collected from consenting subjects in sodium heparin tubes. Whole blood (100 μl) was incubated with the indicated concentrations of LPS for 24 h at 37°C. As indicated, the broad spectrum A Disintegrin And Metalloprotease (ADAM) and MMP inhibitor GM6001 was added 1 h before the addition of LPS at concentrations ranging from 25 to 125 μM. Culture plates were centrifuged at 800 × g to separate plasma from cells. Plasma was collected and prepared for quantitation of soluble CD23 as described below. Pelleted whole blood was treated with ice-cold ACK buffer for lyses of RBC and then stained for FACS analysis of CD23 expression.
Stimuli
Unless otherwise indicated, the following concentrations of each stimulus was used: 5 μg per 1 × 106 cells rabbit anti-IgM μ-chain specific Fab′ 2 (Zymed), 10 μg per 1 × 106 cells LPS from Escherichia coli 011:B4 (Sigma-Aldrich). Poly I:C, 10 μg per 1 × 106 cells; Peptidoglycan, 10 μg per 1 × 106 cells; and CpG DNA, 2 μg per 1 × 106 cells, were provided by Dr. R. Kedl, (University of Colorado, Denver, CO). IL-4 was purchased from BD Pharmingen.
Inhibitors
The following inhibitors were used at the concentrations indicated in the figure legends: actinomycin-D (Sigma-Aldrich), cyclohexamide (Sigma-Aldrich), and GM6001 (U.S. Biological).
Biotinylation of splenocytes
RBC-depleted whole splenocytes were resuspended at 20 × 106/ml in sterile PBS. Biotin at 100 mg/ml (EZ link Sulfo-NHS-LC Biotin from Pierce) in DMSO was added at 1/200 in an Eppendorf tube and rotated at room temperature for 30 min. Forty micromols of glycine (in sterile H2O) was added for 5 min, rotating at room temperature to stop the biotinylation reaction. Cells were washed with complete medium, resuspended at 10 × 106/ml, and plated 1 × 106 cells per sample in a 96-well plate. The cells were then treated with the indicated stimuli for 24 h and the amount of biotinylated sCD23 in the culture supernatants was measured by ELISA as described below.
Biotinylation of murine serum
Murine whole blood was clotted at room temperature for 10 min then centrifuged at 10,000 RPM for 10 min. The serum was removed and the total protein was measured by spectrophotometer. One microgram of total protein was taken from each sample and adjusted to 100 μl total volume in sterile PBS. Ten microliters of 1 M NaHCO3 was added to adjust pH, then 10 μl of EZ-link biotin (100 mg/ml in sterile H2O). Eppendorf tubes were rotated at room temperature for 30 min and the reaction was terminated by the addition of 40 mM Glycine (in sterile H2O) for 5 min at room temperature. The serum was then used directly for the ELISA.
Biotinylation of human plasma
Human plasma was isolated from whole blood as described above. Total protein was approximated by OD280 and 1 μg of total protein was taken from each sample and adjusted to 100 μl total volume in sterile PBS. 10 μl of 1 M NaHCO3 was added to adjust pH then 10 μl of EZ-link biotin (100 mg/ml in sterile H2O) was added. Plasma was rotated at room temperature for 30 min and the reaction was terminated by the addition of 40 mM Glycine (in sterile H2O) for 5 min at room temperature. The serum was then used directly for the ELISA.
Human-soluble CD23 ELISA
Fifty microliters of unconjugated mouse anti-human CD23 at 0.5 μg/ml (Abcam) in carbonate buffer (0.1 M NaHCO3, 0.1 M Na2CO3 (pH 8.8)) was plated in each well of a 96-well plate (Costar plate from Corning) and incubated at 4°C overnight. Plates were washed with washing solution (PBS plus 0.05% Tween 20) three times, and blocked with 100 μl of blocking buffer (0.5% BSA in carbonate buffer) for 4 h at 37°C. Plates were again washed with washing solution three times and 10 μl (1/10 dilution of plasma in sterile PBS) of biotinylated human plasma plus 90 μl of sterile PBS was added to each well. After overnight incubation at 4°C, plates were washed three times with washing solution and 100 μl of streptavadin HRP in PBS (1/5000 from Pierce 10 μg/ml) was added for 1 h at room temperature. The plate was washed three times, and developed with 100 μl of TMB solution (Zymed) for 5 min. The OD was measured at 650 nm in a microplate reader every 4 min for 40 min.
Soluble murine CD23 and CD21 ELISA
Fifty microliters of rat anti-mouse CD23 at 5 μg/ml (BD Pharmingen B3B4) or 50 μl of rat anti-mouse CD21 at 5 μg/ml (BD Pharmingen 7G6) in carbonate buffer (0.1 M NaHCO3, 0.1 M Na2CO3 (pH 8.8)) was adsorbed to each well of a 96-well plate (Costar plate from Corning; cat. No. 9018) by overnight incubation at 4°C. The plate was washed three times with 200 μl washing solution (PBS plus 0.05% Tween 20) and blocked with 100 μl of blocking buffer (0.5% BSA in carbonate buffer) for 4 h at 37°C. The plate was then washed three times with 200 μl washing solution and 20 μl of the culture supernatant from the biotinylated splenocytes plus 80 μl of sterile PBS was added to each well and incubated at 4°C overnight. The plate was washed three times with 200 μl washing solution and 100 μl of Streptavidin HRP in PBS (1/5000 from Pierce 10 μg/ml; cat. no. 21124) was added for 1 h at room temperature. The plate was washed three times and developed with 100 μl of TMB solution (Zymed; cat. no. 00–2023) for 5 min. The absorbance was measured at 650 nm in a microplate reader at 4 min intervals for 40 min.
Staining and flow cytometric analysis
Cells were stained for flow cytometry with PerCp anti-B220 (BD Pharmigen; RA3.3A1), FITC, or PE-conjugated anti-CD23 (BD Pharmingen, clone B3B4), and/or PE anti-CD21 (provided by R. Torres, National Jewish Health) by standard staining procedures. Human PBL were stained as follows: normal human IgG was added (4 mg/ml) to block Fc receptors before incubation with Abs against the pan leukocyte marker CD45 PerCP (BD Pharmingen), CD19 allophycocyanin (eBioscience), and PE anti-CD23 (BD Pharmingen) to identify CD23 expression on B cells. FACS analysis was performed using a FACScan flow cytometer (BD Biosciences) for murine studies and a FACSCalibur flow cytometer for human studies. Flow cytometric analysis was performed using the FlowJo analysis software (CellQuest).
Cell sorting using magnetic beads
Whole splenocytes from C57BL/6 mice were incubated with biotin conjugated rabbit anti-mouse CD93 followed by anti-biotin beads (Miltenyi Biotec) and sorted via magnetic separation using MACS LS columns (Miltenyi Biotec). Cells were sorted for CD23 expressions using PE or FITC conjugated rabbit anti-mouse CD23 followed by anti-PE or anti-FITC magnetic beads and sorted using MACS LS columns via magnetic separation. CD43 and B220 separations were performed using anti-CD43 and anti-B220 magnetic beads (Miltenyi Biotec). Sterile PBS was used as the buffer for all sorting experiments.
Quantitative real-time PCR (qPCR)
All qPCR experiments were conducted and analyzed as follows: Total RNA was isolated from the indicated cells using RNeasy mini kit (Qiagen). The total amount of isolated RNA was measured using a spectrophotometer. First-strand cDNA synthesis was performed using (500 ng total RNA from each sample) Superscript III First-Strand Synthesis kit for qRT-PCR (Invitrogen). Triplicate for each cDNA reaction was used for amplification with a predesigned mouse CD23 (Mm00442792_m1), MMP9 (Mm00442991_m1), ADAM10 (Mm00545742_m1), or β-actin (endogenous control) specific TaqMan primer/probe set (Applied Biosystems) in an ABI PRISM 5700 Sequence Detection System (Applied Biosystems). Relative quantification was determined after establishment of standard curves for CD23, MMP9, ADAM10, and β-actin expression. Quantitative PCR data represent means that were obtained from triplicates of the first-strand cDNA reaction ± SD.
Statistical analysis
Statistical analysis for all of the experiments was performed using the nonparametric Wilcoxon signed-rank test. Analysis of LPS-induced accumulation of sCD23 used the percent change of each treated sample compared with the average of the nontreated controls from five different experiments (each condition was done in duplicate) for a total of ten samples per condition. The data are represented as an average of the ten samples ± SD. The statistical analysis accounted for the false discovery rate by adjusting for multiple comparisons.
Statistical significance for the in vivo accumulation of sCD23 used the percent change of each LPS treated sample (done in duplicate) compared with the average of the PBS-immunized control from three different experiments for a total of six samples per condition using the mice indicated. The data are represented as the average fold change in sCD23 from the six samples ± SD.
The statistical analysis for all of the new surface CD23 experiments used the percent change of each treated sample compared with the average of the nontreated controls from three different experiments (each condition was done in duplicate) for a total of six samples per condition. The data are represented as an average percent change of the six samples ± SD.
Statistical analysis for the real time PCR data used the fold change of each treated sample (each condition done in triplicate) compared with the average of the housekeeper gene for each of two experiments for a total of six samples per condition. The data are represented as the average fold change in the gene indicated ± SD.
Results
Toll-like receptor and BCR agonists induce down modulation of CD23 from the B cell surface
To determine whether Ag or innate immune signals modify CD23 expression, immediately ex vivo splenocytes were stimulated with a panel of BCR and TLR agonists, and changes in B cell surface expression of CD23 was determined by flow cytometry. Peptidoglycan (TLR2,6), poly I:C (TLR3), LPS (TLR4), and CpG DNA (TLR9) stimulation resulted in CD23 down modulation from the B cell surface (Fig. 1a). Although productive TLR signaling can occur via both MyD88-dependent and independent pathways, induction of CD23 down modulation was found to be MyD88 dependent for all TLR stimuli except poly I:C, which was partially independent (Fig. 1a). Ag surrogates, including anti-μ, anti-δ, and anti-κ Abs, also induced CD23 down modulation (Fig. 1a and data not shown). Equivalent effects were seen following treatment with Abs against the B cell Ag receptor’s transducers Ig-α and Ig-β (data not shown). In control experiments, none of these stimuli affected CD21 expression by splenic B cells (data not shown).
FIGURE 1.
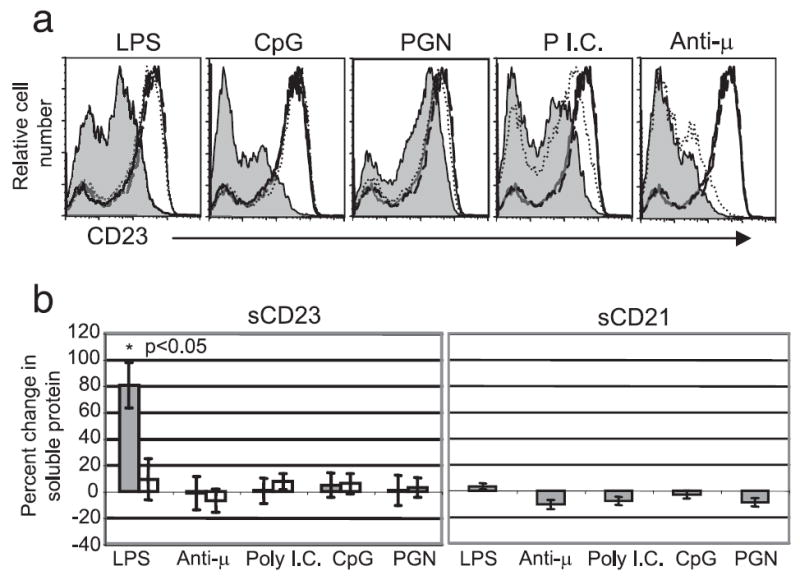
Agonists of TLRs 2, 3, 4, 6, and 9 and the Ag receptor induce CD23 down modulation from B cells, but only LPS treatment results in detectable accumulation of soluble CD23 in culture supernatants. a, Surface expression CD23 by splenic B cells from C57BL/6 and MyD88-deficient mice was determined by flow cytometry 24 h after treatment with the indicated stimuli; solid line-untreated C57BL/6, shaded line-treated C57BL/6, dashed line-untreated MyD88−/−, dotted line-treated MyD88−/−. b, Release of CD23 and CD21 from the cell surface was measured by ELISA of culture supernatants from C57BL/6 ( ) and MyD88−/− (□) splenic B cells 24 h after treatment with the indicated stimuli. Statistical significance was determined using the Wilcoxon signed-rank test. The p value was adjusted for multiple comparisons using the false discovery rate approach.
) and MyD88−/− (□) splenic B cells 24 h after treatment with the indicated stimuli. Statistical significance was determined using the Wilcoxon signed-rank test. The p value was adjusted for multiple comparisons using the false discovery rate approach.
Because sCD23 has been shown to enhance IgE production, we determined whether CD23 down modulation induced by TLR or BCR agonists is a result of CD23 cleavage and release intact into supernatants. To test this, splenocytes from C57BL/6 and MyD88-deficient mice were stimulated with TLR and BCR agonists, and the relative amount of sCD23 in culture supernatants was assayed by ELISA. Only the TLR4 agonist LPS induced the accumulation of sCD23 above that found in nontreated samples (Fig. 1b, left). Furthermore, LPS induced accumulation of sCD23 in culture supernatants was dependent on MyD88 expression. None of the stimuli tested increased the amount of sCD21 in culture supernatants (Fig. 1b, right).
Collectively, these data demonstrate that down modulation of mCD23 can be induced by TLR and BCR agonists. However, only TLR4-mediated signaling leads to significant accumulation of sCD23 in culture supernatants. It is unclear why TLR 4 signals are uniquely able to induce release of CD23 from the cell surface in a form detectable by ELISA. As shown in Fig. 4, this may result from the unique ability of TLR4 signals to mediate activation of MMP9 expression. The ability of TLR4 but not TLR9 to mediate CD23 release from the cell surface indicates that while MyD88 is required for the latter, its activation must not be sufficient to drive this response. Because soluble CD23 levels have been most strongly implicated in regulating IgE production in humans, we focused efforts in the remainder of this manuscript on understanding the basis of TLR4 mediated sCD23 generation.
FIGURE 4.
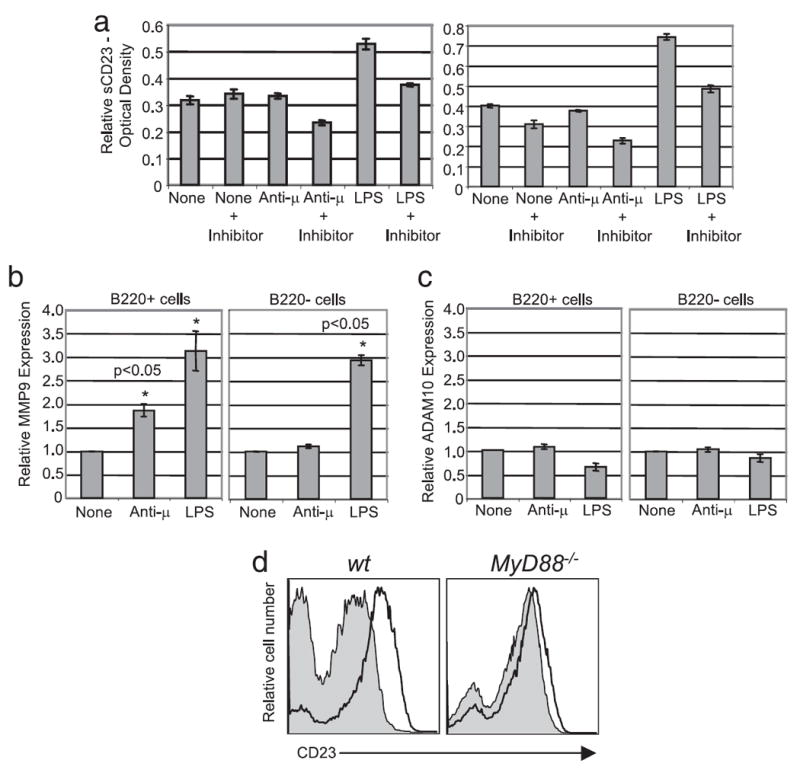
LPS induced modulation of CD23 from the B cell surface is dependent on MMP9 expression. a, Splenocytes from C57BL/6 mice were preincubated for 1 h in presence or absence of 12.5 μM actinomycin-D (left) or 10 μM GM6001 (right) then stimulated as indicated. Soluble CD23 in supernatants was measured by ELISA 24 h after treatment. Relative MMP9 (b) and ADAM10 (c) expression was quantified by real time PCR of treated B220+ B cells and B220− B cells. Statistical significance was determined using the Wilcoxon signed-rank test. p < 0.05. d, Release of CD23 cleaving factors was assayed by transfer of culture supernatants from wild-type (left histogram) or MMP9-deficient (right histogram) untreated (solid black line) or LPS treated (shaded) splenocytes to MyD88-deficient B cell cultures, followed by measurement of cell surface CD23 on these cells by flow cytometry.
LPS induces accumulation of sCD23 in the blood of mice
As shown in Fig. 1, in vitro treatment of whole splenocytes with LPS resulted in the down modulation of mCD23 and the accumulation of sCD23. We next assessed whether LPS also induces mCD23 down modulation in vivo and the accumulation of sCD23 in peripheral blood. C57BL/6 and MyD88-deficient mice were injected i.p. with 50 μg of LPS, and B cell surface expression of CD23 and sCD23 in the sera of mice were determined 30 h later. LPS-induced CD23 modulation from splenic B cells and the accumulation of sCD23 in the sera of mice were analyzed. LPS induced these responses and both were subsequently found to be dependent on MyD88 expression (Fig. 2, a and b). Neither levels of CD21 expression by B cells nor sCD21 generation were altered by LPS treatment (Fig. 2b and data not shown).
FIGURE 2.
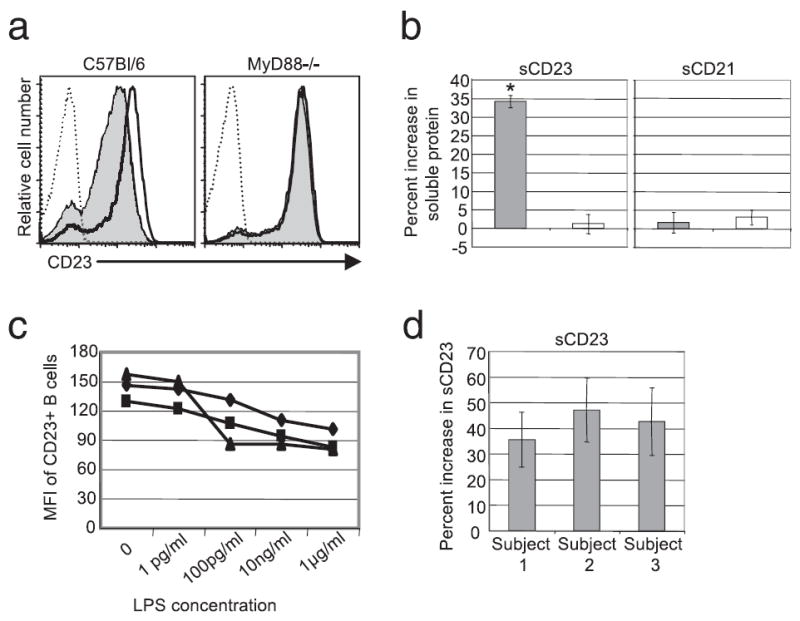
LPS induces CD23 down-modulation by human B cells and the accumulation of sCD23 in vivo. C57BL/6 and MyD88-deficient mice were injected i.p. with 50 μg of LPS. Surface expression of CD23 on splenic B cells (a) and the average percent increase in sCD23 accumulation in the blood of mice immunized with LPS compared with PBS controls; solid line-untreated C57BL/6, shaded line-treated C57BL/6, dashed line-untreated MyD88−/−, dotted line-treated MyD88−/− (b) was determined 30-h post immunization; C57BL/6 () and MyD88−/− (□). c, Human peripheral blood from three patients (◆, patient 1; ▲, patient 2; ■, patient 3) was incubated in the presence of the indicated concentrations of LPS for 24 h. CD23 surface expression on B cells (CD19+) was determined by flow cytometry. d, Supernatants from the cells cultured with 10 ng/ml LPS in c were analyzed by ELISA as described in the Materials and Methods. Shown is the average percent increase in sCD23 in LPS treated cultures relative to unstimulated controls. Statistical significance was determined using the Wilcoxon signed-rank test. *, p < 0.05.
LPS treatment induces the accumulation of sCD23 in human plasma
To determine whether LPS induces accumulation of sCD23 and mCD23 down modulation in humans, peripheral blood was incubated ex vivo with LPS for 24 h. B cells in LPS-treated peripheral blood down modulated mCD23 in a dose dependent manner (Fig. 2c). To assess whether this decrease was a result of mCD23 cleavage, supernatant CD23 levels were assessed by ELISA as described in the Materials and Methods. As shown in Fig. 2d, LPS treatment for 24 h induced sCD23 release from B cells.
LPS, but not other TLR or BCR agonists, induces transcriptional activation of CD23
Both LPS and anti-μ treatment caused down modulation of mCD23, however anti-μ treatment did not induce accumulation of sCD23 above that seen in the untreated sample. To help delineate the mechanisms involved in LPS-induced CD23 down modulation, we performed a kinetic analysis of CD23 expression over a 24-h time period using splenic B cells treated with LPS or anti-μ. Anti-μ treatment, which was shown to operate in a MyD88-independent manner, was used as a control to further understand the mechanism by which LPS induces CD23 modulation.
We found that treatment with LPS induced an initial increase in both the percent of CD23+ B cells and CD23 surface expression on B cells. This increase peaked at 6 h post LPS treatment and was followed by a dramatic decrease in percent CD23+ B cells and mean CD23 surface expression by positive cells (Fig. 3a). This response was not observed in B cells stimulated with anti-μ. These results suggest that LPS treatment induces de novo production of CD23, while BCR signals do not. Other TLR signals also did not induce this early burst in CD23 expression (data not shown).
FIGURE 3.
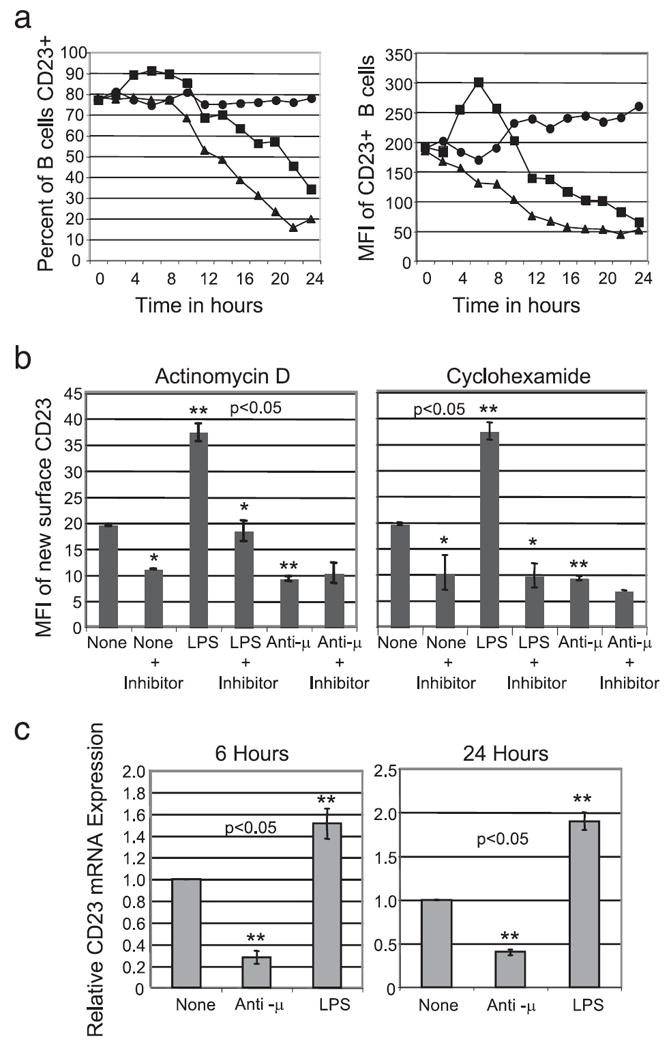
LPS induces transcriptional activation of CD23 expression. a, C57BL/6 splenic B cell surface expression of CD23 was determined by flow cytometry following stimulation in vitro for the times indicated; untreated (●), LPS (■), or anti- μ (▲). b, New surface CD23 was determined by flow cytometry 6 h after in vitro treatment in the presence and absence of 12.5 μM actinomycin-D (left) or 100 ng/ml cyclohexamide (right). c, Relative cellular CD23 mRNA expression was quantified by real time PCR 6 and 24 h following treatment of the indicated stimuli. Statistical significance was determined using the Wilcoxon signed-rank test. (*, statistically significant change in CD23 expression compared with nontreated or treated cells without the inhibitor; **, statistically significant change in CD23 expression compared with the untreated control. p < 0.05).
To determine whether LPS induces de novo production of CD23, whole splenocytes were first incubated with unconjugated anti-CD23 to block existing mCD23, then washed and treated with the stimuli for 6 h. De novo synthesized mCD23 was then measured by staining the cells with PE-labeled anti-CD23. In preliminary experiments, we found that blocking the original surface mCD23 with unconjugated anti-CD23 allowed subsequent detection of only newly expressed CD23 by staining with fluorochrome conjugated anti-CD23. Fig. 3b shows the geometric mean fluorescent intensity of the PE-labeled new CD23 by cells that had been blocked with unconjugated Abs before culture. LPS treatment increased new CD23 surface expression 2-fold while anti-μ treatment decreased CD23 surface expression two-fold relative to spontaneous new expression. De novo LPS-induced production of CD23 was dependent on both transcription and translation, as this effect was blocked by preincubation with either actinomycin-D or cyclohexamide (Fig. 3b). Both inhibitors substantially blocked spontaneous new expression of CD23 as well as the anti-μ effect.
Because LPS treatment induced CD23 expression and anti-μ treatment resulted in decreased CD23 expression, we hypothesized that LPS treatment may increase CD23 message while anti-μ treatment may decrease CD23 message levels. To test this hypothesis, quantitative PCR for CD23 was performed at 6 and 24 h post treatment using cDNA prepared from splenic B cells. LPS treatment caused a 2-fold increase in CD23 message 6 and 24 h post treatment while anti-μ treatment decreased CD23 message 2-fold 6 and 24 h post treatment (Fig. 3c). These findings suggest that the anti-μ effect can be ascribed to protein decay in combination with induced failure to replace CD23 on the cell surface.
TLR-mediated, MyD88-dependent CD23 modulation requires transcriptional activation of MMP9
LPS induction of CD23 synthesis, associated with decreased surface expression and increased accumulation in culture supernatants suggested that LPS must also induce, by a MyD88-dependent mechanism, the expression of a factor that cleaves mCD23. To test this hypothesis, splenocytes were preincubated for 1 h with the transcription inhibitor actinomycin-D before treatment with LPS or anti-μ, and assay of release of pre-existing mCD23. Inhibition of transcription prevented the LPS induced sCD23 generation (Fig. 4a, left). Interestingly, preincubation of splenocytes with actinomycin-D also decreased the amount of sCD23 generated following anti-μ treatment of splenocytes suggesting that both TLR4 and Ag receptor signals induce a cleavage mediator.
Transcription is required for LPS induced accumulation of sCD23 in culture supernatants, so we sought to determine whether an ADAM or MMP is involved in cleavage of mCD23 to sCD23. It has been shown that LPS treatment increases MMP9 expression in culture supernatants from mature splenic B cells (19, 20). Additionally, ADAM10 has been shown to be the principle constitutive CD23 “sheddase” (21, 22). To test the role of these enzymes, splenocytes were preincubated with GM6001, a potent broad spectrum inhibitor of ADAMs and MMPs. GM6001 inhibited the accumulation of sCD23 induced by LPS, suggesting that an ADAM or MMP is involved in CD23 cleavage (Fig. 4a, right).
Quantitative PCR for MMP9 and ADAM10 using cDNA prepared from treated or untreated B220+ splenic B cells and B220− non-B cells was used to establish whether these enzymes are upregulated by LPS and thus are candidate mediators of LPS-induced accumulation of sCD23. LPS induced a 3-fold increase in MMP9 message in both B cells and in non-B cells compared with untreated cells (Fig. 4b). Anti-μ treatment induced a 2-fold increase in MMP9 message in B cells but not alter MMP9 message in B220 negative cells. None of the stimuli affected ADAM10 mRNA expression (Fig. 4c).
To formally establish whether MMP9 is required for LPS induced CD23 modulation, wild type and MMP9 deficient splenocytes were incubated in the presence of LPS for 24 h. Culture supernatants were transferred to MyD88-deficient splenocytes, which do not down modulate CD23 in response to LPS, and the requirement of MMP9 was determined based on down modulation of mCD23. As shown in Fig. 4d, the MyD88-deficient B cells only slightly down modulate mCD23 when incubated in the presence of culture supernatants generated by LPS stimulation of MMP9−/− B cells, suggesting that MMP9 is the primary protease involved in LPS-induced mCD23 down modulation.
Interestingly, as might have been expected, LPS failed to induce sCD23 production from MMP9−/− B cells. However, supernatants derived from cultures of normal splenocytes that had been stimulated with LPS and therefore contained cleavage activity, also failed to mediate cleavage of CD23 from MMP9−/− B cells. The MMP9−/− mice are on the 129 background in which the CD23 gene contains multiple polymorphisms. These may make CD23 insensitive to MMP9-mediated cleavage. Finally, consistent with both their lack of MMP9 and insensitivity of their CD23 to cleavage, B cells from these mice express ~2-fold more CD23 than those from C57BL/6 mice (data not shown).
LPS induces MMP9 expression by transitional B cells and non-B cells selectively
The splenic B cell compartment is composed primarily of transitional 1 and 2 (T1, T2) subsets as well as follicular (FO) and marginal zone (MZ) B cells. As shown in supplemental Fig. 1,5 all of these populations respond to LPS by increasing mCD23 expression within 6 h of stimulation. To determine which of these B cell subpopulations produce MMP9, CD23+ (T2 and FO B cells), and CD23− (T1, MZ and all other cells) splenocytes were isolated using magnetic bead separation. The isolated CD23+ and CD23− splenic populations were cultured for 24 h in the presence of LPS after which mCD23 expression was determined by flow cytometry. It should be noted that some sorted CD23 negative T1/MZ cells spontaneously increase surface expression of CD23 during culture, possibly reflecting maturation. Fig. 5a, left, shows that T1/MZ B cells down modulate mCD23 in response to LPS treatment compared with the untreated control, which up-regulate CD23. Surprisingly, T2/T3/FO B cells treated with LPS increased mCD23 expression compared with the untreated control (Fig. 5a, middle). Down modulation of CD23 by T2/T3/FO was recovered when they were cocultured with T1/MZ cells (Fig. 5a, right). These data indicate that the primary B cell source of MMP9 is T1 and/or MZ B cells.
FIGURE 5.
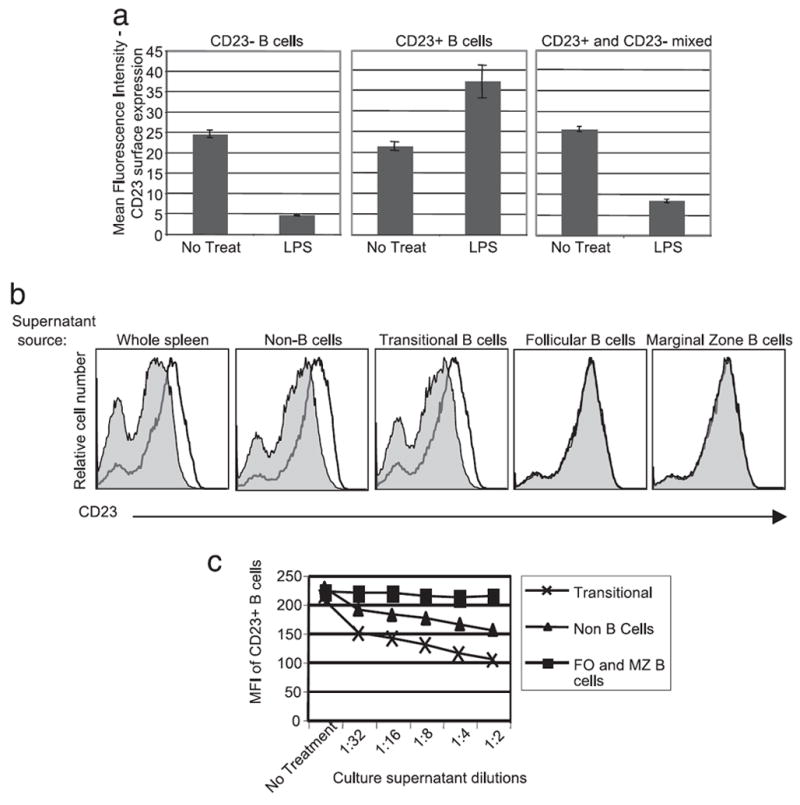
Transitional 1 and non-B cells produce MMP9 in response to LPS treatment. a, CD23 surface expression was determined by flow cytometry 24 h after LPS treatment of the indicated B cell populations. b, Culture supernatants from LPS treated (filled line) or nontreated (solid line) C57BL/6 isolated splenic populations (shown on top of the histograms) were assayed for CD23 cleaving activity as in Fig. 4. Shown are histograms of supernatant treated MyD88−/− B cells. c, LPS-treated culture supernatants from the indicated isolated populations were assayed as in Fig. 4 and data presented as mean CD23 expression by supernatant treated MyD88−/− B cells.
To establish which CD23− splenic B cells produce MMP9 in response to LPS treatment, we isolated CD93+ (transitional B cells) and CD93− cells (FO and MZ B cells, and all other cells) from C57BL/6 splenocytes using positive selection by standard magnetic bead separation techniques. The CD93− splenocytes were further fractionated based on CD23 expression by FITC conjugated anti-CD23 followed by anti-FITC magnetic beads. The isolated CD93−, CD23+ population represents the follicular B cell population. The CD93−, CD23− population was further depleted of CD43+ cells resulting in the isolation of CD93−, CD23−, CD43− marginal zone B cells and CD93−, CD23−, CD43+ non-B cells. The isolated populations were cultured in the presence of LPS for 24 h. Culture supernatants were transferred to cultures of MyD88-deficient splenocytes and, after 24 h incubation, B cell mCD23 expression was determined by flow cytometry. As shown in Fig. 5b, among B cell subpopulations only T1 transitional B cells produced active MMP9 capable of mediating CD23 cleavage.
It is important to note that the non-B cell splenocyte fraction also produced CD23 cleaving activity. To assess the relative amount and thus the relative biologic significance of MMP9 production by non-B cells and transitional 1 B cells, each population was isolated as described previously, cultured at equivalent cell concentration with LPS for 24 h and the culture supernatants were titrated onto MyD88-deficient splenocytes. As shown in Fig. 5c, culture supernatants from transitional B cells contained ~16-fold more activity than the non-B cell fraction indicating that in a per cell basis, T1 B cells are most likely the primary source of LPS-induced MMP9 in the spleen.
IL-4 is well known to induce up-regulation of CD23 on B cells and thus may modify the effects of BCR and TLR4 ligands on CD23 expression. To test this possibility we examined the effects of IL-4 costimulation of responses to these ligands. It is noteworthy that the IL-4 induced CD23 up-regulation seen was modest because unfractionated splenocytes are used. Non-B cells in these cultures, possibly by release of cytokines, induce an increase in CD23 background levels. This notwithstanding, as shown in Fig. 6, both a BCR stimulus and LPS induced efficient down-modulation of CD23 in cultures to which IL-4 was added. Thus IL-4 produced in vivo is likely to increase levels of sCD23 generated upon LPS exposure, thereby potentially enhancing IgE production.
FIGURE 6.
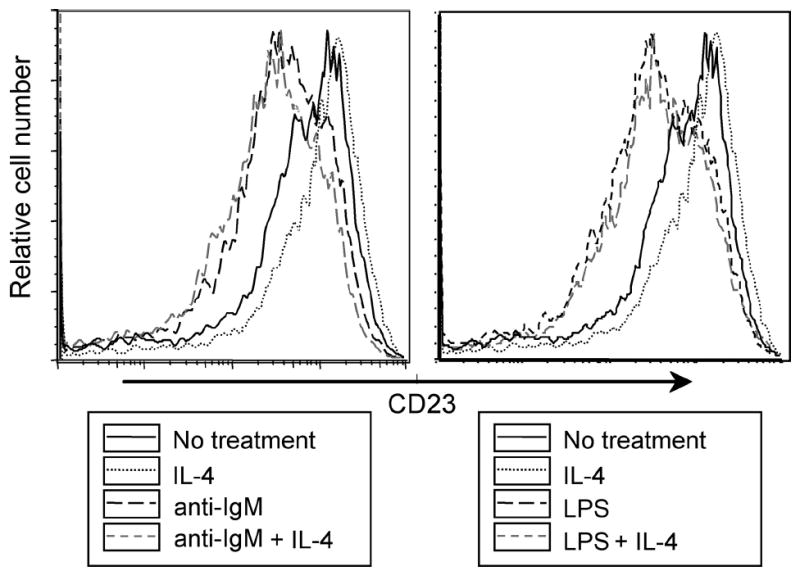
IL-4 stimulation does not affect CD23 modulation in response to TLR4 and BCR ligands. Splenocytes from C57BL/6 mice were cultured for 24 h with or without IL-4 (20 ng/ml) plus F(ab′)2 anti-IgM (1 μg/ml) or LPS(1 μg/ml). CD23 expression by B220+ cells was assayed by flow cytometry.
Discussion
Findings described in this report indicate that MyD88-dependent TLR4 signals dynamically regulate levels of soluble and B cell-associated CD23 in vitro and in vivo. LPS treatment of ex vivo splenocytes induces transcriptional activation of CD23 in all major B cell subsets, and MMP9 expression in transitional B cells as well as a poorly defined non-B cell population. Secreted MMP9 cleaves surface CD23 primarily from a distinct population of B cells, i.e., follicular B cells, resulting in the accumulation of sCD23 in culture supernatants and peripheral blood. Given the role of CD23 in IgE production, these findings suggest a potential mechanism by which LPS enhances IgE synthesis.
Concomitant exposure to LPS and Ag has been shown to enhance IgE responses and allergic disease in murine models. LPS supplementation of ova challenge significantly increases serum ova specific IgE, as well as IL-4 and IL-5 levels in mice. IgG1, IgG2a, IL-13, and IFN-γ serum levels are not increased by addition of LPS to ova challenge (23). LPS has also been shown to enhance latex allergen (24) and cat allergen (25) induced IgE and IgG1. Our findings suggest that LPS may exercise this effect by inducing synthesis of CD23 and MMP9, which enhance IgE production by elevating sCD23 and reducing mCD23.
Although LPS enhances IgE production in adult immune systems, it has been well documented that early exposure to endotoxin reduces the likelihood of developing allergic disease as an adult (26, 27). This paradox might be explained by in depth examination of the theory behind the hygiene hypothesis.
The hygiene hypothesis states that exposure to microbial products during development of the immune system shifts the adult immune repertoire toward a nonallergic phenotype (28). This skewing is manifest by the biased production of Th1 type cytokines IFN-γ and IL-12. Endotoxin or LPS exposure during development of the immune system is believed to inhibit allergic and asthmatic responses in adults and promotes Th1 type cytokine production. Paradoxically, LPS exposure in adult humans and mice induces the production of IgE (29). The mechanism of LPS-induced enhancement of IgE production remains unknown. Our findings suggest that exposure to LPS promotes IgE production indirectly by inducing secretion of MMP9, and consequent shedding of mCD23 from B cells. As previously hypothesized, LPS-induced enhancement of IgE production may be due to either the loss of inhibitory mCD23 or increased systemic sCD23, or both.
LPS promotion of IgE in the mature immune system appears inconsistent with the hygiene hypothesis and a role for early LPS exposure in prevention of IgE-mediated allergic disorders. This inconsistency may be explained by a difference in the response of immature vs mature B cell compartments to LPS. Culture of bone marrow derived B220+ IgM- immature B cells in the presence of LPS or Lipid A (a common component of endotoxin) resulted in a significant increase in CD23 surface expression 72 h post treatment (30). Thus, the alternate effects of endotoxin exposure on IgE production in mature and immature immune compartments could result in part from differences in frequency in those compartments of cells capable of producing MMP9 in response LPS. This hypothesis is consistent with findings of Melamed et al. (20, 31), who showed that B cells from bone marrow cultures of 3–83TgCD19−/− mice did not produce MMP9 while mature B populations from spleens of adult mice secrete MMP9 24 h post LPS treatment.
The relevance of this work to human disease has been demonstrated by several studies that link elevated MMP9 protein to the severity of allergic and asthmatic disorders. MMP9 protein is elevated in the sera of patients with atopic dermatitis (32) and in bronchial fluid of children and adults with asthma (33, 34). Children with stable asthma have less MMP9 protein in bronchial fluid compared with children who are having asthma exacerbations (35). Finally, it was recently reported that chronic asthma in children is associated elevation of both LPS and MMP9 in bronchoalveolar lavage (36).
Non-B lineage immune cells also reportedly produce MMP9 following LPS exposure (37, 38). This is consistent with findings described in this study that non-B cells from the spleen produce MMP9 in response to LPS. However, splenic transitional 1 B cells populations produce considerably more MMP9 than splenic non-B cell populations (Fig. 5c). Transitional B cells, as the name implies, are classically believed to simply represent an intermediary stage in the development of immature B cells to follicular and marginal zone B cells, with no unique function. Recently, it has been reported that transitional 2 B cells could suppress autoimmune symptoms when transferred into collagen-immunized mice (39). Our findings suggest that transitional 1 B cells may also have effector functions because these cells are uniquely competent to produce MMP9 in response to LPS treatment.
In conclusion, the findings reported in this study provide a potential resolution to the LPS paradox. In the context of the mature immune system LPS exposure promotes Th1 type cytokine production while simultaneously increasing IgE synthesis by induction of CD23 transcription concomitant with active MMP9 secretion. The in vitro and in vivo data show that the net result of LPS exposure is decreased mCD23 on splenic B cells and the accumulation of sCD23. Because the lack of inhibitory mCD23 and an increase in sCD23 promote IgE synthesis, these results may provide a mechanism by which systemic LPS exposure enhances IgE synthesis.
Acknowledgments
All of the statistical analysis was performed by Dr. Lening Zhang from the biostatistical department at National Jewish Medical and Research Center. We acknowledge the assistance of Sandra Duran in the preparation of this manuscript. We thank Dr. Ross Kedl for the gift of TLR ligands. JCC is an Ida and Cecil Green Professor of Immunology.
Footnotes
This research was supported by grants from the U.S. National Institutes of Health. L.J.J. was supported in part by the Cancer Research Institute predoctoral emphasis pathway in tumor immunology fellowship. C.C. was supported by a fellowship from National Jewish Health.
Abbreviations used in this paper: m, membrane bound; s, soluble; MMP, matrix metalloprotease; ADAM, A Disintegrin And Metalloprotease; qPCR, quantitative real-time PCR; FO, follicular; MZ, marginal zone.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Gould HJ, Sutton BJ, Beavil AJ, Beavil RL, McCloskey N, Coker HA, Fear D, Smurthwaite L. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21:579–628. doi: 10.1146/annurev.immunol.21.120601.141103. [DOI] [PubMed] [Google Scholar]
- 2.Hibbert RG, Teriete P, Grundy GJ, Beavil RL, Reljic R, Holers VM, Hannan JP, Sutton BJ, Gould HJ, McDonnell JM. The structure of human CD23 and its interactions with IgE and CD21. J Exp Med. 2005;202:751–760. doi: 10.1084/jem.20050811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fremeaux-Bacchi V, Fischer E, Lecoanet-Henchoz S, Mani JC, Bonnefoy JY, Kazatchkine MD. Soluble CD21 (sCD21) forms biologically active complexes with CD23: sCD21 is present in normal plasma as a complex with trimeric CD23 and inhibits soluble CD23-induced IgE synthesis by B cells. Int Immunol. 1998;10:1459–1466. doi: 10.1093/intimm/10.10.1459. [DOI] [PubMed] [Google Scholar]
- 4.Ford JW, Kilmon MA, Haas KM, Shelburne AE, Chan-Li Y, Conrad DH. In vivo murine CD23 destabilization enhances CD23 shedding and IgE synthesis. Cell Immunol. 2006;243:107–117. doi: 10.1016/j.cellimm.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Gagro A, Rabatic S. Allergen-induced CD23 on CD4+ T lymphocytes and CD21 on B lymphocytes in patients with allergic asthma: evidence and regulation. Eur J Immunol. 1994;24:1109–1114. doi: 10.1002/eji.1830240515. [DOI] [PubMed] [Google Scholar]
- 6.Jung CM, Prinz JC, Rieber EP, Ring J. A reduction in allergen-induced FcεR2/CD23 expression on peripheral B cells correlates with successful hyposensitization in grass pollinosis. J Allergy Clin Immunol. 1995;95:77–87. doi: 10.1016/s0091-6749(95)70155-9. [DOI] [PubMed] [Google Scholar]
- 7.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poole JA, Meng J, Reff M, Spellman MC, Rosenwasser LJ. Anti-CD23 monoclonal antibody, lumiliximab, inhibited allergen-induced responses in antigen-presenting cells and T cells from atopic subjects. J Allergy Clin Immunol. 2005;116:780–788. doi: 10.1016/j.jaci.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Rosenwasser LJ, Busse WW, Lizambri RG, Olejnik TA, Totoritis MC. Allergic asthma and an anti-CD23 mAb (IDEC-152): results of a phase I, single-dose, dose-escalating clinical trial. J Allergy Clin Immunol. 2003;112:563–570. doi: 10.1016/s0091-6749(03)01861-x. [DOI] [PubMed] [Google Scholar]
- 10.Hewitt CR, Brown AP, Hart BJ, Pritchard DI. A major house dust mite allergen disrupts the immunoglobulin E network by selectively cleaving CD23: innate protection by antiproteases. J Exp Med. 1995;182:1537–1544. doi: 10.1084/jem.182.5.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulz O, Laing P, Sewell HF, Shakib F. Der p I, a major allergen of the house dust mite, proteolytically cleaves the low-affinity receptor for human IgE (CD23) Eur J Immunol. 1995;25:3191–3194. doi: 10.1002/eji.1830251131. [DOI] [PubMed] [Google Scholar]
- 12.Takigawa M, Tamamori T, Horiguchi D, Sakamoto T, Yamada M, Yoshioka A, Toda K, Imamura S, Yodoi J. Fcε receptor II/CD23-positive lymphocytes in atopic dermatitis. I. The proportion of FcεRII+ lymphocytes correlates with the extent of skin lesion. Clin Exp Immunol. 1991;84:275–282. [PMC free article] [PubMed] [Google Scholar]
- 13.Wehrmann W, Reinhold U, Pawelec G, Wernet P, Kreysel HW. In vitro generation of IFN-γ in relationship to in vivo concentration of IgE and IgG subclasses and FcεRl/CD23 positive circulating lymphocytes in patients with severe atopic dermatitis (AD) Acta Derm Venereol Suppl. 1989;144:127–130. doi: 10.2340/00015555144127130. [DOI] [PubMed] [Google Scholar]
- 14.Davidsson A, Karlsson MG, Hellquist HB. Allergen-induced changes of B-cell phenotypes in patients with allergic rhinitis. Rhinology. 1994;32:184–190. [PubMed] [Google Scholar]
- 15.Di Lorenzo G, Drago A, Pellitteri ME, Candore G, Colombo A, Potestio M, Di Salvo A, Mansueto S, Caruso C. Serum levels of soluble CD23 in patients with asthma or rhinitis monosensitive to Parietaria: its relation to total serum IgE levels and eosinophil cationic protein during and out of the pollen season. Allergy Asthma Proc. 1999;20:119–125. doi: 10.2500/108854199778612590. [DOI] [PubMed] [Google Scholar]
- 16.Batterman J, Mazza DS, Meriney DK, Cleveland WL, Reddy MM, Grieco MH. In vitro release of soluble CD23 by human lymphocytes in ragweed-sensitive versus nonatopic patients following stimulation with ragweed antigen E. Ann Allergy Asthma Immunol. 1996;76:359–362. doi: 10.1016/S1081-1206(10)60038-5. [DOI] [PubMed] [Google Scholar]
- 17.Jarvinen KM, Aro A, Juntunen-Backman K, Suomalainen H. Large number of CD19+/CD23+ B cells and small number of CD8+ T cells as early markers for cow’s milk allergy (CMA) Pediatr Allergy Immunol. 1998;9:139–142. doi: 10.1111/j.1399-3038.1998.tb00360.x. [DOI] [PubMed] [Google Scholar]
- 18.Michel O, Kips J, Duchateau J, Vertongen F, Robert L, Collet H, Pauwels R, Sergysels R. Severity of asthma is related to endotoxin in house dust. Am J Respir Crit Care Med. 1996;154:1641–1646. doi: 10.1164/ajrccm.154.6.8970348. [DOI] [PubMed] [Google Scholar]
- 19.Norn S, Baek L, Jensen C, Skov PS, Permin H, Jarlov JO, Koch C. Influence of bacterial endotoxins on basophil histamine release: potentiation of antigen- and bacteria-induced histamine release. Allergy. 1986;41:125–130. doi: 10.1111/j.1398-9995.1986.tb00288.x. [DOI] [PubMed] [Google Scholar]
- 20.Melamed D, Messika O, Glass-Marmor L, Miller A. Modulation of matrix metalloproteinase-9 (MMP-9) secretion in B lymphopoiesis. Int Immunol. 2006;18:1355–1362. doi: 10.1093/intimm/dxl068. [DOI] [PubMed] [Google Scholar]
- 21.Lemieux GA, Blumenkron F, Yeung N, Zhou P, Williams J, Grammer AC, Petrovich R, Lipsky PE, Moss ML, Werb Z. The low affinity IgE receptor (CD23) is cleaved by the metalloproteinase ADAM10. J Biol Chem. 2007;282:14836–14844. doi: 10.1074/jbc.M608414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weskamp G, Ford JW, Sturgill J, Martin S, Docherty AJ, Swendeman S, Broadway N, Hartmann D, Saftig P, Umland S, et al. ADAM10 is a principal “sheddase” of the low-affinity immunoglobulin E receptor CD23. Nat Immunol. 2006;7:1293–1298. doi: 10.1038/ni1399. [DOI] [PubMed] [Google Scholar]
- 23.Murakami D, Yamada H, Yajima T, Masuda A, Komune S, Yoshikai Y. Lipopolysaccharide inhalation exacerbates allergic airway inflammation by activating mast cells and promoting Th2 responses. Clin Exp Allergy. 2007;37:339–347. doi: 10.1111/j.1365-2222.2006.02633.x. [DOI] [PubMed] [Google Scholar]
- 24.Slater JE, Paupore EJ, Elwell MR, Truscott W. Lipopolysaccharide augments IgG and IgE responses of mice to the latex allergen Hev b 5. J Allergy Clin Immunol. 1998;102:977–983. doi: 10.1016/s0091-6749(98)70336-7. [DOI] [PubMed] [Google Scholar]
- 25.Ormstad H, Groeng EC, Duffort O, Lovik M. The effect of endotoxin on the production of IgE, IgG1 and IgG2a antibodies against the cat allergen Fel d 1 in mice. Toxicology. 2003;188:309–318. doi: 10.1016/s0300-483x(03)00078-7. [DOI] [PubMed] [Google Scholar]
- 26.Prescott SL, Macaubas C, Smallacombe T, Holt BJ, Sly PD, Loh R, Holt PG. Reciprocal age-related patterns of allergen-specific T-cell immunity in normal vs. atopic infants. Clin Exp Allergy 28 Suppl. 1998;5:39–44. doi: 10.1046/j.1365-2222.1998.028s5039.x. discussion 50–31. [DOI] [PubMed] [Google Scholar]
- 27.Yabuhara A, Macaubas C, Prescott SL, Venaille TJ, Holt BJ, Habre W, Sly PD, Holt PG. TH2-polarized immunological memory to inhalant allergens in atopics is established during infancy and early childhood. Clin Exp Allergy. 1997;27:1261–1269. [PubMed] [Google Scholar]
- 28.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lapa e Silva JR, Possebon da Silva MD, Lefort J, Vargaftig BB. Endotoxins, asthma, and allergic immune responses. Toxicology. 2000;152:31–35. doi: 10.1016/s0300-483x(00)00289-4. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi EA, Akira S, Nobrega A. Role of TLR in B cell development: signaling through TLR4 promotes B cell maturation and is inhibited by TLR2. J Immunol. 2005;174:6639–6647. doi: 10.4049/jimmunol.174.11.6639. [DOI] [PubMed] [Google Scholar]
- 31.Melamed D, Benschop RJ, Cambier JC, Nemazee D. Developmental regulation of B lymphocyte immune tolerance compartmentalizes clonal selection from receptor selection. Cell. 1998;92:173–182. doi: 10.1016/s0092-8674(00)80912-5. [DOI] [PubMed] [Google Scholar]
- 32.Devillers AC, van Toorenenbergen AW, Klein Heerenbrink GJ, Muldert PG, Oranje AP. Elevated levels of plasma matrix metalloproteinase-9 in patients with atopic dermatitis: a pilot study. Clin Exp Dermatol. 2007;32:311–313. doi: 10.1111/j.1365-2230.2007.02378.x. [DOI] [PubMed] [Google Scholar]
- 33.Ko FW, Diba C, Roth M, McKay K, Johnson PR, Salome C, King GG. A comparison of airway and serum matrix metalloproteinase-9 activity among normal subjects, asthmatic patients, and patients with asthmatic mucus hypersecretion. Chest. 2005;127:1919–1927. doi: 10.1378/chest.127.6.1919. [DOI] [PubMed] [Google Scholar]
- 34.Tang LF, Du LZ, Chen ZM, Zou CC. Levels of matrix metalloproteinase-9 and its inhibitor in bronchoalveolar lavage cells of asthmatic children. Fetal Pediatr Pathol. 2006;25:1–7. doi: 10.1080/15227950600701396. [DOI] [PubMed] [Google Scholar]
- 35.Doherty GM, Kamath SV, de Courcey F, Christie SN, Chisakuta A, Lyons JD, Heaney LG, Ennis M, Shields MD. Children with stable asthma have reduced airway matrix metalloproteinase-9 and matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio. Clin Exp Allergy. 2005;35:1168–1174. doi: 10.1111/j.1365-2222.2005.02326.x. [DOI] [PubMed] [Google Scholar]
- 36.Hauk PJ, Krawiec M, Murphy J, Boguniewicz J, Schiltz A, Goleva E, Liu AH, Leung DY. Neutrophilic airway inflammation and association with bacterial lipopolysaccharide in children with asthma and wheezing. Pediatr Pulmonol. 2008;43:916–923. doi: 10.1002/ppul.20880. [DOI] [PubMed] [Google Scholar]
- 37.Corbel M, Lagente V, Theret N, Germain N, Clement B, Boichot E. Comparative effects of betamethasone, cyclosporin and nedocromil sodium in acute pulmonary inflammation and metalloproteinase activities in bronchoalveolar lavage fluid from mice exposed to lipopolysaccharide. Pulm Pharmacol Ther. 1999;12:165–171. doi: 10.1006/pupt.1999.0178. [DOI] [PubMed] [Google Scholar]
- 38.Min D, Moore AG, Bain MA, Breit SN, Lyons JG. Activation of macrophage promatrix metalloproteinase-9 by lipopolysaccharide-associated proteinases. J Immunol. 2002;168:2449–2455. doi: 10.4049/jimmunol.168.5.2449. [DOI] [PubMed] [Google Scholar]
- 39.Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, Mauri C. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. 2007;178:7868–7878. doi: 10.4049/jimmunol.178.12.7868. [DOI] [PubMed] [Google Scholar]