

Abstract
Background
Striate keratodermas (PPKS) are a group of rare autosomal dominant palmoplantar keratodermas, characterized by a thickening of the skin on the palms and soles. PPKS is characterized by hyperkeratosis extending along the length of each finger and on the palm of the hand, as well as by patches of hyperkeratosis on the soles.
Objective
We report a four-generation Pakistani kindred in which eleven members were affected with PPKS.
Methods
Based on previous reports of DSG1 mutations in PPKS, we performed direct DNA sequencing analysis.
Results
Clinically, these patients presented with hyperkeratotic palms and with linear hyperkeratosis on the fingers. Additionally, focal hyperkeratosis was seen on the sole of the toes as well as the ball and heel of the foot. DNA sequencing analysis revealed a heterozygous G-to-T transversion in the 3′ splice acceptor site of intron 11 of the DSG1 gene designated 1688 -1 G > T. We predict that this mutation will lead to the skipping of exon 12 which is out of frame (134 nt), subsequent degradation of the mutant mRNA by nonsense mediated RNA decay, and haploinsufficiency for DSG1.
Conclusion
We report a novel splice site mutation in the DSG1 gene in PPKS, which further underscores the significance of the desmoglein gene family in diseases of epidermal integrity.
Keywords: desmoglein 1, cell adhesion, palmoplantar keratoderma, desmosome, splice site mutation
Introduction
Palmoplantar keratodermas (PPK) are a genetically and phenotypically heterogeneous group of genodermatoses characterized by hyperkeratosis on the palms of the hands and/or soles of the feet. In some cases, this hyperkeratosis may be associated with other epidermal or extra-epidermal disorders [1]. PPK is further sub-classified according to the pattern of hyperkeratosis of the lesions. The striate form of palmoplantar keratoderma (PPKS) is a rare autosomal dominant disorder characterized by linear hyperkeratotic streaks along the volar surfaces of fingers and focal keratoderma on the soles [2].
There are 12 reported cases of PPKS in the literature in which the molecular lesion has been identified, and mutations can reside in at least three different genes: desmoglein 1, desmoplakin, and keratin 1. PPKS1 and PPKS2 are associated with mutations in the genes coding for the desmosomal proteins desmoglein 1 and desmoplakin, respectively [3, 4, 5]. A third variant, PPKS3, has recently been reported in a large British kindred and is associated with a frameshift mutation within the V2 domain of keratin 1 [6]. The skin lesions seen in PPKS are thought to result from impaired desmosomal function at sites exposed to a high degree of mechanical stress, such as palms and soles, where these cell-cell junctions are critical for epidermal integrity [7, 8].
Desmosomes are a type of anchoring junction that serve to anchor cells to one another via the intermediate filament cytoskeleton network, thereby conferring an additional degree of strength to tissues that undergo high levels of mechanical stress [9]. Desmosomes are comprised of members from three protein families—the cadherin family, the armadillo family, and the plakin family. The cadherin family is represented by desmogleins (DSG 1–4) and desmocollins (DSC1–3), while the armadillo family is represented by plakoglobin (PG) and plakophilins (PKP 1–3 and p0071), and the plakin family is represented by desmoplakin (DP) [10, 11, 12]. Desmogleins and desmocollins play a critical role in the formation of desmosomes. Extracellularly, desmogleins and desmocollins on opposing cells interact heterophilically via their cadherin repeat domains in a calcium dependent manner. Intracellularly, the cadherins bind to plakoglobin and plakophilin, which in turn bind to desmoplakin. Desmoplakin then binds to intermediate filaments, thereby securing the entire structure to the cytoskeleton. Interestingly, PKKS can result from a perturbation in either the intermediate filament network, (K1), the desmosomal plaque (DP) or the transmembrane cadherin (DSG1).
In this study, we report a novel splice site mutation in the DSG1 gene in a four-generation Pakistani kindred in which eleven members were affected with PPKS. This further underscores the significance of the desmoglein gene family in diseases of epidermal integrity.
Materials and Methods
Polymerase chain reaction and DNA sequencing
Genomic DNA was isolated from peripheral blood lymphocytes following institutional review board approval. Direct sequence analysis of the DSG1 gene was performed as previously described [8, 13]. PCR prod ucts were electrophoresed in 0.8% agarose gels, and purified using a standard PCR purification protocol (Concert, Gibco BRL, Marlingen Bioscience Inc., Ijamsville, MD, USA) and sequenced using an ABI Prism 310 automated sequencing system (PE-Applied Biosystems, Foster City, CA, USA) in both directions using the same primers used for initial PCR. Sequence comparisons between patient samples and controls were performed by visual inspection.
Mutation identification and confirmation by use of mismatch PCR
The tranversion mutation neither created nor destroyed a restriction site directly, therefore we introduced a site using mismatch PCR. DSG1 ex12mutF1 primer (5′-TACTATCCCTCCACCACTAGT-3′) was used to introduce a G-to-T transversion at the penultimate nucleotide (nt1666) directly upstream from the G-to-T transversion mutation (nt1668), thereby creating a novel Bfa1 (CT/AG) restriction site only when the transversion mutation 1668-1 G→T was not present. After PCR amplification, the fragments were digested by the restriction enzyme Bfa1 (New England Biolabs, Beverly, MA, USA) and separated on a 1% agarose gel in the presence of ethidium bromide and visualized by UV light.
Results
Clinical features
We studied a four-generation Pakistani kindred in which eleven individuals were affected with PPKS (Figure 1A). Clinically, these patients presented with hyperkeratosis on the palm of the hands, with particular prominence on the creases of the palm, and linear hyperkeratosis along the flexor aspect of the fingers. Additionally, focal hyperkeratosis was seen on the sole of the toes as well as the ball and heel of the foot, areas that undergo a particularly high degree of mechanical stress (Figure 1C–E). Affected individuals did not have a history of blister formation, and no phenotypes other than the hyperkeratosis were observed. Hair was normal in appearance and texture, with no evidence of woolly hair or cardiac abnormalities reported by family members, although all family members were not examined by a clinician.
Figure 1.
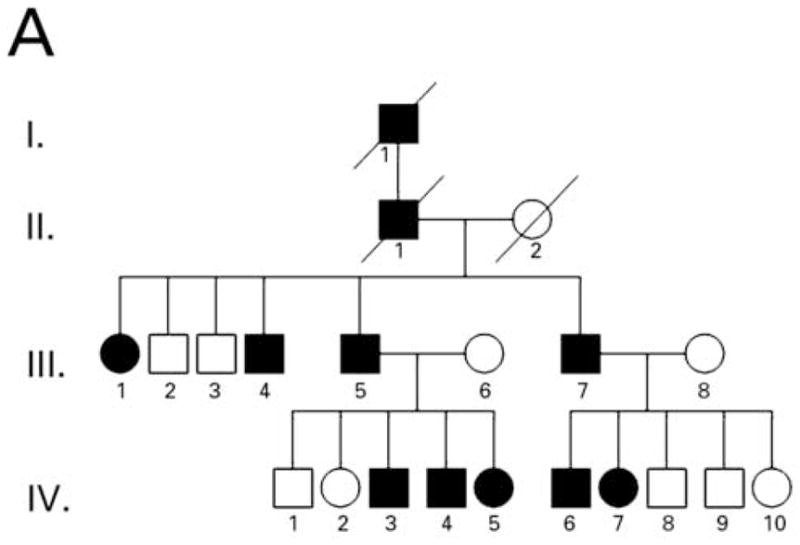
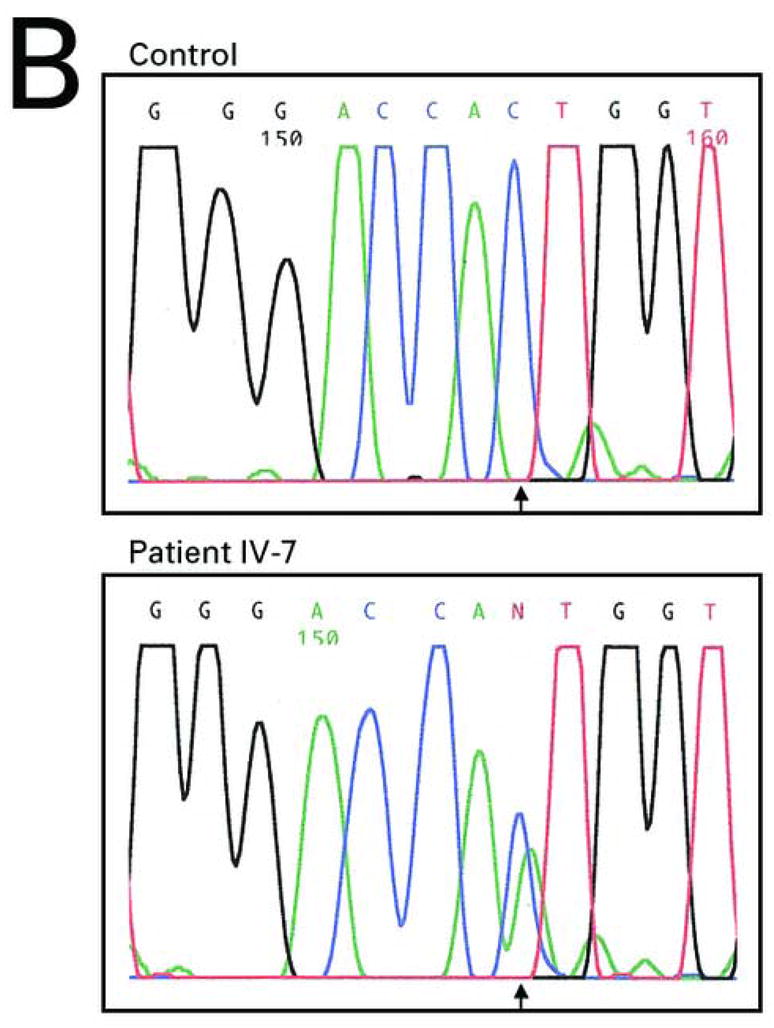
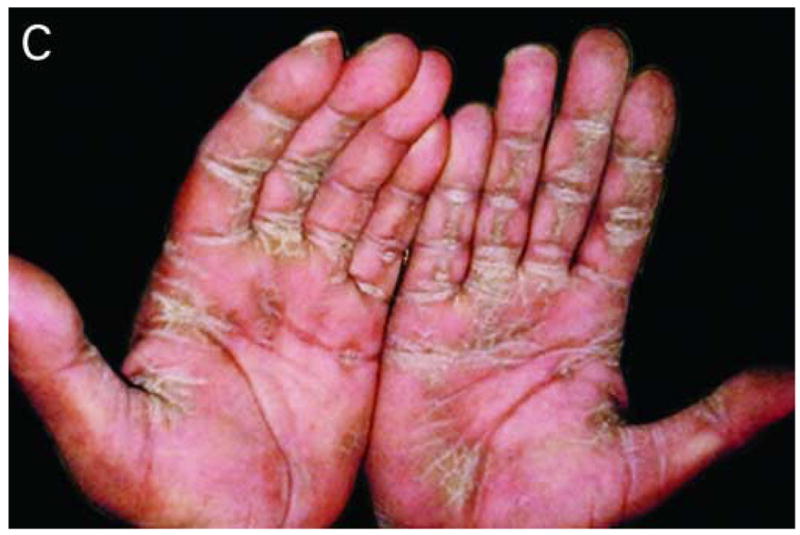
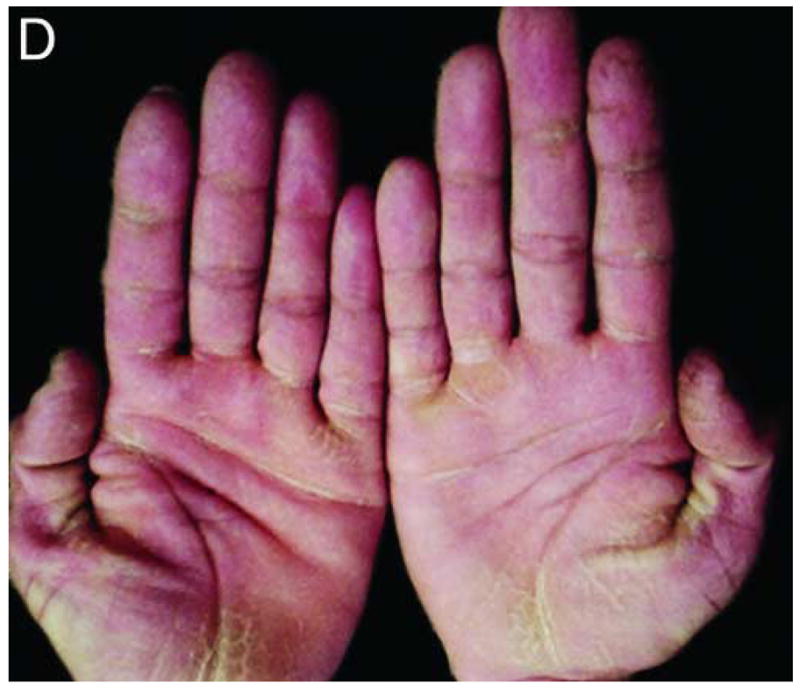
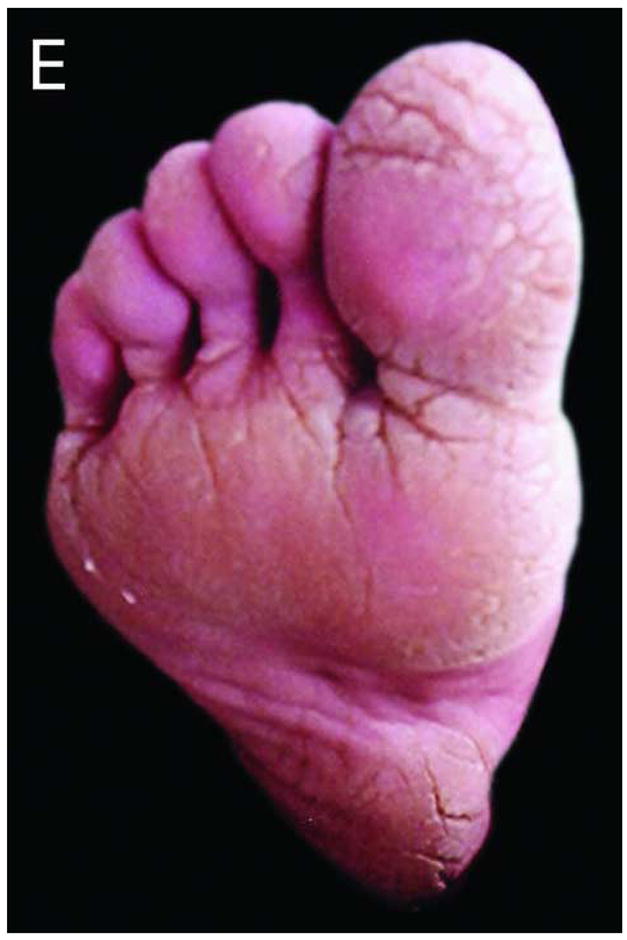
Pedigree, sequence analysis, and clinical features of PKKS family. A) Pedigree reveals autosomal dominant inheritance with 11 affected members in 4 generations. Filled circles and squares represent affected females and males respectively. B) Sequence analysis of intron 11 of an affected individual (IV-7) and control. The arrows indicate the heterozygous G-to-T transversion in the 3′-splice acceptor site of intron 11 of the DSG1 gene. The sequence is shown in reverse orientation (antisense strand). C–E) Clinical presentation of focal hyperkeratosis on the toes, ball, and heel of the foot of an affected individual. Clinical presentation of hyperkeratosis on the palms of the hands, most prominently in creases of the palm, and in linear streaks along the flexor aspect of the fingers of two affected individuals.
Identification and confirmation of a novel mutation in DSG1
Direct sequence analysis of the intron 11/exon 12 splice junction region of DSG1 gene in affected individuals revealed a heterozygous G-to-T transversion in the 3′-splice acceptor site of intron 11 designated 1668-1 G→T (Figure 1B). In order to confirm the presence of this mutation, we utilized mismatch PCR for DNA samples from affected and unaffected family members. This analysis confirmed presence of the mutation 1668-1 G→T in all affected individuals, and lack of the mutation in all of the unaffected individuals (data not shown). These results suggest that the mutation 1668-1 G→T is indeed the pathogenic mutation.
Discussion
We identified a novel heterozygous mutation in the DSG1 gene of a four-generation Pakistani kindred with striate palmoplantar keratoderma. The mutation is a G-to-T tranversion at the splice acceptor site of intron 11 of DSG1. We predict that this mutation will lead to skipping of exon 12, which is out of frame (134 nt), leading to degradation of the mutant mRNA by nonsense mediated RNA decay, and subsequent haploinsufficiency.
In the literature to date, there are 12 reported cases of PPKS in which mutations have been identified. Of these, 7 are in the DSG1 gene, 4 are in the DP gene and 1 is in keratin 1 (Table 1). In all but one of the reported cases, PPKS is dominantly inherited.
Table 1.
Summary of mutations associated with PPKS.
| Gene | Mutation | Zygosity | Location | Result | Predicted Effect | Phenotype | Reference |
|---|---|---|---|---|---|---|---|
| DSG1 | IVS 2–1 G→A | Heterozygous | Intron 2 | In-frame deletion | Haploinsufficiency | PPKS | Rickman et al., (1999) |
| DSG1 | R26X | Heterozygous | Exon 2 | PTC | Haploinsufficiency | PPKS | Hunt et al., (2001) |
| DSG1 | S132X | Heterozygous | Exon 5 | PTC | Haploinsufficiency | PPKS | Kljuic et al., (2003) |
| DSG1 | 1079insC | Heterozygous | Exon 9 | PTC | Haploinsufficiency or Dominant-negative | PPKS | Hunt et al., (2001) |
| DSG1 | Y365X | Heterozygous | Exon 9 | PTC | Haploinsufficiency or Dominant-negative | PPKS | Hunt et al., (2001) |
| DSG1 | 1189delA | Heterozygous | Exon 9 | PTC | Haploinsufficiency or Dominant-negative | PPKS | Hunt et al., (2001) |
| DSG1 | 1627delA | Heterozygous | Exon 11 | PTC | Haploinsufficiency or Dominant-negative | PPKS | Hunt et al., (2001) |
| DSG1 | 121insT | Heterozygous | Exon 3 | PTC | Haploinsufficiency | Focal, non- striated PPKa | Milingou et al., (2006) |
| DSG1 | 1688 –1 G→T | Heterozygous | Intron 11 | Out-of- frame deletion | Haploinsufficiency | PPKS | Present study |
| DP | 8804 C→T | Heterozygous | Exon 4 | PTC | Haploinsufficiency | PPKS | Armstrong et al., (1999) |
| DP | 939+1 G→A | Heterozygous | Intron 7 | PTC | Haploinsufficiency | PPKS | Whittock et al., (1999) |
| DP | 7901delG | Homozygous | Exon 24 | PTC | Impaired protein function | Generalized PPKS, WH, DLVC | Norgett et al., (2000) |
| DP | 608ins30bp | Heterozygous | Exon 14 | In-frame insertion | Haploinsufficiency or Dominant-negative | PPKS, WH, Cardiomyopathy | Norgett et al, (2006) |
| K1 | 1628delG | Heterozygous | Exon 9 | Out-of- frame deletion | Dominant negative | PPKS | Whittock et al., (2002) |
Though this case involves a DSG1 mutation associated with PPK, this is the only case listed in this table that is not a case of PPKS.
Abbreviations: DSG1 = Desmoglein 1; DP = Desmoplakin; K1 = Keratin 1; PTC = Premature Termination Codon; PPKS = Striate Palmoplantar Keratoderma; WH = Wooly Hair; DLVC = Dialated Left Ventricular Cardiomyopathy.
The prevalence of mutations in DSG1 associated PPKS highlights the critical role of desmoglein 1 in the desmosome. Interestingly, there has also been a heterozygous DSG1 mutation reported by Milingou et al. that results in focal, non-striated palmoplantar keratoderma [1] (Table 1). Unlike previously reported DSG1 associated PPKS cases in which the mutation is located either in an exon or splice site within the extracellular domain of DSG1, the splice site mutation reported here affects exon 12 which encodes a portion of the transmembrane domain and intracellular linker region of DSG1.
In all cases where DSG1 mutations have been reported in PPKS, there is evidence that haploinsufficiency for the affected protein leads to the disease phenotype (Table 1). In most cases, the DSG1 mutation leads to the formation of a premature termination codon, however an in-frame deletion has also been observed [14]. The mutations resulting in a premature termination codon are expected to trigger nonsense mediated mRNA decay (NMRD) leading to the degradation of the mutant transcript [15, 16]. In the case of the in-frame deletion, the mutation results in the excision of exon 3, thereby removing a portion of the pro-sequence, the mature protein cleavage site, and part of the first extracellular domain of DSG1. While activation of the NMRD pathway is not the likely cause of haploinsufficiency, Rickman et al. report that haploinsufficiency is most likely due to an insufficient amount of wild-type DSG1 protein synthesis [14].
Similar to DSG1 associated PPKS, a dominantly inherited mutation resulting in the formation of a premature termination codon is responsible for the majority of desmoplakin (DP) associated PPKS cases. Again, the introduction of a premature termination codon is expected to trigger NMRD and subsequent haploinsufficiency. The only exception being in a case reported by Norgett et al. where a homozygous recessive mutation in exon 24 of DP results in a PTC, but the resulting truncated protein is synthesized [17]. In this case the disease is a result of impaired protein function. Recently, Norgett et al. reported another case of DP associated PPKS in which the disease results from a 30 base pair in-frame insertion. This mutation causes an insertion of 10 amino acids in the N-terminus of the protein, and the authors speculate that the resulting pathogenicity may be due to either haploinsufficiency or a dominant negative effect of the mutant protein [18]. Interestingly, in the latter two cases reported by Norgett et al. woolly hair and cardiomyopathy are observed in addition to PPKS, suggesting phenotypic overlap with Naxos disease.
While PPKS is most often associated with DSG1 or DP mutations, there is one case in which keratin 1 (K1) is associated with PPKS [6]. In this case, an out-of-frame deletion results in the formation of a mutant protein that lacks a portion of a glycine loop motif in the V2 domain and gains 70 amino acids. The authors stated that pathogenicity is due to a dominant negative effect caused by the loss of the V2 domain which inhibits proper formation of the intermediate filament cytoskeleton, however since the mutation is out-of-frame, NMRD may also be possible.
In contrast to other dominantly inherited keratodermas, where dominant negative interference is suspected and RNAi could be used therapeutically, the same is not true of PPKS, where haploinsufficiency seems to be the predominant mechanism [19] (Table 1). In our present study we report a novel desmoglein 1 splice site mutation which results in PPKS. This finding adds to the body of desmosomal mutations known to cause epidermal disease, thereby enhancing our understanding of the significance of desmosomes in epidermal integrity.
Acknowledgments
We are grateful to the family members for their participation in this study. We thank Helen Lam for excellent technical assistance. This work was supported in part by USPHS NIH/NIAMS R01 AR44924 (to AMC).
Abbreviations
- DSG1
human desmoglein 1 gene/protein
- PPKS
striate palmoplantar keratoderma
- DP
desmoplakin gene/protein
- K1
keratin 1 gene/protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Milingou M, Wood P, Masouye I, McLean WH, Borradori L. Focal palmoplantar keratoderma caused by an autosomal dominant inherited mutation in the desmoglein 1 gene. Dermatology. 2006;212:117–22. doi: 10.1159/000090651. [DOI] [PubMed] [Google Scholar]
- 2.Fartasch M, Vigneswaran N, Diepgen TL, Hornstein OP. Abnormalities of keratinocyte maturation and differentiation in keratosis palmplantaris striata. Immunohistochemical and ultrastructural study before and during etretinate therapy. Am J Dermatopathol. 1990;12:275–82. doi: 10.1097/00000372-199006000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Whittock NV, Bower C. Targetting of desmoglein 1 in inherited and acquired skin diseases. Clin Exp Dermatol. 2003;28:410–5. doi: 10.1046/j.1365-2230.2003.01311.x. [DOI] [PubMed] [Google Scholar]
- 4.Whittock NV, Ashton GH, Dopping-Hepenstal PJ, Gratian MJ, Keane FM, Eady RA, et al. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol. 1999;113:940–6. doi: 10.1046/j.1523-1747.1999.00783.x. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong DKB, McKenna KE, Purkis PE, Green KJ, Eady RAJ, Leigh IM, Hughes AE. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Human Molecular Genetics. 1999;8:143–148. doi: 10.1093/hmg/8.1.143. [DOI] [PubMed] [Google Scholar]
- 6.Whittock NV, Smith FJ, Wan H, Mallipeddi R, Griffiths WA, Dopping-Hepenstal P, Ashton GH, Eady RA, McLean WHI, McGrath JA. Frameshift mutation in the V2 domain of human Keratin 1 results in striate palmoplantar keratoderma. J Invest Dermatol. 2002;118:838–44. doi: 10.1046/j.1523-1747.2002.01750.x. [DOI] [PubMed] [Google Scholar]
- 7.Hunt DM, Sahota VK, Taylor K, Simrak D, Hornigold N, Arnemann J, et al. Clustered cadherin genes: a sequence-ready contig for the desmosomal cadherin locus on human chromosome 18. Genomics. 1999;62:445–55. doi: 10.1006/geno.1999.6036. [DOI] [PubMed] [Google Scholar]
- 8.Hunt DM, Rickman L, Whittock NV, Eady RA, Simrak D, Dopping-Hepenstal PJ, et al. Spectrum of dominant mutations in the desmosomal cadherin desmoglein 1, causing the skin disease striate palmoplantar keratoderma. Eur J Hum Genet. 2001;9:197–203. doi: 10.1038/sj.ejhg.5200605. [DOI] [PubMed] [Google Scholar]
- 9.Yin T, Green KJ. Regulation of desmosome assembly and adhersion. Semin Cell Dev Biol. 2004;15:665–77. doi: 10.1016/j.semcdb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol. 2000;1:208–16. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- 11.Kljuic A, Gilead L, Martinez-Mir A, Frank J, Christiano AM, Zlotogorski A. A nonsense mutation in the desmoglein 1 gene underlies striate keratoderma. Exp Dermatol. 2003;12:523–7. doi: 10.1034/j.1600-0625.2003.00017.x. [DOI] [PubMed] [Google Scholar]
- 12.Hatzfeld M, Green KJ, Sauter H. Targeting pf p0071 to desmosomes and adherens junctions is mediated by different protein domains. J Cell Sci. 2003;116:1219–33. doi: 10.1242/jcs.00275. [DOI] [PubMed] [Google Scholar]
- 13.Frank J, Cserhalmi-Friedman PB, Ahmad W, Panteleyev AA, Aita VM, Christiano AM. Characterization of the desmosomal cadherin gene family: Genomic organization of two desmoglein genes on human chromosome 18q12. Exp Dermatol. 2001;10:90–94. doi: 10.1034/j.1600-0625.2001.010002090.x. [DOI] [PubMed] [Google Scholar]
- 14.Rickman L, Simrak D, Stevens HP, Hunt DM, King IA, Bryant SP, Eady RAJ, Leigh IM, Arnemann J, Magee AI, Kelsell DP, Buxton RS. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderm. Hum Mol Genet. 1999;8:971–6. doi: 10.1093/hmg/8.6.971. [DOI] [PubMed] [Google Scholar]
- 15.Culbertson MR RNA surveillance. Unforseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet. 1999;15:74–80. doi: 10.1016/s0168-9525(98)01658-8. [DOI] [PubMed] [Google Scholar]
- 16.Cui Y, Hagan KW, Zhang S, Peltz SW. Identification and characterization of genes that are required for the accelerated degradation of mRNAs containing a premature translational termination codon. Genes Dev. 1995;9:423–36. doi: 10.1101/gad.9.4.423. [DOI] [PubMed] [Google Scholar]
- 17.Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JCR, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, wooly hair and keratoderma. Hum Mol Genet. 2000;9:2761–66. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 18.Norgett EE, Lucke TW, Bowers B, Munro CS, Leigh IM, Kelsell DP. Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantar keratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. J Invest Dermatol. 2006;126:1651–4. doi: 10.1038/sj.jid.5700291. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi S, Tanaka T, Matsuyoshi N, Imamura S. Keratin 9 point mutation in the pedigree of epidermolytic hereditary palmoplantar keratoderma perturbs keratin intermediate filament network formation. FEBS Lett. 1996;386:149–55. doi: 10.1016/0014-5793(96)00393-6. [DOI] [PubMed] [Google Scholar]