

Abstract
During the last decade, several causative genes for hereditary hair diseases have been identified, which have disclosed the molecular mechanisms involved in hair follicle morphogenesis and cycling. We and others recently reported that mutations in the P2RY5 gene, encoding an orphan G protein-coupled receptor, underlie autosomal recessive woolly hair and/or hypotrichosis. Although these findings clearly reveal the involvement of P2RY5 mutations in hereditary hair diseases, the clinical manifestations of P2RY5 mutations have not completely been elucidated due to limited information to date. In this study, we ascertained a consanguineous family of Iranian origin with an affected girl showing sparse and hypopigmented scalp hair. She had woolly hair phenotype with normal hair density at birth, but progressed with age to have hypotrichosis. Direct sequencing analysis resulted in the identification of a novel homozygous mutation in the P2RY5 gene of the patient, which results in a non-conservative amino acid change, G146R, at the protein level. Our findings extend the mutation spectrum of P2RY5 mutations, and further support a crucial role of P2Y5 in hair growth in humans.
Keywords: P2RY5, lipase H, woolly hair, hypotrichosis
INTRODUCTION
The mammalian hair follicle (HF) is a complex multicellular organ, which undergoes dynamic cell kinetics and rearrangements throughout life in the form of a periodic hair cycle (1). Recent advances in molecular genetics have enabled the identification of numerous genes that are expressed in the HF during development and cycling (2). There are several types of hereditary hypotrichosis for which causative genes have been identified. For example, atrichia with papular lesions (OMIM 209500) is caused by mutations in hairless gene (HR) (3–5). Mutations in desmoglein 4 gene (DSG4) cause localized autosomal recessive hypotrichosis/monilethrix (OMIM 607903/252200) (6–9). Furthermore, it has recently been shown that mutations in the lipase H gene (LIPH) underlie an autosomal recessive form of hypotrichosis (OMIM 604379) (10–12). These findings provide insight into mechanisms of hair cycle regulation, hair shaft differentiation, and hair growth. However, mechanisms involved in determining the hair texture, which is variable among individuals and different populations, remain largely unknown.
Woolly hair (WH) is a group of hair disorders characterized by tightly curled hair (13). There are some syndromes that show WH as a part of the disease, such as Naxos disease (MIM 601214) (14) and Carvajal syndrome (MIM 605676) (15). In addition, isolated WH without systemic symptoms also exists and can show either an autosomal dominant (ADWH; OMIM 194300) (16) or recessive (ARWH; OMIM 278150) (16, 17) inheritance pattern. Importantly, it has been suggested that hypotrichosis can be an associated feature of ARWH (16, 17). We and others recently performed linkage studies (18–20) and identified pathogenic mutations in P2RY5 gene in several families with ARWH or hypotrichosis (19, 20). The P2RY5 gene encodes a G protein-coupled receptor P2Y5, which is abundantly expressed in the inner root sheath of the HF, and is likely to be involved in the maintenance of the structural integrity of the hair shaft (19).
In this study, we ascertained a consanguineous family of Iranian origin with an affected girl showing sparse and hypopigmented hairs. Direct sequencing analysis led to the identification of a novel homozygous mutation G146R in the P2RY5 gene of the affected individual. These data enable a better understanding of the clinical features resulting from P2RY5 mutations, and underscore the crucial role of P2Y5 in HF development and hair growth in humans.
MATERIALS AND METHODS
Mutation analysis of P2RY5 gene
After obtaining informed consent, we collected peripheral blood samples from the family members and 100 unrelated healthy control individuals in EDTA-containing tubes (under institutional approval and in adherence to the Declaration of Helsinki Principles). Genomic DNA was isolated from these samples according to standard techniques. The P2RY5 gene was screened for mutations as described previously (19). The mutation 436G>A generates a BclI restriction enzyme site, which was used to screen parents and control individuals. A part of the coding sequence of the P2RY5 gene was amplified by PCR using a forward primer (5′-CACGGAATTGGCCATTTGGA-3′) and a reverse primer (5′-GTTTTCCATGTGGCTTCTGG-3′). The amplification conditions were 94°C for 2 min, followed by 35 cycles of 94°C for 30 sec, 57°C for 30 sec, and 72°C for 30 sec, with a final extension at 72°C for 7 min. The amplified PCR products, 301 bp in size, were digested with the BclI at 50°C overnight, and run on 1.5% agarose gels.
RESULTS
Clinical features
The 8-year-old proband is the child of first cousin parents of Iranian origin (Fig. 1A). At birth, she had tightly curled hair on her scalp which was of normal density (Fig. 1B). Since the age of 1 year, her hair loss has gradually progressed. Upon examination, she exhibited sparse, thin, short, and hypopigmented scalp hairs (Fig. 1C, D). Perifollicular papules were detected only in the occipital region (Fig. 1D). The eyebrows were also sparse, whereas the eyelashes were normal. Under light microscopy, the hair shafts showed dystrophic features (Fig. 1E), and the distal portion of the hair shafts were frequently broken (Fig. 1F). Teeth, nails, and sweating were normal, and palmoplantar hyperkeratosis was absent. There was no family history of either heart diseases or neurologic abnormalities.
Figure 1. Clinical features of the proband with ARWH.

(A) Pedigree of the family. Clinical appearance at the ages of 1 (B) and 8 (C, D). (E, F) Appearance of the hair shafts under light microscopy. Scale bars: 100 μm.
Identification of a novel mutation in P2RY5 gene
We first checked for mutations in DSG4 and LIPH genes which are known to underlie autosomal recessive forms of hypotrichosis (6, 10), but found no sequence variants in either gene. Based on the recent findings of P2RY5 mutations in ARWH or hypotrichosis (19, 20), we performed direct sequencing analysis of P2RY5 gene, and identified a novel mutation in the P2RY5 in the family. The affected individual is homozygous for a G- to -A transition at position 436 of the P2RY5 gene, designated 436G>A, which results in a substitution of glycine by an arginine residue at amino acid 146 (G146R) (Fig. 2A). Both parents are heterozygous for this nucleotide change (Fig. 2A). Screening assays using the BclI restriction enzyme excluded the existence of this mutation in 100 unrelated healthy control individuals (200 chromosomes) (Fig. 2B; data not shown).
Figure 2. Identification of a mutation in the P2RY5 gene.

(A) Homozygous mutation 436G>A (G146R) in the P2RY5 gene of the patient. (B) Screening assays for the mutation 436G>A (G146R). PCR products from the mutant allele were digested into 196 bp and 105 bp fragments with the BclI restriction enzyme. MWM, molecular weight marker; F, father; M, mother; Pt, patient.
Discussion
In this study, we studied a consanguineous Iranian family with an affected girl showing sparse, thin and depigmented scalp hairs, and identified a novel homozygous mutation 436G>A (G146R) in the P2RY5 of the patient (Fig. 2A). The P2RY5 gene is a nested gene residing within intron 17 of the retinoblastoma 1 (RB1) gene on human chromosome 13q14, and encodes P2Y5 protein which is a heptaspan transmembrane G protein-coupled receptor (GPCR) (21). The glycine residue at codon 146 is located within the fourth transmembrane domain in P2Y5 protein and is highly conserved between different species (Fig. 3). The non-conservative amino acid change G146R is most likely to severely affect the structure of P2Y5 protein.
Figure 3. Multiple amino acid sequence alignment of P2Y5 between different species.
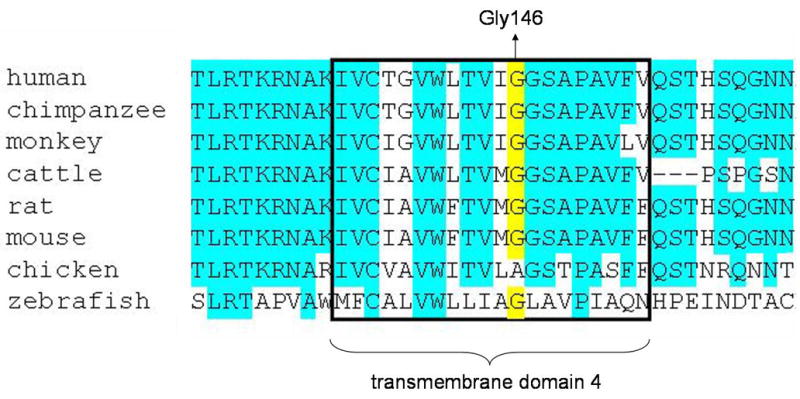
Residue Gly146 is indicated in yellow. Residues that are conserved among at least six species are colored blue. The fourth transmembrane domain is boxed.
We previously identified five distinct pathogenic mutations in the P2RY5 genes in several consanguineous Pakistani families with affected individuals showing tightly curled hair (19) (Fig. 4). Although WH is a unifying feature among all affected individuals, hair density is variable from normal to sparse, and such variations in severity are detected even within a single family (19). In addition, we recently identified a homozygous mutation C278Y in the P2RY5 in a consanguineous Brazilian family with several affected individuals with a severe hypotrichosis (Fig. 4) (22). It is noteworthy that all affected individuals in the Brazilian family showed WH phenotype at birth and lost their hair during the first decade of life. In addition to our findings, Pasternack et al. recently identified two distinct mutations in the P2RY5 gene in Saudi Arabian families with affected individuals characterized by progressive hair loss that were diagnosed as autosomal recessive hypotrichosis simplex (MIM 146520), but display abnormal hair texture (Fig. 4) (20). Collectively, it is evident that the phenotypes resulted from mutations in the P2RY5 gene are highly variable (23). A similar phenomenon has been noted with mutations in the DSG4 gene, which display an overlap in phenotype between hypotrichosis and monilethrix (7–9, 24).
Figure 4. Schematic representation of P2Y5 protein and the P2RY5 mutations.
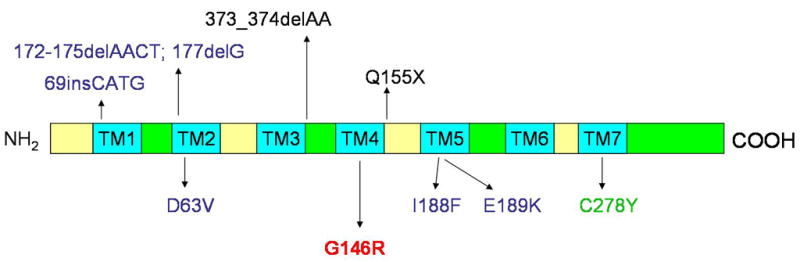
Premature termination codon mutations are shown above the schematic, and missense mutations are shown below. The mutation G146R reported in this study is indicated in red. Mutations identified in Pakistani families and Saudi Arabian families are colored in blue and black, respectively. The mutation C278Y found in a Brazilian family is indicated in green. Transmembrane domains (TM1-7) are colored in blue.
Mutations in LIPH gene were recently shown to cause an autosomal recessive form of hypotrichosis (10–12), as well as ARWH (Shimomura et al., submitted). Interestingly, clinical features of affected individuals with LIPH mutations are indistinguishable from those with P2RY5 mutations. The LIPH gene encodes a phospholipase A1 family member and is known to produce lysophosphatidic acid (LPA) (25). LPA is an extracellular mediator of many biological functions and has been reported to promote hair growth in vivo (26), and P2Y5 is a receptor of LPA (20). Our work further suggests that the LIPH/LPA/P2Y5 signaling pathway plays a crucial role in the hair structure in humans, and also provides a new therapeutic target for hair disorders.
Acknowledgments
We gratefully acknowledge the family members for having participated in this study. We thank Dr. Leonard Kristal (State University of New York) for expert consultation. We thank Ha Mut Lam for excellent technical assistance. This work was supported by US Public Health Service National Institute of Health grant R01AR44924 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (to A.M.C.).
References
- 1.Paus R, Foitzik K. In search of the “hair cycle clock”: a guided tour. Differentiation. 2004;72:489–511. doi: 10.1111/j.1432-0436.2004.07209004.x. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt-Ullrich R, Paus R. Molecular principles of hair follicle induction and morphogenesis. Bioessays. 2005;27:247–261. doi: 10.1002/bies.20184. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad W, Faiyaz ul Haque M, et al. Alopecia universalis associated with a mutation in the human hairless gene. Science. 1998;279:720–724. doi: 10.1126/science.279.5351.720. [DOI] [PubMed] [Google Scholar]
- 4.Cichon S, Anker M, Vogt IR, et al. Cloning, genomic organization, alternative transcripts and mutational analysis of the gene responsible for autosomal recessive universal congenital alopecia. Hum Mol Genet. 1998;7:1671–1679. doi: 10.1093/hmg/7.11.1671. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad W, Zlotogorski A, Panteleyev AA, et al. Genomic organization of the human hairless gene (HR) and identification of a mutation underlying congenital atrichia in an Arab Palestinian family. Genomics. 1999;56:141–148. doi: 10.1006/geno.1998.5699. [DOI] [PubMed] [Google Scholar]
- 6.Kljuic A, Bazzi H, Sundberg JP, et al. Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell. 2003;113:249–260. doi: 10.1016/s0092-8674(03)00273-3. [DOI] [PubMed] [Google Scholar]
- 7.Shimomura Y, Sakamoto F, Kariya N, Matsunaga K, Ito M. Mutations in the desmoglein 4 gene are associated with monilethrix-like congenital hypotrichosis. J Invest Dermatol. 2006;126:1281–1285. doi: 10.1038/sj.jid.5700113. [DOI] [PubMed] [Google Scholar]
- 8.Schaffer JV, Bazzi H, Vitebsky A, et al. Mutations in the desmoglein 4 gene underlie localized autosomal recessive hypotrichosis with monilethrix hairs and congenital scalp erosions. J Invest Dermatol. 2006;126:1286–1291. doi: 10.1038/sj.jid.5700237. [DOI] [PubMed] [Google Scholar]
- 9.Zlotogorski A, Marek D, Horev L, et al. An autosomal recessive form of monilethrix is caused by mutations in DSG4: clinical overlap with localized autosomal recessive hypotrichosis. J Invest Dermatol. 2006;126:1292–1296. doi: 10.1038/sj.jid.5700251. [DOI] [PubMed] [Google Scholar]
- 10.Kazantseva A, Goltsov A, Zinchenko R, et al. Human hair growth deficiency is linked to a genetic defect in the phospholipase gene LIPH. Science. 2006;314:982–985. doi: 10.1126/science.1133276. [DOI] [PubMed] [Google Scholar]
- 11.Ali G, Chishti MS, Raza SI, John P, Ahmad W. A mutation in the lipase H (LIPH) gene underlie autosomal recessive hypotrichosis. Hum Genet. 2007;121:319–325. doi: 10.1007/s00439-007-0344-0. [DOI] [PubMed] [Google Scholar]
- 12.Jelani M, Wasif N, Ali G, Chishti MS, Ahmad W. A novel deletion mutation in LIPH gene causes autosomal recessive hypotrichosis (LAH2) Clin Genet. 2008 doi: 10.1111/j.1399-0004.2008.01011.x. in press. [DOI] [PubMed] [Google Scholar]
- 13.Chien AJ, Valentine MC, Sybert VP. Hereditary woolly hair and keratosis pilaris. J Am Acad Dermatol. 2006;54:S35–39. doi: 10.1016/j.jaad.2005.01.092. [DOI] [PubMed] [Google Scholar]
- 14.McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 15.Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 16.Hutchinson PE, Cairns RJ, Wells RS. Woolly hair. Clinical and general aspects. Trans St Johns Hosp Dermatol Soc. 1974;60:160–177. [PubMed] [Google Scholar]
- 17.Salamon T. Über eine familie mit recessiver Kraushaarigkeit, hypotrichose und anderen anomalien. Hautarzt. 1963;14:540–544. [PubMed] [Google Scholar]
- 18.Wali A, Chishti MS, Ayub M, et al. Localization of a novel autosomal recessive hypotrichosis locus (LAH3) to chromosome 13q14.11-q21.32. Clin Genet. 2007;72:23–29. doi: 10.1111/j.1399-0004.2007.00818.x. [DOI] [PubMed] [Google Scholar]
- 19.Shimomura Y, Wajid M, Ishii Y, et al. Disruption of P2RY5, an orphan G protein-coupled receptor, underlies autosomal recessive woolly hair. Nat Genet. 2008;40:335–339. doi: 10.1038/ng.100. [DOI] [PubMed] [Google Scholar]
- 20.Pasternack SM, von Kügelgen I, Aboud KA, et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet. 2008;40:329–334. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- 21.Webb TE, Kaplan MG, Barnard EA. Identification of 6H1 as a P2Y purinoceptor: P2Y5. Biochem Biophys Res Commun. 1996;219:105–110. doi: 10.1006/bbrc.1996.0189. [DOI] [PubMed] [Google Scholar]
- 22.Petukhova L, Sousa EC, Martinez-Mir A, et al. Genome-wide linkage analysis of an autosomal recessive hypotrichosis identifies a novel P2RY5 mutation. Genomics. 2008 doi: 10.1016/j.ygeno.2008.06.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sprecher E. Disentangling the roots of inherited hair disorders. Nat Genet. 2008;40:265–266. doi: 10.1038/ng0308-265. [DOI] [PubMed] [Google Scholar]
- 24.Schweizer J. More than one gene involved in monilethrix: intracellular but also extracellular players. J Invest Dermatol. 2006;126:1216–1219. doi: 10.1038/sj.jid.5700266. [DOI] [PubMed] [Google Scholar]
- 25.Sonoda H, Aoki J, Hiramatsu T, et al. A novel phosphatidic acid-selective phospholipase A1 that produces lysophosphatidic acid. J Biol Chem. 2002;277:34254–34263. doi: 10.1074/jbc.M201659200. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi T, Kamimura A, Hamazono-Matsuoka T, Honda S. Phosphatidic acid has a potential to promote hair growth in vitro and in vivo, and activates mitogen-activated protein kinase/extracellular signal-regulated kinase kinase in hair epithelial cells. J Invest Dermatol. 2003;121:448–456. doi: 10.1046/j.1523-1747.2003.12426.x. [DOI] [PubMed] [Google Scholar]