

Abstract
Polymorphisms of DNA repair genes RAD51 and XRCC3 increase susceptibility to acute myeloid leukemia (AML) in adults, an effect enhanced by deletion of the glutathione-S-transferase M1 (GSTM1) gene. In this study, we genotyped 452 children with de novo AML treated on CCG protocols 2941 and 2961 and compared genotype frequencies with those of normal blood donors, and analyzed the impact of genotype on outcome of therapy. XRCC3 Thr241Met, RAD51 G135C and GSTM1 genotypes did not increase susceptibility to AML when assessed singly. In contrast, when XRCC3 and RAD51 genotypes were examined together a significant increase in susceptibility to AML was seen in children with variant alleles. Analysis of outcome of therapy showed that patients heterozygous for the XRCC3 Thr241Met allele had improved post-induction disease-free survival compared to children homozygous for the major or minor allele, each of whom had similar outcomes. Improved survival was due to reduced relapse in the heterozygous children, and this effect was most marked in children randomized to therapy likely to generate DNA double-strand breaks (etoposide, daunomycin), compared with anti-metabolite (fludarabine, cytarabine) based therapy. In contrast, RAD51 G135C and the GSTM1 deletion polymorphism did not influence outcome of AML therapy in our study population.
Keywords: AML, polymorphisms, outcome, DNA repair
Introduction
DNA is at constant risk of damage from both endogenous and exogenous sources. Cells have highly complex pathways to accomplish repair of DNA damage and maintain genomic integrity.1,2 An inability to respond adequately to DNA damage or failure to repair DNA damage accurately leads to genetic instability that may lead to an increased susceptibility to cancer. The capacity to respond to and repair DNA damage in an accurate manner varies among individuals. Genetic polymorphisms have been described for multiple genes associated with DNA repair and may contribute to this interindividual variation.3
DNA double-strand breaks (DSBs) can form in response to a variety of exogenous agents like ionizing radiation and certain chemotherapeutic drugs as well as endogenous agents like reactive oxygen species, defective metabolism of telomeres or replication forks encountering a single-strand break. Repair of DSBs is accomplished in one of two ways: homologous recombination, a high-fidelity process that utilizes DNA from the intact homologous chromosome as a template, and non-homologous end joining, a less accurate process by which the two broken ends are ligated after limited resection at each end. An early step in homologous recombination is the resection of the 3′ ends of the DSB to form single stranded tails that invade the intact homologous DNA double helix forming a Holliday junction.4,5 The RAD51 protein plays a central role in this process by facilitating the initial step of strand invasion.6 The XRCC3 protein interacts directly with and stabilizes the RAD51 protein during the homologous recombination process.7-10 Cell lines lacking RAD51 or its paralogs (for example, XRCC3) demonstrate increased sensitivity to DNA damaging agents like ionizing radiation.11-13 In addition, RAD51 deficient cell lines show an increased frequency of spontaneous chromosomal aberrations contributing to genetic instability.14 These data suggest that RAD51 as well as XRCC3 play an important role in maintaining genomic stability.15-17
A previous study has associated the G/C polymorphism at position −135 in the 5′ untranslated region of the RAD51 gene and the C/T polymorphism in exon 7 of the XRCC3 gene that results in a threonine-to-methionine amino acid substitution (XRCC3 Thr241Met) with susceptibility to acute myeloid leukemia (AML) in adults.18 The authors reported increased susceptibility to both de novo and therapy-related AML when both variant alleles were present. This effect was further increased in persons with a deletion polymorphism in the gene coding for the detoxification enzyme glutathione-S-transferase M1 (GSTM1). The RAD51 G135C polymorphism has also been associated with increased risk of breast cancer in BRCA2 mutation carriers,19 and epidemiological studies noted positive associations between XRCC3 Thr241Met and melanomas,20 bladder cancer,21 breast cancer,22 lung cancer23 and gliomas.24 The functional importance of deletion of the GSTM1 is well described. The biochemical consequences of the XRCC3 Thr241Met polymorphism have not been precisely delineated. However, a number of studies have shown biological differences in response to DNA damage in persons with a variant genotype. For example, increased DNA adducts, increased chromosomal damage and increased micronuclei have all been demonstrated in persons with variant genotypes.25-29 In addition, protein conservation analysis predicts a significant change in function of XRCC3 alleles carrying the variant genotype.30 Data regarding the functional significance of the RAD51 polymorphism is limited. Hasselbach et al.31 have reported an increase in promoter activity with the RAD51 G135C polymorphism.
Consequently, we hypothesized that polymorphisms in RAD51, XRCC3 and GSTM1 might also lead to an increased susceptibility and/or altered outcome in childhood AML and analyzed outcomes with respect to genotype. There are no prior data describing the impact of the XRCC3 and RAD51 variants on outcome of therapy for childhood AML. However, in previous work using a different dataset, we have shown that GSTM1 deletion alone does not impact outcome of therapy.32
Patients and methods
Patients
The study population consisted of children with de novo AML treated on CCG protocols 2941 and 2961 between 1995 and 2002. Children with therapy-related AML were excluded, as numbers were few. Clinical data, including age, sex, white blood cell (WBC) count at diagnosis, race, presence of chloroma, presence of central nervous system (CNS) disease, immunophenotype and cytogenetic abnormalities were collected prospectively (data not shown). Cases were classified on the basis of criteria established and revised by the French–American–British (FAB) Cooperative Study Group by central pathology review. All FAB categories except acute promyelocytic leukemia (APL-AML M3) were eligible for enrollment and were treated on the same chemotherapy regimens.33,34 All patients consented to enrollment on the therapeutic studies after approval of the study by the IRB of each participating institution, and to submission of samples for biological studies. The genotyping study and analysis was approved by the IRB of Cincinnati Children’s Hospital and Medical Center. Blood samples obtained from 646 healthy blood donors (487 Caucasian, 146 African American, 13 unknown race) served as controls for genotype frequencies in the normal population.
Chemotherapy treatment regimen
CCG-2961 was a phase III randomized trial for patients age <21 years with previously untreated AML or myelodysplastic syndrome (MDS) conducted between August 1996 and December 2002. CCG-2941 was a feasibility pilot study for the successor CCG-2961 trial and had a similar treatment plan. All patients received intensively timed induction therapy with idarubicin, dexamethasone, cytarabine, thioguanine, etoposide and daunomycin (IDA-DCTER) given on days 0–3 followed by DCTER given on days 10–13. On recovery of WBC and platelet counts, patients were randomly assigned to receive consolidation therapy with Regimen A, that is, IDA-DCTER or Regimen B, that is, idarubicin, fludarabine, cytarabine and granulocyte-colony stimulating factor (IDA-FLAG). Intrathecal cytarabine was used for CNS prophylaxis. Following consolidation, patients with a matched related donor proceeded to allogeneic marrow transplant with ablative conditioning (busulfan and cytoxan). Those without a related donor received intensification with high-dose cytarabine and l-asparaginase (Capizzi II) and additional intrathecal cytarabine. Patients on the Capizzi II arm were further randomized to receive immune modulation with interleukin-2 or standard follow-up care. Bone marrow transplant recipients were excluded from this randomization.33,34
Genotyping
DNA was extracted from diagnostic marrow samples using the TRIzol (Invitrogen, Carlsbad, California, USA) reagent according to the manufacturer’s directions and normalized to 10 ng μl−1. Genotyping for RAD51 G135C, XRCC3 Thr241Met and GSTM1 deletion polymorphisms was performed using a fluorescence-based allelic discrimination assay (Taqman, Applied Biosystems, Foster City, CA, USA). PCR cycling reactions were performed in 96-well microtiter plates in a GeneAmp PCR System 9600 (Perkin-Elmer, Waltham, Massachusetts, USA). Thermocycling conditions and PCR conditions are described below for each polymorphism.
RAD51 G135C
Probes and primers were as described previously by Auranen et al.35 The reaction included an initial two-step incubation at 50 °C for 2 min and 95 °C for 10 min followed by denaturation at 95 °C for 15 s and annealing and extension at 60 °C for 1 min. The last two steps were repeated for 40 cycles for a final PCR product size of 157 bp. For each 25 μl reaction, 10 ng DNA template was added to the reaction mixture containing wild-type VIC and variant FAM probe, PCR mastermix (Applied Biosystems) and forward and reverse primers (final concentration 0.3 mm).
XRCC3 Thr241Met
Probes and primers are forward primer GGAGTGTGTGAATAAGAAGGTCCCC; reverse primer TCCGCATCCTGGCTAAAAATAC; VIC-probe (wild type) CCACGCTGCGTGAG and FAM-probe (variant) CCATGCTGCGTG. The reaction consisted of an initial two-step incubation at 50 °C for 2 min and 95 °C for 10 min followed by denaturation at 95 °C for 15 s and annealing and extension at 62 °C for 1 min. The last two steps were repeated for 40 cycles for a final PCR product size of 258 bp. For each 25 μl reaction, 10 ng DNA template was added to the reaction mixture containing wild-type VIC and variant FAM probe, PCR mastermix (Applied Biosystems) and forward and reverse primers (final concentration 0.3 mm).
GSTM1
The probes and primers and thermocycling conditions are as described previously by Kiffmeyer et al.36
Results were analyzed by the automated TaqMan allelic discrimination assay using sequence detection system 2.1 software (ABI TaqMan 7700, Applied Biosystems). Genotyping results were repeated in 10% of samples; concordance between repeats was 100%. For XRCC3 Thr241Met, the accuracy of genotyping was confirmed with direct sequencing of randomly selected samples.
Genomic DNA was extracted from peripheral blood samples from our control population (normal blood donors) using QIAamp blood DNA isolation kits (Qiagen Sciences, Maryland, USA) as per the manufacturer’s protocol. Genotyping was performed as described for the patient population.
Statistical Analysis
The observed genotype frequencies of the XRCC3 Thr241Met and RAD51 G135C polymorphisms in the control population were compared with those calculated by the Hardy–Weinberg equilibrium (p2 + q2 + 2pq = 1, where q is the variant allele frequency). Genotype frequencies were compared in the control and patient population and tested for significant differences using odds ratios (ORs) and their 95% confidence intervals (CIs) calculated by logistic regression analysis.
Data obtained from CCG-2941 through 14 April 2005 and from CCG-2961 through 10 June 2005 were used for analyses. The Kaplan-Meier method was used to calculate estimates of overall survival (OS), event-free survival (EFS) and disease-free survival (DFS).37 Estimates are reported with their Greenwood standard errors.38 Differences in these estimates were tested for significance using the log-rank statistic.39 Cumulative incidence estimates were used to determine treatment-related mortality (TRM) and relapse-free survival (RFS). Differences between TRM and RFS estimates were tested for significance using Gray’s test.40 Patients lost to follow-up were censored at their date of last known contact or at 6 months prior to the cut-off date in order to prevent deaths and relapses being reported sooner than ongoing follow-up. The significance of observed differences in proportions was tested using the χ2 squared test and Fisher’s exact test when data were sparse.
Definitions
OS is defined as time from study entry to death from any cause. EFS is defined as time from study entry to failure at the end of two courses, relapse or death from any cause. DFS is defined as time from the end of one course of therapy to failure at the end of two courses, relapse or death from any cause. TRM is defined as the time from study entry to death from non-progressive disease where failures at the end of two courses, relapses and deaths from progressive disease were competing events. RFS is defined as the time from the end of one or two courses of therapy to death from progressive disease, failure at the end of two courses or relapse where deaths from non-progressive disease were competing events.
Results
Susceptibility to childhood AML
Genotype frequencies for the XRCC3 Thr241Met, RAD51 G135C and GSTM deletion polymorphisms were examined in controls and patients with de novo AML. Genotype frequencies were in Hardy-Weinberg equilibrium in both cases and controls and were not different between cases and controls. ORs did not differ significantly from the reference wild-type genotype for any of the polymorphisms when examined singly (Table 1). In work by others in adult AML, a cumulative effect of more than one variant genotype has been demonstrated, with increased risk of AML in persons with both XRCC3 Thr241Met and RAD51 G135C variant alleles.18 To determine if a similar effect occurred in our pediatric AML cases, we examined combinations of XRCC3 Thr241Met and RAD51 G135C genotypes (Table 2). The data show a doubling in risk of AML in children with a variant RAD5 G135C genotype if they carry a wild-type XRCC3 Thr241Met genotype. In addition, risk of AML was significantly increased in children with a variant XRCC3 Thr241Met genotype if they carried a wild-type RAD51 G135C genotype. GSTM genotype was added to the model, examining the cumulative effect of one, two and three variants. Susceptibility to AML was increased in children with two variant alleles (one variant (controls n = 308, (95% cases n = 189; OR 1.20 (95% CI 0.85-1.69) P = 0.298; two variants (controls n = 174, cases n = 136) OR 1.53 (95% CI 1.06-2.21), P = 0.024). The OR changed very little with inclusion of three variant genotypes (OR 1.58; 95% CI 0.78-3.20; P = 0.2); however, it should be noted that the number of children with variant alleles of each of XRCC3, RAD51 and GSTM was small (controls n = 21, cases n = 17).
Table 1.
Frequencies of XRCC3 Thre241Met, RAD51 G135Cand GSTM1 null polymorphisms in AML and control populations and the relative risk of AML associated with those genotypes
| Genotype | Controls | AML | OR (95% CI) | P-value | |
|---|---|---|---|---|---|
| n | n | ||||
| XRCC3 Thr241Met | 646 | 413 | |||
| Thr/The | 253 | 168 | 1.0 (Ref) | ||
| Thr/Met | 309 | 190 | 0.93 (0.71–1.21) | 0.571 | |
| Met/Met | 84 | 55 | 0.99 | 0.944 | |
| Thr/Met+Met/Met | 393 | 245 | 0.94 | 0.623 | |
| RAD51 G135C | 646 | 452 | |||
| G/G | 555 | 374 | 1.0 (Ref) | ||
| G/C | 85 | 73 | 1.27 (0.91–1.79) | 0.161 | |
| C/C | 6 | 5 | 1.24 (0.37–4.08) | 0.727 | |
| G/C+C/C | 91 | 78 | 1.27 (0.91–1.77) | 0.153 | |
| GSTM1 | |||||
| Mu+ | 340 | 243 | 1.0 (Ref) | ||
| Mu− | 299 | 218 | 1.07 (0.84–1.36) | 0.591 | |
Abbreviations: AML, acute myeloid leukemia; CI, confidence intervals.
Table 2.
Combined logistic regression analysis to study the association between DNA double-strand break repair genotypes and AML risk
| RAD 51 | XRCC3 | Controls | AML | OR (95% CI) | P-value |
|---|---|---|---|---|---|
| n | n | ||||
| 644 | 413 | ||||
| WTa | WT | 273 | 130 | 1.0 (Ref) | |
| Va | WT | 36 | 38 | 2.22 (1.24–3.66) | 0.002 |
| WT | V | 280 | 211 | 1.58 (1.20–2.08) | 0.001 |
| V | V | 55 | 34 | 1.30 (0.81–2.09) | 0.282 |
Abbreviations: AML, acute myeloid leukemia; CI, confidence intervals.
WT, denotes a homozygous wild-type genotype; V, denotes at least one variant allele, either heterozygous or homozygous.
XRCC3 Thr241Met genotype and outcome
Genotype frequencies for the XRCC3 Thr241Met polymorphism were compared with known AML prognostic factors and no association was found with WBC count at presentation, FAB type or cytogenetic abnormality (Table 3). However, black children were more likely to have the XRCC3 C241T homozygous genotypes (CC or TT) than white children (74 vs 49%, P = 0.01).
Table 3.
XRCC3 genotype and correlation with known AML prognostic factors
| Characteristic |
CT |
CC |
TT |
CT vs CC+TT | |||
|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | P-value | |
| Study | |||||||
| 2941 | 19 | 10 | 7 | 4 | 7 | 13 | |
| 2961 | 171 | 90 | 161 | 96 | 48 | 87 | 0.23 |
| Gender | |||||||
| Male | 109 | 57 | 92 | 55 | 28 | 51 | |
| Female | 81 | 43 | 76 | 45 | 27 | 49 | 0.53 |
| Race | |||||||
| White | 144 | 76 | 94 | 56 | 44 | 80 | <0.01 |
| Black | 9 | 5 | 23 | 14 | 3 | 5 | 0.02 |
| Hispanic | 26 | 14 | 34 | 20 | 7 | 13 | 0.24 |
| Asian | 4 | 2 | 8 | 5 | 0 | 0 | 0.54 |
| Other | 7 | 4 | 8 | 5 | 1 | 2 | 0.95 |
| Unknown | 0 | 0 | 1 | 1 | 0 | 0 | |
| FAB | |||||||
| M0 | 10 | 5 | 7 | 4 | 6 | 11 | 0.97 |
| M1 | 28 | 15 | 32 | 19 | 7 | 13 | 0.53 |
| M2 | 55 | 29 | 45 | 27 | 21 | 38 | 0.97 |
| M4 | 50 | 26 | 45 | 27 | 12 | 22 | 0.95 |
| M5 | 31 | 16 | 26 | 15 | 7 | 13 | 0.77 |
| M6 | 4 | 2 | 6 | 4 | 0 | 0 | 0.76 |
| M7 | 12 | 6 | 2 | 1 | 2 | 4 | 0.03 |
| Denovo (NOS) | 0 | 0 | 5 | 3 | 0 | 0 | 0.06 |
| Cytogenetics | |||||||
| Normal | 29 | 25 | 25 | 22 | 4 | 15 | 0.48 |
| t(8;21) | 19 | 17 | 20 | 18 | 4 | 15 | 0.97 |
| Abn 16 | 15 | 13 | 9 | 8 | 3 | 11 | 0.34 |
| Abn 11 | 25 | 22 | 22 | 19 | 6 | 22 | 0.85 |
| t(6;9) | 2 | 2 | 3 | 3 | 0 | 0 | 1.00 |
| −7/7− | 3 | 3 | 4 | 4 | 1 | 4 | 0.73 |
| −5/5− | 2 | 2 | 2 | 2 | 0 | 0 | 1.00 |
| +8 | 7 | 6 | 6 | 5 | 2 | 7 | 0.89 |
| +21 | 1 | 1 | 1 | 1 | 0 | 0 | 1.00 |
| Pseudodiploid | 8 | 7 | 15 | 13 | 4 | 15 | 0.13 |
| Hyperdiploid | 2 | 2 | 3 | 3 | 3 | 11 | 0.30 |
| Hypodiploid | 2 | 2 | 3 | 3 | 0 | 0 | 1.00 |
| Unknown | 75 | 39 | 55 | 33 | 28 | 51 | |
| CNS involvement at presentation | 10 | 5 | 8 | 5 | 2 | 4 | 0.89 |
| Chloroma at presentation | 18 | 9 | 19 | 11 | 5 | 9 | 0.79 |
| Age in years (median, range) | 11.0 | (0.01–19.5) | 10.3 | (0.2–19.8) | 10.0 | (0.3–20.9) | 0.61 |
| WBC count at presentation (median, range) |
19 300 | (300–684000) | 24 500 | (1000–860000) | 14 400 | (1300–252500) | 0.22 |
| Bone marrow blast % (median, range) |
69.5 | (2–100) | 68.5 | (0–100) | 80 | (9–100) | 0.56 |
Abbreviations: AML, acute myeloid leukemia; CNS, central nervous system; FAB, French–American–British; WBC, white blood cell.
We observed a trend toward improved 5-year OS among the patients heterozygous for the XRCC3 Thr241Met polymorphism (CT genotype; n = 190) compared to the CC (n = 168) and TT (n = 55) genotypes (53 ± 8 vs 47 ± 7%; P = 0.08). There was no significant difference in outcome between the homozygous wild type (CC) and homozygous variant (TT) genotypes; hence, these patients are described as a single group for the remainder of this analysis. EFS was improved in children with the heterozygote CT genotype compared to the homozygous CC and TT genotypes (43 ± 8 vs 34 ± 7%; P = 0.08). To further analyze the reason for the observed differences we analyzed induction outcomes and found no differences in induction success rates according to XRCC3 genotype (Table 4). In contrast, when post-induction events were analyzed, we observed a significant difference in 5-year DFS from end of two courses with superior survival in heterozygotes (56 ± 9% for CT vs 44 ± 9% for CC + TT genotypes; P = 0.05). The difference in outcome was due to an increased frequency of relapse in the homozygotes as compared to the heterozygote patient population (5-year RFS from end of two courses was 64 ± 9% for CT vs 51 ± 9% for CC + TT genotypes; P = 0.03, Figure 1). TRM from end of two courses was not significantly different between the genotype groups (8 ± 5% for CT vs 7 ± 4% for CC + TT; P = 0.68). Comparison of the heterozygous children with children with only the homozygous wild-type genotype (CC) showed that OS was increased in the heterozygous children (53 ± 8% for CT vs 46 ± 8% for CC; P = 0.079). As described above, inferior survival in the homozygous wild-type children seemed largely due to increased relapse in the homozygous cases (RFS from end of course 64 ± 9% for CT vs 52 ± 10% for CC; P = 0.046).
Table 4.
Induction events according to XRCC3 Thr241Met genotype
|
Response at the end of first course |
XRCC3 Thr241Met genotype |
CT vs CC+TT | |||||
|---|---|---|---|---|---|---|---|
|
CT |
CC |
TT |
P-value | ||||
| n | % | n | % | n | % | ||
| Remission | 160 | 86 | 142 | 88 | 46 | 87 | 0.694 |
| Death | 12 | 6 | 9 | 6 | 2 | 4 | 0.729 |
| Failure | 14 | 8 | 10 | 6 | 5 | 9 | 0.995 |
| Inevaluable | 4 | 7 | 2 | ||||
Figure 1.
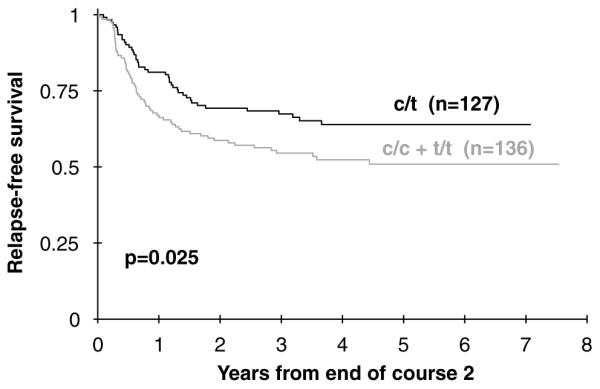
Relapse-free survival from end of course 2 for XRCC3 genotypes C/T vs C/C + T/T.
The frequency of the variant XRCC3 allele differed significantly by race and the statistical significance of the observation was reduced somewhat (DFS (P = 0.098) and RFS (P = 0.067)) in a multivariate regression model that adjusted for race. These data raise the possibility that the inferior outcome in black children compared with white children previously reported from the COG 2941 and 2961 studies might be due to differences in XRCC3 genotype frequency.33 However, univariate and multivariate hazard ratios for XRCC3 and race for all endpoints in this study are quite similar, suggesting that XRCC3 genotype alone is unlikely to explain the differences in outcome seen in that study. In addition, comparison of outcomes for black and white children homozygous for the XRCC3 genotype showed inferior outcomes for black children for all endpoints analyzed, as did a similar analysis of heterozygous children (for example, RFS from end of course 2 for white children was 65% compared with 33% for black children; P = 0.025). It should be noted, however, that there were only 35 black children included in the current study, so power to detect modest differences was highly limited.
We then analyzed post-induction outcome measures in patients randomized to Regimen A (IDA-DCTER) and those randomized to receive Regimen B (IDA-FLAG). A total of 5-year DFS from end of two courses was improved in the heterozygous children in the IDA-DCTER arm (59 ± 14% for the CT group as compared to 43 ± 12% for the CC + TT group (P = 0.06) but did not differ significantly by genotype in the IDA-FLAG arm (5-year DFS 54 ± 13% for CT vs 45 ± 14% for CC + TT group; P = 0.35; Table 5). Similarly, RFS was significantly different by XRCC3 genotype in the IDA-DCTER arm (67 ± 14 vs 49 ± 12%; P = 0.04) but not in the IDA-FLAG arm (64 ± 12 vs 53 ± 14%; P = 0.30)). TRM did not differ significantly by genotype in either arm (8 ± 7 vs 6 ± 6% in the IDA-DCTER arm; P = 0.69 and 10 ± 7 vs 9 ± 7% in the IDA-FLAG arm; P = 0.86). Outcomes were similar for the entire patient cohort (n = 574) treated on CCG protocols 2941 and 2961 and the patient population genotyped for XRCC3 Thr241Met, suggesting that the genotyped population was representative of the entire patient population (data not shown).
Table 5.
Outcome data post-randomization according to XRCC3 Thr241Met genotype
| Endpoint at 5 years |
IDA-DCTER |
IDA-FLAG |
||||
|---|---|---|---|---|---|---|
| C/T (%) | C/C+T/T (%) | P-value | C/T (%) | C/C+T/T (%) | P-value | |
| OS on study | 68 ± 6 | 56 ± 5 | 0.03 | 55 ± 6 | 47 ± 6 | 0.38 |
| RFS post-randomization | 65 ± 7 | 46 ± 6 | 0.03 | 60 ± 7 | 49 ± 7 | 0.26 |
| Treatment-related mortality | 11 ± 4 | 14 ± 4 | 0.43 | 17 ± 4 | 20 ± 4 | 0.51 |
Abbreviations: IDA-DCTER, idarubicin, dexamethasone, cytarabine, thioguanine, etoposide and daunomycin; IDA-FLAG, idarubicin, fludarabine, cytarabine and granulocyte-colony stimulating factor; OS, overall survival; RFS, relapse-free survival.
To determine whether the reduced relapse rate in the heterozygous children might be due to a greater use of bone marrow transplant in those children, we compared the utilization of BMT by genotype and found no difference (CC: 28 of 117 patients transplanted, (24%) CT: 29 of 133 patients (22%) and TT: 9 of 36 patients (25%), P = 0.88). These data show patients actually transplanted; results were similar if analyzed by intent to treat (donor vs no donor; data not shown). XRCC3 Thr241Met genotype did not appear to influence OS or DFS among the patients who underwent bone marrow transplantation (5-years OS 75.9 ± 8.7% for CC, 65.6 ± 9.3% for CT, 66.6 ± 15.7% for TT, P = 0.5) and (5-years DFS 68.1 ± 9.7% for CC, 65.8 ± 9.3% for CT, 66.7 ± 15.7% for CC, P = 0.85).
RAD51 G135C genotype and outcome
RAD51 G135C genotype did not significantly influence any clinical outcome. OS at 5 years was 60 ± 44 vs 49 ± 13 vs 49 ± 6%; P = 0.95) and 5-year EFS was similar in all genotypes (40 ± 44 vs 41 ± 13 vs 38 ± 5%; P = 0.64). TRM from study entry was also similar among the three groups (20 ± 36 vs 17 ± 9 vs 16 ± 4%; P = 0.99).
GSTM1 genotype and outcome
OS did not differ significantly in children with no copy of GSTM1 (GSTM1 null) and those with at least one copy (GSTM1 positive) (51 ± 7 vs 49 ± 7%; P = 0.96). Similar results were observed for EFS (39 ± 7 vs 38 ± 7%; P = 0.90), TRM from study entry (16 ± 5 vs 17 ± 5%; P = 0.62) and RFS from the end of one course (61 ± 8 vs 58 ± 8%; P = 0.49) between the two groups.
Discussion
In this study, we have analyzed the frequency of polymorphisms in DNA repair genes previously linked to susceptibility to AML in adults to address the effects of these variants on susceptibility to childhood AML.18 In addition, we investigated the influence of these polymorphisms on the outcome of therapy for childhood AML, which has not previously been studied. Our data show similar genotype frequencies in control and patient populations for the RAD51 G135C, XRCC3 Thr241Met and GSTM1 polymorphisms, suggesting that these variants, when assessed singly, do not play a role in the etiology of childhood AML. Though the control population was dissimilar with respect to age, genotype frequencies were in Hardy-Weinberg equilibrium and corresponded with previously reported genotype frequencies for each of these polymorphisms, suggesting that these subsets are truly representative. These data contrast with our previous observation of increased risk of AML in children with GSTM1 deletion, demonstrating the importance of replicating positive findings in genetic association studies in independent datasets for confirmation.41 It should be noted that our patient population included only de novo childhood AML, not therapy-related AML.
A similar study in adults by Seedhouse et al.18 showed that the presence of the variant genotype for both RAD51 and XRCC3 significantly increased the risk of developing both de novo and therapy-related AML (OR 3.77 and 8.11, respectively). These authors also showed that with the addition of the GSTM1 deletion polymorphism, the risk of developing AML was notably increased (OR 15.26). Our study showed a doubling of risk of AML in children with a RAD 51 G135C variant allele and a wild-type XRCC3 Thre241Met genotype. In addition risk of AML was significantly increased in children with at least one variant XRCC3 Thr241Met allele. In contrast, risk was not significantly elevated in children with variant alleles at both wild-type XRCC3 Thre241Met and RAD51 G135C. Addition of GSTM1 genotype to the model did not further increase risk of AML. These data indicate the importance of examining multiple genes in the same pathway to identify the role of genotype. Our study is in agreement with the findings of Seedhouse et al. to the extent that we demonstrated interaction between genotypes at different loci. Our study differs, however, in that the largest effect was seen in children with a variant allele at one locus and a wild-type allele at the second locus. These findings may be a consequence of biological differences in the etiology of childhood AML, compared with adult AML. The difference in the spectrum of cytogenetic abnormalities seen in childhood AML compared with adult AML supports the hypothesis that the biological mechanisms resulting in childhood AML may be somewhat distinct from those causing AML in adults.42 It is also possible that these are chance observations and replication in an independent pediatric AML dataset will be important to determine the reliability of this finding.
In addition to studying the influence of variant genotypes involved in homologous recombination on susceptibility to AML, we examined the effect of genotype on the outcome of therapy, as one of the cytotoxic effects of AML chemotherapy is the generation of DNA DSBs. The data show that the XRCC3 Thr241Met polymorphism has a significant influence on the post-induction outcomes for our patient population. Surprisingly, the best outcome was found in heterozygotes with improved 5-year DFS for C/T vs CC + TT genotypes (P = 0.05). An increased frequency of relapse in the homozygotes led to inferior outcomes in these children while, TRM was similar in homozygotes and heterozygotes.
Our patient population was randomized to two different post-induction chemotherapy regimens–Regimen A consisted of IDA-DCTER and Regimen B consisted of IDA-FLAG. When outcomes of children with AML treated on CCG2961 and CCG2941 were examined as a whole, without consideration of genotype, no significant difference was observed among the patients treated on Regimen A (IDA-DCTER) and Regimen B (IDA-FLAG). While IDA-FLAG consists primarily of anti-metabolite-based therapy, IDA-DCTER includes etoposide and daunomycin therapy that induces double-strand DNA breaks, requiring homologous recombination for accurate repair. Since XRCC3 plays a key role in homologous recombination, we examined the outcomes of patients treated on Regimen A and B separately with respect to XRCC3 Thr241Met genotype. The heterozygous patients had improved survival in both treatment arms compared to the homozygotes. However, this difference in outcome was statistically significant only in the patients who were treated on Regimen A (IDA-DCTER).
While it is believed that XRCC3 is a key player in the initial strand invasion and nucleoprotein filament during the process of homologous recombination, its precise mechanism of action is still not clear. Previous epidemiological and in vitro biological studies have suggested that XRCC3 Thr241Met is a functionally important polymorphism.30,43-45 A number of studies of adult malignancies have shown associations of XRCC3 Thr241Met genotype with cancer susceptibility, including melanoma,20 breast cancer,22 MDS, chronic gastritis and gastric cancers,46 and aerodigestive cancers.47 Functional studies examining the role of the variant allele have also been reported in the literature. Matullo et al.21,25 identified that XRCC3 Thr241Met polymorphism resulted in reduced DNA repair activity when using p32-labelling to measure DNA adduct levels as a measure of DNA repair capacity. In addition, Lindh et al.48 reported an increase in mitotic defects in cells expressing only XRCC3 Thr241Met. In contrast to these findings, Araujo et al.49 were not able to detect a significant difference between wild type and variant proteins in their ability to correct the hypersensitivity of the irsISF cell line to DNA damage using Mitomycin C or to complement the homologous recombination defect in XRCC3-deficient cells.
One possible hypothesis to explain why the outcome was statistically different only in the patients who were treated on Regimen A (IDA-DCTER) could be that DSB repair is inferior in heterozygous children. This would potentially allow superior killing of blasts when homologous recombination is required to repair damage, and the effect of genotype is most evident in children receiving therapy that requires homologous recombination for repair. Unfortunately, in the studies mentioned before, homozygous and heterozygous genotypes were not compared. It might be expected that if the variant allele is truly associated with reduced homologous recombination, the effect of the polymorphism would be most evident in children homozygous for the variant allele. It is surprising, therefore, that the heterozygous group has improved survival as compared to the wild type and the variant homozygotes.
In summary, in our study, we observed a significant interaction between XRCC3 and RAD51 genotypes in susceptibility to childhood AML. In addition, we have shown a difference in the post-induction outcome of childhood AML with respect to the XRCC3 Thr241Met genotype with heterozygous children showing superior survival. The survival difference was most significant in children receiving chemotherapy that we would expect to generate DSBs. While these findings are of interest, like all such studies, these observations need to be replicated in additional independent datasets to validate the importance of this polymorphism in predicting response to chemotherapy. Additionally, molecular and biochemical studies are needed to clarify the functional significance of the XRCC3 Thr241Met polymorphism, particularly in heterozygotes.
Acknowledgements
This work was supported by R01CA93552-01 (SMD), R21 CA10262-01, R01 CA114563-01, (SM) and COG Grant CA 98543. A complete listing of grant support for research conducted by CCG and POG before initiation of the COG grant in 2003 is available online at: http://www.childrensoncologygroup.org/admin/grantinfo.htm
References
- 1.Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24:3799–3808. doi: 10.1200/JCO.2005.05.4171. Review. [DOI] [PubMed] [Google Scholar]
- 2.Ishikawa K, Ishii H, Saito T. DNA damage-dependent cell cycle checkpoints and genomic stability. DNA Cell Biol. 2006;25:406–411. doi: 10.1089/dna.2006.25.406. Review. [DOI] [PubMed] [Google Scholar]
- 3.Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:1513–1530. [PubMed] [Google Scholar]
- 4.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 5.O’Driscoll M, Jeggo PA. The role of double-strand break repair–insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 6.Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 7.Bishop DK, Ear U, Bhattacharyya A, Calderone C, Beckett M, Weichselbaum RR, et al. Xrcc3 is required for assembly of Rad51 complexes in vivo. J Biol Chem. 1998;273:21482–21488. doi: 10.1074/jbc.273.34.21482. [DOI] [PubMed] [Google Scholar]
- 8.Schild D, Lio Y, Collins DW, Tsomondo T, Chen DJ. Evidence for simultaneous protein interactions between human Rad51 paralogs. J Biol Chem. 2000;275:16443–16449. doi: 10.1074/jbc.M001473200. [DOI] [PubMed] [Google Scholar]
- 9.Masson JY, Stasiak AZ, Stasiak A, Benson FE, West SC. Complex formation by the human RAD51C and XRCC3 recombination repair proteins. Proc Natl Acad Sci USA. 2001;98:8440–8446. doi: 10.1073/pnas.111005698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada NA, Hinz JM, Kopf VL, Segalle KD, Thompson LH. XRCC3 ATPase activity is required for normal XRCC3-Rad51C complex dynamics and homologous recombination. J Biol Chem. 2004;279:23250–23254. doi: 10.1074/jbc.M402247200. Epub 2004 Mar 22. [DOI] [PubMed] [Google Scholar]
- 11.Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell. 1998;1:783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- 12.Kurumizaka H, Ikawa S, Nakada M, Eda K, Kagawa W, Takata M, et al. Homologous-pairing activity of the human DNA-repair proteins Xrcc3.Rad51C. Proc Natl Acad Sci USA. 2001;98:5538–5543. doi: 10.1073/pnas.091603098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, et al. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell. 2003;11:1109–1117. doi: 10.1016/s1097-2765(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 14.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshihara T, Ishida M, Kinomura A, Katsura M, Tsuruga T, Tashiro S, et al. XRCC3 deficiency results in a defect in recombination and increased endoreduplication in human cells. EMBO J. 2004;23:670–680. doi: 10.1038/sj.emboj.7600087. Epub 2004 Jan 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bleuyard JY, White CI. The Arabidopsis homologue of Xrcc3 plays an essential role in meiosis. EMBO J. 2004;23:439–449. doi: 10.1038/sj.emboj.7600055. Epub 2004 Jan 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei C, Skopp R, Takata M, Takeda S, Price CM. Effects of double-strand break repair proteins on vertebrate telomere structure. Nucleic Acids Res. 2002;30:2862–2870. doi: 10.1093/nar/gkf396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seedhouse C, Faulkner R, Ashraf N, Das-Gupta E, Russell N. Polymorphisms in genes involved in homologous recombination repair interact to increase the risk of developing acute myeloid leukemia. Clin Cancer Res. 2004;10:2675–2680. doi: 10.1158/1078-0432.ccr-03-0372. [DOI] [PubMed] [Google Scholar]
- 19.Levy-Lahad E, Lahad A, Eisenberg S, Dagan E, Paperna T, Kasinetz L, et al. A single nucleotide polymorphism in the RAD51 gene modifies cancer risk in BRCA2 but not BRCA1 carriers. Proc Natl Acad Sci USA. 2001;98:3232–3236. doi: 10.1073/pnas.051624098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winsey SL, Haldar NA, Marsh HP, Bunce M, Marshall SE, Harris AL, et al. A variant within the DNA repair gene XRCC3 is associated with the development of melanoma skin cancer. Cancer Res. 2000;60:5612–5616. [PubMed] [Google Scholar]
- 21.Matullo G, Guarrera S, Carturan S, Peluso M, Malaveille C, Davico L, et al. DNA repair gene polymorphisms, bulky DNA adducts in white blood cells and bladder cancer in a case-control study. Int J Cancer. 2001;92:562–567. doi: 10.1002/ijc.1228. [DOI] [PubMed] [Google Scholar]
- 22.Kuschel B, Auranen A, McBride S, Novik KL, Antoniou A, Lipscombe JM, et al. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet. 2002;11:1399–1407. doi: 10.1093/hmg/11.12.1399. [DOI] [PubMed] [Google Scholar]
- 23.Popanda O, Schattenberg T, Phong CT, Butkiewicz D, Risch A, Edler L, et al. Specific combinations of DNA repair gene variants and increased risk for non-small cell lung cancer. Carcinogenesis. 2004;25:2433–2441. doi: 10.1093/carcin/bgh264. Epub 2004 Aug 27. [DOI] [PubMed] [Google Scholar]
- 24.Wang LE, Bondy ML, Shen H, El-Zein R, Aldape K, Cao Y, et al. Polymorphisms of DNA repair genes and risk of glioma. Cancer Res. 2004;64:5560–5563. doi: 10.1158/0008-5472.CAN-03-2181. [DOI] [PubMed] [Google Scholar]
- 25.Matullo G, Palli D, Peluso M, Guarrera S, Carturan S, Celentano E, et al. XRCC1, XRCC3, XPD gene polymorphisms, smoking and (32)P-DNA adducts in a sample of healthy subjects. Carcinogenesis. 2001;22:1437–1445. doi: 10.1093/carcin/22.9.1437. [DOI] [PubMed] [Google Scholar]
- 26.Au WW, Salama SA, Sierra-Torres CH. Functional characterization of polymorphisms in DNA repair genes using cytogenetic challenge assays. Environ Health Perspect. 2006;111:1843–1850. doi: 10.1289/ehp.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aka P, Mateuca R, Buchet JP, Thierens H, Kirsch-Volders M. Are genetic polymorphisms in OGG1, XRCC1 and XRCC3 genes predictive for the DNA strand break repair phenotype and genotoxicity in workers exposed to low dose ionising radiations? Mutat Res. 2004;556:169–181. doi: 10.1016/j.mrfmmm.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Angelini S, Kumar R, Carbone F, Maffei F, Forti GC, Violante FS, et al. Micronuclei in humans induced by exposure to low level of ionizing radiation: influence of polymorphisms in DNA repair genes. Mutat Res. 2005;570:105–117. doi: 10.1016/j.mrfmmm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Jiao L, Chang P, Firozi PF, Lai D, Abbriuzzese JL, Li D. Polymorphisms of phase II xenobiotic-metabolizing and DNA repair genes and in vitroN-ethyl-N-nitrosourea-induced 06-ethylguanine levels in human lymphocytes. Mutat Res. 2007;627:146–157. doi: 10.1016/j.mrgentox.2006.11.001. Epub 2006 Dec 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savas S, Kim DY, Ahmad MF, Shariff M, Ozcelik H. Identifying functional genetic variants in DNA repair pathways using protein conservation analysis. Cancer Epidemiol Biomarkers Prev. 2004;13:801–807. [PubMed] [Google Scholar]
- 31.Hasselbach L, Haase S, Fischer D, Kolberg HC, Sturzbecher HW. Characterisation of the promoter region of the human DNA-repair gene Rad51. Eur J Gynaecol Oncol. 2005;26:589–598. [PubMed] [Google Scholar]
- 32.Davies SM, Robison LL, Buckley JD, Tjoa T, Woods WG, Radloff GA, et al. Glutathione-s-transferase polymorphisms and outcome of chemotherapy in childhood AML. J Clin Oncol. 2001;19:1279–1287. doi: 10.1200/JCO.2001.19.5.1279. [DOI] [PubMed] [Google Scholar]
- 33.Lange BJ, Dinndorf P, Smith FO, Arndt C, Barnard D, Feig S, et al. Pilot study of idarubicin-based intensive-timing induction therapy for children with previously untreated acute myeloid leukemia: Children’s Cancer Group Study 2941. J Clin Oncol. 2004;22:150–156. doi: 10.1200/JCO.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Aplenc R, Alonzo TA, Gerbing RB, Smith FO, Meshinchi S, Ross JA, et al. Ethnicity and survival in childhood acute myeloid leukemia: a report from the Children’s Oncology Group. Blood. 2006;108:74–80. doi: 10.1182/blood-2005-10-4004. Epub 2006 Mar 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auranen A, Song H, Waterfall C, Dicioccio RA, Kuschel B, Kjaer SK, et al. Polymorphisms in DNA repair genes and epithelial ovarian cancer risk. Int J Cancer. 2005;117:611–618. doi: 10.1002/ijc.21047. [DOI] [PubMed] [Google Scholar]
- 36.Kiffmeyer WR, Langer E, Davies SM, Envall J, Robison LL, Ross JA. Genetic polymorphisms in the Hmong population: implications for cancer etiology and survival. Cancer. 2004;100:411–417. doi: 10.1002/cncr.11913. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan E, Meier P, Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 38.Greenwood M. The Natural Duration of Cancer. His Majesty’s Stationery Office; London, England: 1926. pp. 1–26. Report on Public Health and Medical Subjects. [Google Scholar]
- 39.Kalbfleisch J, Prentice R. The Statistical Analysis of Failure Time Data. John Wiley; New York: 1980. pp. 20–23. [Google Scholar]
- 40.Gray R. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 41.Davies SM, Robison LL, Buckley JD, Radloff GA, Ross JA, Perentesis JP. GST polymorphisms in children with myeloid leukemia: a Children’s Cancer Group (CCG) study. Cancer Epidemiol Biomarkers. 2000;9:563–566. [PubMed] [Google Scholar]
- 42.Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001;98:1312–1320. doi: 10.1182/blood.v98.5.1312. [DOI] [PubMed] [Google Scholar]
- 43.Ryk C, Kumar R, Sanyal S, Verdier PJ, Hemminki K, Larsson P, et al. Influence of polymorphism in DNA repair and defence genes on p53 mutations in bladder tumours. Cancer Lett. 2006;241:142–149. doi: 10.1016/j.canlet.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 44.Han S, Zhang HT, Wang Z, Xie Y, Tang R, Mao Y, et al. DNA repair gene XRCC3 polymorphisms and cancer risk: a meta-analysis of 48 case-control studies. Eur J Hum Genet. 2006;14:1136–1144. doi: 10.1038/sj.ejhg.5201681. [DOI] [PubMed] [Google Scholar]
- 45.Savas S, Kim DY, Ahmad MF, Shariff M, Ozcelik H. Identifying functional genetic variants in DNA repair pathway using protein conservation analysis. Cancer Epidemiol Biomarkers Prev. 2004;13:801–807. [PubMed] [Google Scholar]
- 46.Duarte MC, Colombo J, Rossit AR, Caetano A, Borim AA, Wornrath D, et al. Polymorphisms of DNA repair genes XRCC1 and XRCC3, interaction with environmental exposure and risk of chronic gastritis and gastric cancer. World J Gastroenterol. 2005;11:6593–6600. doi: 10.3748/wjg.v11.i42.6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gal TJ, Huang WY, Chen C, Hayes RB, Schwartz SM. DNA repair gene polymorphisms and risk of second primary neoplasms and mortality in oral cancer patients. Laryngoscope. 2005;115:2221–2231. doi: 10.1097/01.mlg.0000183736.96004.f7. Erratum in: Laryngoscope. 2006 116(3):507. [DOI] [PubMed] [Google Scholar]
- 48.Lindh AR, Rafii S, Schultz N, Cox A, Helleday T. Mitotic defects in XRCC3 variants T241M and D213N and their relation to cancer susceptibility. Hum Mol Genet. 2006;15:1217–1224. doi: 10.1093/hmg/ddl037. Epub 2006 Feb 27. [DOI] [PubMed] [Google Scholar]
- 49.Araujo FD, Pierce AJ, Stark JM, Jasin M. Variant XRCC3 implicated in cancer is functional in homology-directed repair of double-strand breaks. Oncogene. 2002;21:4176–4180. doi: 10.1038/sj.onc.1205539. [DOI] [PubMed] [Google Scholar]