

Abstract
We report a sensitive peptide pull-down approach in combination with protein identification by LC-MS/MS and qualitative abundance measurements by spectrum counting to identify proteins binding to histone H3 tail containing dimethyl lysine 4 (H3K4me2), dimethyl lysine 9 (H3K9me2), or acetyl lysine 9 (H3K9ac). Our study identified 86 nuclear proteins that associate with the histone H3 tail peptides examined, including seven known direct binders and 16 putative direct binders with conserved PHD finger, bromodomain, and WD40 domains. The reliability of our proteomic screen is supported by the fact that more than one-third of the proteins identified were previously described to associate with histone H3 tail directly or indirectly. To our knowledge, the results presented here are the most comprehensive analysis of H3K4me2, H3K9me2, and H3K9ac associated proteins and will provide a useful resource for researchers studying the mechanisms of histone code effector proteins.
Keywords: Epigenetics, Histone code effectors, Histone modifications, Proteomics
1 Introduction
Histone proteins are extensively modified by an array of PTMs that include phosphorylation, acetylation, methylation, ADP-ribosylation, proline isomerization, ubiquitinylation, sumoylation, citrullination, butyrylation, and propionylation [1–4]. Many of these PTMs have been associated with distinct biological processes, such as gene activation, silencing, replication, mitosis, and DNA damage response. Recent progress in MS has allowed for unbiased identification of many novel histone PTMs [5–13] to reveal important insights into the complexity of histone regulation.
The functions of histone PTMs have been the subject of extensive investigations over the past decade. For example, acetylation of histone H3 tail has been generally correlated with transcriptional activation [14], in contrast, distinct H3 methylation states are associated with different regulatory outcomes, for example, methylation of H3K4 is associated with gene activation, whereas methylation of H3K9 and H3K27 with gene repression [15,16]. Although specific modifications have been correlated with distinct biological processes, the precise mechanisms by which histone modifications transmit their biological signals into meaningful biological readouts are poorly understood. There is mounting evidence that histone modifications transmit biological signal through the recruitment of effector proteins, or “readers” [17]. Association of the downstream effectors may lead to changes in the accessibility of the DNA template to the transcriptional machinery, recruitment of enzymatic activities such as ATP-dependent chromatin remodeling complexes, or changes in the higher order structure of chromatin, all of which would lead to specific regulatory outcomes [2, 17].
The binding to histone PTMs by effector protein is mediated by conserved binding modules, such as the canonical bromodomain binding to acetylated lysines [18]. The binding modules for methylated lysines include chromodomains, Tudor domains, MBT-repeats, WD40-repeats, and PHD fingers [19]. It has also become apparent that multiple effector molecules can recognize a single modification, but the actual in vivo binding is dependent on a broader biological context. These observations underscore the need for a more comprehensive analysis of epigenetic readers and highlight the importance of understanding how histone PTMs regulate biological processes.
Here, we report a comprehensive, unbiased screen for proteins associated with three H3 PTMs: dimethylated K4 (H3K4me2); dimethylated K9 (H3K9me2), and acetylated K9 (H3K9ac). Our studies led to the identification of 86 proteins that bind, directly or indirectly, to the amino terminus of histone H3. Among the identified proteins, one-third represents previously described direct effectors of H3, H3K4me2, or H3K9me2, as well as their known associated proteins. Importantly, we have identified many novel modification-specific binders, including PHD finger-, WD40-, and bromodomain-containing proteins. Results presented here offer a rich source of candidate effector molecules for further downstream mechanistic analysis. To our knowledge, this analysis is the most comprehensive study to identify novel histone PTM binders in an unbiased manner.
2 Materials and methods
2.1 Peptides
The H3 amino terminal peptides containing amino acids 1–21 coupled to a biotin linker, ARTKQTARKSTGGKAP-RKQLA-GGK-biotin (H3 peptide), ART(dimethyl-K]QTAR-KSTGGKAPRKQLA-GGK-biotin (H3K4me2), ARTKQTAR (dimethyl-K)STGGKAPRKQLA-GGK-biotin (H3K9me2), and ARTKQTAR(acetyl-K)STGGKAPRKQLA-GGK-biotin (H3K9ac) were purchased from Upstate-Millipore (Billerica, MA).
2.2 PHF2 antibody
PHF2 antiserum was generated by immunizing rabbits with purified recombinant GST-PHF2 C-terminal fragment corresponding amino acids 830–1098 of PHF2.
2.3 HeLa nuclear extracts
HeLa S3 cells were purchased from National Cell Culture Center (Minneapolis, MN). The cytoplasmic (S100) and nuclear extracts (NE) were prepared as previously described [20, 21]. Briefly, HeLa cells were washed in cold PBS, and resuspended in five times the packed cell volume (PCV) with hypotonic buffer (10 mM Tris, pH 7.3, 1.5 mM MgCl2, 10 mM KCl, 10 mM β-mercaptoethanol, and 0.5 mM PMSF). The cells were incubated on ice for 10 min and then pelleted by centrifugation at 1900×g for 10 min. The swollen cell pellet was then resuspended in half the PCV with hypotonic buffer and homogenized with 15 strokes in a glass dounce homogenizer. The lysate was centrifuged at 5000×g for 20 min at 4°C to separate the cytoplasmic proteins from the nuclear pellet. The nuclear pellet volume (NPV) was determined and the pellet was resuspended in 0.5 mL of extraction buffer (20 mM Tris, pH 7.3, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 10 mM β-mercaptoethanol, and 0.5 mM PMSF) per milliliter of NPV. The nuclei pellet was then homogenized by ten strokes in a glass dounce homogenizer. While stirring, 0.5 mL of extraction buffer containing 1.2 M KCl was added per milliliter of NPV drop wise to the homogenized nuclear extract. The extract was further stirred for 30 min at 4°C. The sample was then centrifuged for 30 min at 20 000×g. The NE supernatant was dialyzed against dialysis buffer (20 mM Tris, pH 7.3, 100 mM KCl, 0.2 mM EDTA, 20% glycerol, 10 mM β-mercaptoethanol, and 0.5 mM PMSF) and then centrifuged at 20 000×g for 30 min. The NE supernatant was aliquoted, snap frozen in liquid nitrogen, and stored at −80°C.
2.4 Peptide pull-down
The peptides were prebound to streptavidin agarose beads (Invitrogen, Carlsbad, CA) in 50 μL of NETN buffer (20 mM Tris, pH 8, 1 mM EDTA, and 0.5% NP-40) containing 100 mM NaCl. Five milligrams of HeLa nuclear extracts was added to each of the peptide-bound agarose beads and rotated for 5 h at 4°C. The beads were then washed five times with 1 mL of NETN buffer containing 200 mM NaCl. The washed beads were boiled with 60 μL of Laemmli buffer, analyzed by SDS-PAGE on a 4–20% gel (Invitrogen), and stained with colloidal CBB.
2.5 In-gel digestion and MS
Sample preparation and MS analysis on a Finnigan LTQ mass spectrometer (Thermo Finnigan, San Jose, CA) were as previously described in ref. [22]. Briefly, each lane of the gel was divided into eight equal size gel slices. The gel slices were destained with 50 mM ammonium bicarbonate in 50% methanol and washed overnight with HPLC grade water. The gel slices were then crushed and digested with 200 ng of trypsin in 50 mM ammonium bicarbonate at 37°C for 4 h. Digested peptides were extracted with 200 μL of ACN and dried by speed vac. Each sample was dissolved in 20 μL of 5% methanol/95% water/0.1% formic acid and analyzed on a C18 column (100 mm×75 μm, 300 Å pore diameter, PicoFrit; New Objective, Woburn, MA). The mobile phase A (0.1% formic acid in water) and B (0.1% formic acid in methanol) was used with a gradient of 10–80% mobile phase B over 10 min followed by 80% B for 10 min at a flow rate of 200 nL/min. Peptides were directly electrosprayed into the mass spectrometer using a nano-spray source. The LTQ was operated in a data-dependent mode, acquiring fragmentation spectra of the 20 strongest ions.
2.6 Database search and protein identification
Raw MS data were searched using BioWorks 3.2 Sequest software (Thermo Electron, Waltham, MA) against the NCBI human protein RefSeq database. Results from the eight bands for each pull-down/MS were loaded into the multi-consensus files with the following filter threshold settings: XCorr of 1.50 (1+), 1.80 (2+), and 3.00 (3+) and probability of 1 × 10−2 for peptides and XCorr of 10.0 and probability of 1× 10−3 for proteins. All protein identifications were verified by manual inspection of MS/MS assignments. The number of spectra acquired from the peptide mixture was used as a measure of protein abundance within the protein pull-down as described previously in ref. [23].
2.7 Data visualization and cluster analysis
Protein abundance data derived by spectrum counting were subjected to hierarchical cluster analyses in Spotfire Decision Site v.7.0 with correlations determined by magnitude and shape (Euclidean distance) [24].
3 Results
3.1 Peptide pull-down strategy
To identify the effector proteins that associate with H3 modifications at K4 and K9, we used C-terminally biotinylated peptides corresponding to the N-terminal H3 tail (aa 1–21) either unmodified (H3), dimethylated at K4 (H3K4me2), dimethylated at K9 (H3K9me2), or acetylated at K9 (H3K9ac). The peptides were immobilized on streptavidin agarose beads and incubated with HeLa nuclear extracts (Fig. 1). Bound proteins were eluted and separated by SDS-PAGE. CBB staining revealed both distinct and similar protein bands eluting from the four H3 peptide pull-downs. As expected, there were far fewer bands in the streptavidin beads control lane, indicating that nonspecific binding to the agarose beads was relatively low (Fig. 2A). While protein stains provide a good visualization of proteins present, low abundance proteins may not be readily visible. For unbiased and sensitive proteomic analysis, we divided each lane into eight equal size slices and digested each in situ with trypsin for protein identification and quantification.
Figure 1.

Strategy to isolate binders to modified K4 and K9 of histone H3. Unmodified H3 or modified H3 peptides containing dimethylated K4, dimethylated K9 or acetylated K9 were incubated with HeLa nuclear extracts and washed with low stringency buffers to enhance the isolation of low affinity binding proteins. The resulting proteins were resolved by SDS-PAGE for protein identification and quantification.
Figure 2.

SDS-PAGE of binders isolated in the H3 pull-downs. (A) Biotinylated H3 peptides containing amino acids 1–21 and the indicated modifications (me2 = dimethylation; ac = acetylation) were used to isolate binders from HeLa nuclear extracts. The isolated proteins were analyzed by SDS-PAGE separation and stained with CBB. (B) The unmodified H3 peptide pull-down is highlighted to show the identity of the major proteins bound to this peptide.
3.2 Identification and quantification of proteins bound to modified histone H3 peptides
The 40 tryptic peptide samples were analyzed individually by HPLC-MS/MS on an LTQ mass spectrometer. To assess the extent of nonspecific binding, we also analyzed the streptavidin beads control pull-down. The majority of proteins that bound to the control agarose beads consisted mainly of common and abundant nonspecific proteins that we often identify in immunoprecipitation assays (see Table 1 of Supporting Information). All proteins identified in the four H3 peptide pull-downs are listed in Table 1.
Table 1.
Proteins identified each pull-down
| Ordera) | HGNC symbol | GeneID | Controlb) | H3b) | K4me2b) | K9me2b) | K9acb) | Description | Known interactionc) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | MYST2 | 11143 | 0 | 6 | 8 | 7 | 2 | C2HC type zinc finger, acetyltransferase domain | No |
| 2 | WDR61 | 80349 | 0 | 8 | 28 | 27 | 17 | WD40 domain | No |
| 3 | UBTF | 7343 | 0 | 11 | 36 | 34 | 5 | HMG box | No |
| 4 | LOC650865 | 650865 | 0 | 1 | 22 | 17 | 0 | Similar to SET, subunit of INHAT complex | Yes, direct binder to H3 |
| 5 | SET | 6418 | 6 | 6 | 126 | 124 | 45 | Subunit of INHAT complex | Yes, direct binder to H3 |
| 6 | WDR5 | 11091 | 0 | 14 | 94 | 65 | 25 | WD40 domain | Yes, direct binder to K4me2 |
| 7 | PAF1 | 54623 | 5 | 15 | 52 | 34 | 17 | Transcription elongation | Yes, direct binder to K4me2 |
| 8 | CDC73 | 79577 | 0 | 18 | 52 | 21 | 7 | Component of PAF1 complex | Yes, via interactions with PAF1 |
| 9 | CDK9 | 1025 | 0 | 0 | 3 | 2 | 0 | Protein kinase, phoshporylate Pol II CTD | No |
| 10 | PHF2 | 5253 | 0 | 0 | 3 | 0 | 0 | PHD finger and JmjC domains | No |
| 11 | RANBP5 | 3843 | 0 | 0 | 4 | 0 | 1 | No | |
| 12 | SIN3A | 25942 | 0 | 0 | 5 | 0 | 0 | HDAC1 complex | Yes, via interactions with HDAC1-NuRD complex |
| 13 | PHF8 | 23133 | 0 | 0 | 6 | 0 | 0 | PHD finger and JmjC domains | No |
| 14 | RRP12 | 23223 | 0 | 0 | 4 | 2 | 2 | No | |
| 15 | C1orf149 | 64769 | 0 | 0 | 6 | 0 | 2 | No | |
| 16 | HTATSF1 | 27336 | 0 | 0 | 5 | 1 | 3 | No | |
| 17 | LEO1 | 123169 | 0 | 6 | 26 | 8 | 9 | Component of PAF1 complex | Yes, via interactions with PAF1 |
| 18 | CTR9 | 9646 | 0 | 9 | 32 | 12 | 18 | Component of PAF1 complex | Yes, via interactions with PAF1 |
| 19 | SENP3 | 26168 | 0 | 0 | 4 | 0 | 0 | Peptidase domain | No |
| 20 | CSDA | 8531 | 0 | 0 | 3 | 0 | 0 | Cold shock domain | No |
| 21 | CCDC86 | 79080 | 0 | 0 | 4 | 0 | 0 | No | |
| 22 | PHF16 | 9767 | 0 | 0 | 2 | 0 | 0 | PHD finger domain | No |
| 23 | PHF12 | 57649 | 0 | 0 | 2 | 0 | 0 | PHD finger and FHA domain | No |
| 24 | RUVBL1 | 8607 | 0 | 0 | 3 | 0 | 0 | No | |
| 25 | IWS1 | 55677 | 0 | 0 | 8 | 0 | 0 | No | |
| 26 | PHF23 | 79142 | 0 | 0 | 10 | 0 | 0 | PHD finger domain | No |
| 27 | DKC1 | 1736 | 0 | 0 | 8 | 0 | 0 | No | |
| 28 | TRIM26 | 7726 | 0 | 2 | 9 | 3 | 2 | Zinc-binding and RING domains | No |
| 29 | LOC144097 | 144097 | 0 | 0 | 7 | 0 | 0 | No | |
| 30 | SPIN1 | 10927 | 0 | 0 | 10 | 0 | 0 | No | |
| 31 | CHD1 | 1105 | 0 | 0 | 29 | 0 | 7 | Chromodomain | Yes, direct binder to H3K4me2 |
| 32 | RUVBL2 | 10856 | 0 | 1 | 5 | 3 | 2 | No | |
| 33 | PELP1 | 27043 | 0 | 0 | 6 | 1 | 0 | No | |
| 34 | RBBP5 | 5929 | 0 | 3 | 28 | 12 | 3 | WD40 domain, complex with WDR5 | Yes, via interactions with WDR5 |
| 35 | TEX10 | 54881 | 0 | 2 | 7 | 1 | 0 | No | |
| 36 | CBX1 | 10951 | 0 | 5 | 2 | 25 | 1 | HP1β homolog, chromodomain | Yes, direct H3K9me2 binder |
| 37 | CHAF1B | 8208 | 2 | 1 | 0 | 42 | 2 | WD40 domain, CAF1 complex | Yes, via interactions with HP1 |
| 38 | CBX5 | 23468 | 0 | 14 | 2 | 188 | 22 | HP1α homolog, chromodomain | Yes, direct H3K9me2 binder |
| 39 | YAF2 | 10138 | 0 | 0 | 0 | 2 | 0 | Zinc finger domain | No |
| 40 | LOC653972 | 653972 | 2 | 259 | 91 | 416 | 225 | HP1γ homolog, Chromodomain | Yes, direct H3K9me2 binder |
| 41 | DSN1 | 79980 | 0 | 0 | 0 | 4 | 0 | Component of kinetochore complex | No |
| 42 | TRIM28 | 10155 | 21 | 4 | 2 | 47 | 0 | Zinc finger, bromo-, and PHD finger domains | Yes, via interactions with HP1 |
| 43 | SUZ12 | 23512 | 0 | 0 | 0 | 2 | 0 | No | |
| 44 | DPF2 | 5977 | 0 | 0 | 0 | 2 | 0 | Two PHD finger domains | No |
| 45 | SSRP1 | 6749 | 0 | 0 | 0 | 3 | 0 | Component of FACT complex | No |
| 46 | UHRF2 | 115426 | 0 | 0 | 0 | 6 | 0 | PHD finger and RING domains | No |
| 47 | ANP32B | 10541 | 0 | 0 | 0 | 10 | 0 | Subunit of INHAT complex | Yes, via interactions with SET |
| 48 | ANP32A | 8125 | 0 | 0 | 0 | 6 | 0 | Subunit of INHAT complex | Yes, via interactions with SET |
| 49 | MECP2 | 4204 | 0 | 60 | 67 | 16 | 79 | MBD domain | No |
| 50 | UHRF1 | 29128 | 7 | 5 | 1 | 0 | 9 | PHD finger and RING domains | No |
| 51 | SMCHD1 | 23347 | 0 | 2 | 0 | 0 | 5 | SMC proteins flexible hinge domain | No |
| 52 | BAZ1B | 9031 | 1 | 1 | 0 | 5 | 18 | PHD finger and bromodomains | No |
| 53 | CHAF1A | 10036 | 3 | 0 | 0 | 5 | 53 | Component of CAF1 complex | Yes, via interactions with HP1 |
| 54 | MTA3 | 57504 | 1 | 38 | 3 | 20 | 49 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 55 | GATAD2B | 57459 | 5 | 181 | 15 | 84 | 245 | Component of MeCP1 complex | Yes, component of NuRD/MeCP1 complex |
| 56 | GATAD2A | 54815 | 7 | 98 | 9 | 50 | 185 | Component of MeCP1 complex | Yes, component of NuRD/MeCP1 complex |
| 57 | HMG20A | 10363 | 0 | 8 | 1 | 7 | 7 | HMG box | No |
| 58 | RBBP4 | 5928 | 2 | 41 | 1 | 25 | 38 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 59 | HDAC2 | 3066 | 1 | 119 | 4 | 23 | 105 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 60 | MBD2 | 8932 | 0 | 57 | 2 | 14 | 42 | Component of MeCP1 complex | Yes, component of NuRD/MeCP1 complex |
| 61 | HDAC1 | 3065 | 0 | 63 | 4 | 20 | 51 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 62 | MTA1 | 9112 | 0 | 97 | 5 | 30 | 65 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 63 | MTA2 | 9219 | 2 | 345 | 12 | 143 | 214 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 64 | MBD3 | 53615 | 0 | 204 | 7 | 116 | 170 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 65 | RBBP7 | 5931 | 4 | 218 | 17 | 125 | 166 | component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 66 | LOC648695 | 648695 | 0 | 11 | 0 | 4 | 8 | Similar to RBBP4 | Yes, component of NuRD/MeCP1 complex |
| 67 | PHF14 | 9678 | 0 | 11 | 0 | 9 | 4 | PHD finger domains | No |
| 68 | MST101 | 114825 | 0 | 8 | 1 | 7 | 0 | No | |
| 69 | ERCC6 | 2074 | 0 | 11 | 0 | 11 | 2 | DNA helicase domain | Yes, direct binder to H3 |
| 70 | KBTBD6 | 89890 | 0 | 3 | 0 | 3 | 1 | POZ domain | No |
| 71 | CHD3 | 1107 | 29 | 42 | 4 | 53 | 30 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 72 | HAT1 | 8520 | 4 | 10 | 0 | 17 | 7 | Acetyl transferase | No |
| 73 | RAI1 | 10743 | 2 | 8 | 0 | 9 | 6 | Predicted PHD finger domain | no |
| 74 | TRIM33 | 51592 | 0 | 7 | 0 | 8 | 4 | PHD finger and bromodomains | no |
| 75 | CHD4 | 1108 | 55 | 93 | 10 | 101 | 0 | Component of NuRD complex | Yes, component of NuRD/MeCP1 complex |
| 76 | CDK2AP2 | 10263 | 0 | 12 | 0 | 0 | 0 | No | |
| 77 | HIST1H2BO | 8348 | 0 | 12 | 2 | 0 | 0 | Histone H2B member | No |
| 78 | BAZ2A | 11176 | 0 | 6 | 0 | 0 | 0 | PHD finger and bromodomains | No |
| 79 | NIPBL | 25836 | 0 | 0 | 0 | 0 | 5 | No | |
| 80 | POGZ | 23126 | 2 | 6 | 0 | 0 | 16 | No | |
| 81 | C13orf8 | 283489 | 0 | 0 | 0 | 0 | 18 | No | |
| 82 | ADNP | 23394 | 0 | 0 | 0 | 6 | 25 | Homeodomain | No |
| 83 | WIZ | 58525 | 0 | 8 | 5 | 2 | 8 | No | |
| 84 | EHMT2 | 10919 | 2 | 5 | 7 | 0 | 7 | Histone lysine-methyltransferase | No |
| 85 | HIST1H1C | 3006 | 0 | 10 | 7 | 8 | 7 | Histone H1 family | No |
| 86 | EBNA1BP2 | 10969 | 0 | 7 | 4 | 3 | 2 | No |
The proteins are listed in the same order as in Fig. 3.
The numbers correspond to the number of MS/MS spectra of the protein that was detected in the entire experiment.
This column refers to whether the protein is known to bind directly or associate with direct binders to the unmodified or modified H3 amino-terminal tail.
It has been demonstrated that stable isotope labeling with amino acids in cell culture (SILAC) in combination with high-resolution MS can provide an accurate method for quantifying the relative amount of each protein identified in the pull-downs [25–27]. Indeed, such an approach has been used to identify histone modification-specific binding proteins and protein interaction partners [28, 29]. However, a major limitation of the isotopic amino acid labeling method is the limited number of samples that can be used for relative quantification, which requires pair wise comparisons. Therefore, it is not convenient to use such methods to assess the binding of proteins in the five pull-downs conducted in this study. Instead, we opted for spectral counting as described by Yates and coworkers [23] which serves as a simple and convenient method to estimate the abundance of each protein. We combined the results of the eight MS analyses for each pull-down and the total number of spectra were tallied, Table 1.
To compare and contrast the composition of proteins pulled down by the various modified histone H3 peptides, we used hierarchical clustering to analyze and heat maps to visualize the spectral counts as simple estimates of the relative abundance of each protein (Fig. 3) [24, 30]. As expected, the streptavidin beads pull-down contained the least amount of proteins overall. In a few instances, a significant number of peptides for a protein were detected in the control pull-down, such as in CHD3, CHD4, and TRIM28; however, the marked increase in the peptide spectral counts in the H3 pull-downs suggests that these proteins likely also bound to H3 or to the modified H3 sites (Table 1). In all, we identified 86 distinct proteins that preferentially bound to at least one of the four H3 peptides examined relative to the control beads. Of these 86 proteins, 65 were not recovered in the control beads pull-down; for the 21 proteins that were detected in the control pull-down, they were significantly enriched with greater than 70% increase in the number of peptides identified in at least one of the H3 pull-downs (Table 1).
Figure 3.

Cluster analysis of protein abundance among modified H3 pull-downs. For each protein, spectral counts from Table 1 are scaled to Z-scores, calculated across the various H3 modifications and control beads listed below. As shown in the legend, red indicates abundance that is above average among the eight pull-downs; green indicates below-average abundance, with various shades indicating intermediary abundance levels. To highlight proteins present at similar levels in the same samples, Z-scores for each protein were clustered along the horizontal axis.
3.3 Recovery of known H3 tail binding partners
If our approach captured bona fide interactions, we would expect to identify known binders to the H3 peptides used in this study. Indeed, our screen identified seven proteins previously shown to recognize H3 tail directly including known readers of H3K4me2, H3K9me2, and unmodified H3 tail (Tables 2 and 3). The major protein complex isolated with the unmodified H3 peptide is the NuRD/MeCP1 protein complex (Fig. 2B and Table 2), as previously reported [31–37]. In addition, we also recovered known interacting proteins of these direct effectors, suggesting that a subset of the detected interactions is indirect. These data are discussed below in more detail (Section 3.4). Remarkably, 33 proteins (more than one-third of the proteins identified in our screen) were previously reported to bind directly or associate with direct binders to the H3 tails examined. These results support the validity of our approach and strongly suggest that the remaining 55 proteins are relevant, novel binding partners that directly or indirectly associate with the H3 peptides examined in this study.
Table 2.
Known H3 binding proteins identified
| HGNC symbol | GeneID | Alternative name | Description | Reference |
|---|---|---|---|---|
| MTA3 | 57504 | Component of NuRD complex | [33] | |
| GATAD2B | 57459 | p66β | Component of MeCP1 complex | [31] |
| GATAD2A | 54815 | p66α | Component of MeCP1 complex | [31] |
| RBBP4 | 5928 | RbAp48 | Component of NuRD complex | [36] |
| HDAC2 | 3066 | Component of NuRD complex | [36] | |
| MBD2 | 8932 | Component of MeCP1 complex | [32] | |
| HDAC1 | 3065 | Component of NuRD complex | [36] | |
| MTA1 | 9112 | Component of NuRD complex | [35] | |
| MTA2 | 9219 | Component of NuRD complex | [36] | |
| MBD3 | 53615 | Component of NuRD complex | [37] | |
| RBBP7 | 5931 | RbAp46 | Component of NuRD complex | [36] |
| CHD3 | 1107 | Mi2-α | Component of NuRD complex | [34] |
| CHD4 | 1108 | Mi2-β | Component of NuRD complex | [36] |
| ERCC6 | 2074 | CSB | NER repair protein | [50] |
Table 3.
Summary of direct and putative direct binders to the H3 peptides identified
| Protein | Domain(s) | Binding site(s) | Reference |
|---|---|---|---|
| Known binders | |||
| CBX5 (HP1α) | Chromodomain | K9me2/me3 | [15] |
| CBX1 (HP1β) | Chromodomain | K9me2/me3 | [15] |
| LOC653972 (HP1γ) | Chromodomain | K9me2/me3 | [15] |
| CHD1 | Chromodomain | K4me2/me3 | [16] |
| WDR5 | WD40 domain | K4me2 | [40] |
| ERCC6 | DNA helicase domain | unmodified H3 | [50] |
| SET | WD40 domain | K9me2 or K9me3 | [49] |
| Putative binders | |||
| PHF2 | PHD finger and JmjC | K4me2 | This study |
| PHF8 | PHD finger and JmjC | K4me2 | This study |
| PHF12 | PHD finger | K4me2 | This study |
| PHF16 | PHD finger | K4me2 | This study |
| PHF14 | PHD finger | K9me2, unmethylated at K4 | This study |
| PHF23 | PHD finger | K4me2 | This study |
| UHRF2 | PHD finger | K4me2 | This study |
| TRIM28 | PHD finger and bromodomain | K9me2 | This study |
| RAI1 | PHD finger | H3 unmethylated at K4 | This study |
| TRIM33 | PHD finger and bromodomain | H3 unmethylated at K4 | This study |
| BAZ1B | PHD finger and bromodomain | K9ac | This study |
| BAZ2A | PHD finger and bromodomain | H3 | This study |
| DPF2 | PHD finger | K9me2 | This study |
| UHRF1 | PHD finger and RING domain | K9me2 | This study |
| WDR61 | WD40 | K9me2 and K4me2 | This study |
| CHAF1B | WD40 | K9me2 | This study |
3.4 Known effectors of H3K9me2 and H3K4me2 identified
The three isoforms of HP1 (α, β, and γ), which bind specifically to H3K9me2, were identified in our screen. The chromodomain of HP1 mediates the interaction with H3 di- and trimethylated at K9 for gene silencing and maintenance of heterochromatin. Two of the HP1 isoforms (α and β) displayed strong binding preference for H3K9me2 as previously reported [15], whereas the HP1γ homolog (LOC653972) also bound to unmodified H3 and H3K9ac to a lesser degree. Accordingly, our screen identified proteins known to associate with HP1, including a component of the CAF1 complex CHAF1B [38], and corepressor protein TRIM28/KAP-1 [39]. CHAF1B and TRIM28/KAP-1 exhibited preferential binding to the H3K9me2 peptide, suggesting that the association is HP1-dependent. It is also noteworthy that TRIM28/KAP-1 contains a PHD finger that may contribute to its specificity of binding to H3K9me2.
WDR5 was identified to preferentially bind to H3K4me2 using a similar peptide pull-down assay [40]. In agreement with this result, WDR5 was significantly enriched in the H3K4me2 pull-down in comparison to the unmodified H3 tail. WDR5 contains a WD40 domain and is a common component of the MLL family of H3K4 methyltransferase complexes. WDR5 associate with RbBP5, ASH2 and a SET domain protein to form the “core complex” that is sufficient for methylation on nucleosomal substrates [41]. RbBP5, which is known to tightly associate with WDR5, was also recovered in our study, and although less abundant in terms of peptide recovery, it exhibited binding specificity closely mimicking that of WDR5. Thus, it is likely that RbBP5 binding to H3K4me2 peptide is via interactions with WDR5.
CHD1 is an ATP-dependent chromatin remodeling protein that binds to H3 tails containing di- or trimethylated K4 via its tandem chromodomains [42]. We confirmed CHD1 interaction with H3K4me2 in our peptide pull-downs (Tables 1 and 3). We also detected weak binding of CHD1 to the H3K9ac peptide, although the biological significance of this interaction is not clear.
Interestingly, we also identified multiple subunits of PAF1 elongation complex: PAF1, LEO1, CTR9, CDC73 that preferentially associated with H3K4me2 tail, although binding to other H3 peptides was also observed. PAF1 promotes transcriptional elongation and regulates methylation of H3 at K4. Preference for H3K4me2 may be explained by the previously reported association of the PAF1 complex with CHD1 and H3K4 methyltransferase core complex [43, 44]. Alternatively, one or more PAF1 components can directly bind to the histone tail.
Finally, we wish to comment on the lack of recovery of BPTF, ING2, and TAF3, three proteins that have been reported to recognize methylated H3K4 via PHD finger motif. The PHD fingers of these proteins show a higher preference for binding to trimethyllysines over dimethyllysines in pull-down assays, which likely accounts for their absence in our H3K4me2 pull-down [45, 46]. In future studies, we will perform similar proteomic screen to search for novel proteins associated with specific mono-, and trimethyllysines, which will expand our knowledge on how the different methylation states may be distinguished by the downstream effectors.
3.5 Known binders discriminating against H3K4me2 identified
Histone modifications can exert their function not only via recruitment of specific readers, but also through exclusion of certain effector molecules from the histone tails. One example includes the NuRD complex, whose binding to the H3 tail is perturbed by methylation at K4 [47, 48]. In agreement with these reports, our results indicate that the binding of multiple components of the NuRD complex, and the related MeCP1 complex, are dramatically reduced in the H3K4me2 pull-down, however to a much lesser degree, H3K9me2 also reduced binding of this complex (Fig. 2A and Table 1).
3.6 Other identified proteins previously implicated in association with H3 tail
Our study also identified two proteins previously reported to directly bind H3, albeit without clearly defined modification-dependent specificity. The histone chaperone SET/TAF-1β was shown to directly recognize unmodified and methylated H3 tails [49]. However in our assay, SET/TIF-1β does not significantly bind to the unmodified H3 tail, but instead strongly associates with H3K4me2, H3K9me2 and, to a lesser extent, with H3K9Ac.
The second protein, ERCC6, is an ATP-dependent chromatin remodeler involved in DNA excision repair and was previously reported to bind histone tails [50]. In our assay ERCC6 exhibited the highest spectral counts to the unmodified H3 and H3K9me2 peptides; however binding to H3K4me2 was not detectable, whereas binding to the H3K9Ac peptide was very weak, Table 1.
In sum, our screen identified the NuRD/MeCP1 complex and seven proteins (HP1α, HP1β, HP1γ, CHD1, WDR5, PAF1, SET/TAF-1β, and ERCC6) which were previously shown to directly associate with modified or unmodified H3 tail. These direct binders and their associated proteins constituted 33 of the 86 proteins identified in these four peptide pull-down assays. Importantly, the protein partners that associate with the direct binders displayed similar binding preference to that of the direct binders. Notably however, not all protein partners of known binders had analogous association patterns. For example, ANP32A and ANP32B, which associate with SET/TIF-1β in the INHAT complex, appear to bind specifically to H3K9me2, whereas SET/TIF-1β recognizes both methylated peptides equally well. These data indicate that different complexes formed by a given protein may exhibit different histone-binding properties.
Taken together, results discussed above (section 3.3–3.6) demonstrate not only the feasibility of the approach undertaken in this study, but also suggest that the majority of the remaining 53 identified proteins represents novel direct and indirect H3 tail binders.
3.7 Identification of novel binders to modified H3 lysines
Consistent with the role of K4 and K9 methylation in gene activation and silencing, respectively, many of the identified K4 binders are involved in transcriptional activation, whereas K9 binders are involved in transcriptional repression. It is likely that many of the detected proteins associate with H3 indirectly. However, it is striking that 13 of the identified novel binders contain PHD finger domain, a structural fold implicated in specific methyllysine recognition on H3 tail, and more recently, in recognizing H3 tail with an unmethylated K4 [51]. Thus, many of the detected PHD finger proteins may in fact represent direct H3 binders. Five novel PHD finger binders associated specifically with H3K4me2 peptide (PHF2, PHF8, PHF12, PHF16, and PHF23) and two with H3K9me2 peptide (DPF2 and UHRF2) (Table 3). Another PHD finger protein, TRIM28/KAP-1 also preferentially bound to H3K9me2; however, as discussed above (section 3.4), this interaction may be mediated by its association with HP1. Additionally, three identified PHD finger proteins discriminated against methylation at K4 (PHF14, RAI1, and TRIM33) in manner similar to the PHD finger of BHC80 [51], and one recognized only the unmodified tail (BAZ2A). Although the identification of these PHD finger proteins was based on a low number of peptides identified for each of these proteins (Table 1), we were able to confirm the binding of PHF2 to H3K4me2 by western blotting analysis of H3 peptide pull-downs containing various methylation states at K4 and K9, Fig. 4.
Figure 4.
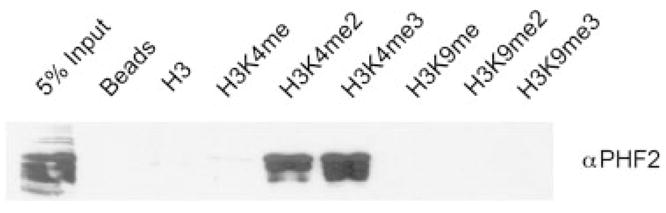
Confirmatory binding of PHF2 to H3K4me2. Western blot for PHF2 in H3 peptide pull-downs from HeLa nuclear extracts with the various methylations states in K4 and K9.
Two of the identified H3K4me2 binding proteins (PHF2 and PHF8) contained a PHD finger and a JmjC domain, a conserved histone demethylase domain, Table 3. If association of PHF2 and PHF8 with H3K4me2 indeed occurs directly via the PHD finger, it would provide a first example of a JmjC domain protein that specifically recognize this site.
PHD fingers and bromodomains are often present and adjacent to each other in many chromatin-associated proteins. These binding modules have been shown to functionally cooperate, possibly through the combinatorial reading of methyl–acetyl modification patterns [52]. Four of the identified PHD finger proteins also contain a bromodomain (TRIM28/KAP-1, BAZ1B, BAZ2A, and TRIM33), but only BAZ1B preferentially bound to the H3K9ac peptide. However, given the very limited set of modifications we used in this study, our results cannot preclude the possibility of combinatorial methyl–acetyl reading by these factors.
WD40 is another domain implicated in the recognition of the methylated proteins. WDR61 was prominently detected in both the H3K4me2 and H3K9me2 pull-downs (Tables 1 and 3), with weaker association with the unmodified and acetylated peptides. This binding pattern is similar to that of WDR5, although the number of WDR61 peptides detected in the H3K4me2 and the H3K9me2 pull-downs were comparable, suggesting that the WD40 domain of this protein may not have the ability to discriminate between these two methylated lysines. We are currently investigating a possible mechanism of methylated H3 recognition by WDR61.
4 Discussion
In this study, we have identified 86 proteins that bind to his-tone H3 amino-terminal peptides, including H3 tail peptides with dimethylated K4 and K9, and acetylated K9. Given the importance of H3K4 and H3K9 methylation in gene expression regulation, the identification and catalog of effector protein complexes recognizing these marks is an important first step in understanding the molecular mechanisms of epigenetic regulation. To our knowledge, this report is the most comprehensive study to date on the identification of proteins bound to these commonly modified residues on histone H3.
We used the number of tryptic peptides identified for each of the 86 proteins in the pull-down as a rough estimate of the relative amount of proteins identified. While many variables will affect the number of peptides that can be identified for each protein, such as protein abundance, efficiency of protein digestion and peptide extraction, and how well the peptide “fly” in the mass analyzer, there is however a general correlation between protein abundance and the number of spectra identified [23]. It is important to emphasize that spectral counting is not truly quantitative, though it does provide a useful approximation of the relative amounts of each protein identified in the four pull-downs. For proteins that are identified based on large number of spectra, the relative difference in protein abundance more closely correlates with the difference in the number spectra identified in each pull-down. For proteins that are identified with very low number of peptides, it is more difficult to draw meaningful conclusions about how significant the difference in the spectra number correlates with relative difference in protein abundance between different pull-downs. We caution that for all the new binders identified in this study, further biochemical validations are warranted. Despite these limitations, for proteins that were identified based on a very low number of peptides, such as PHF2, we were able to confirm its specific interaction with H3K4me2 by Western blotting analysis (Fig. 4) thus at least for the PHF2, spectral counting as a means of semi-quantification is valid.
Two previous studies that used a similar peptide pull-down approach to identify novel binders to H3K4me2 and H3K4me3 readers in 293 nuclear extracts lead to the identification of WDR5 and BPTF, respectively [40, 46]. In this study, in addition to WDR5, many more H3K4me2 associated proteins were identified possibly because we further optimized the experimental procedure, used less stringent wash conditions and performed a more detailed mass spec analysis of the recovered proteins (see Section 2 for details). A significant advantage of this approach over the candidate approach with recombinant candidate proteins is that novel effectors are discovered in an unbiased manner. However, a resulting caveat is that we are unable to distinguish direct binding to the modified lysine from indirect interactions. Confirmation of direct binding requires further downstream biochemical characterization, such as in vitro binding assays and in vivo co-localization studies. Nevertheless, the over-representation of proteins containing methyllysine binding modules among the newly identified binders, as well as high recovery of the previously characterized H3K4me2 and H3K9me2 methylation effectors in our screen suggests that this study will significantly expand the spectrum of histone PTMs effector proteins.
The diversity of proteins identified in our screen, many of which in a modification-dependent manner, will provide a rich source of biological leads and provide a stepping stone for future studies.
Supplementary Material
Acknowledgments
We thank Joanna Wysocka and Eric Chan for critical comments on the manuscript. J.W. is partially supported by the Research Platform of Cell Signaling Networks from the Science and Technology Commission of Shanghai Municipality. This work was supported by National Institutes of Health Grant 1R43CA132680-01 to D.W.C.
Abbreviation
- NPV
nuclear pellet volume
Footnotes
The authors have declared no conflict of interest.
5 References
- 1.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Sprung R, Tang Y, Ball H, et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hake SB, Xiao A, Allis CD. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br J Cancer. 2007;96:R31–R39. [PubMed] [Google Scholar]
- 5.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 6.Johnson L, Mollah S, Garcia BA, Muratore TL, et al. Mass spectrometry analysis of Arabidopsis histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Res. 2004;32:6511–6518. doi: 10.1093/nar/gkh992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu F, Nusinow DA, Chalkley RJ, Plath K, et al. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol Cell Proteomics. 2006;5:194–203. doi: 10.1074/mcp.M500285-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Wisniewski JR, Zougman A, Kruger S, Mann M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics. 2007;6:72–87. doi: 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Taverna SD, Ueberheide BM, Liu Y, Tackett AJ, et al. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc Natl Acad Sci USA. 2007;104:2086–2091. doi: 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 11.Beck HC, Nielsen EC, Matthiesen R, Jensen LH, et al. Quantitative proteomic analysis of post-translational modifications of human histones. Mol Cell Proteomics. 2006;5:1314–1325. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Medzihradszky KF, Zhang X, Chalkley RJ, Guan S, et al. Characterization of Tetrahymena histone H2B variants and posttranslational populations by electron capture dissociation (ECD) Fourier transform ion cyclotron mass spectrometry (FT-ICR MS) Mol Cell Proteomics. 2004;3:872–886. doi: 10.1074/mcp.M400041-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Su X, Zhang L, Lucas DM, Davis ME, et al. Histone H4 acetylation dynamics determined by stable isotope labeling with amino acids in cell culture and mass spectrometry. Anal Biochem. 2007;363:22–34. doi: 10.1016/j.ab.2006.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung WL, Briggs SD, Allis CD. Acetylation and chromosomal functions. Curr Opin Cell Biol. 2000;12:326–333. doi: 10.1016/s0955-0674(00)00096-x. [DOI] [PubMed] [Google Scholar]
- 15.Bannister AJ, Zegerman P, Partridge JF, Miska EA, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 16.Sims RJ, III, Reinberg D. Histone H3 Lys 4 methylation: caught in a bind? Genes Dev. 2006;20:2779–2786. doi: 10.1101/gad.1468206. [DOI] [PubMed] [Google Scholar]
- 17.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhalluin C, Carlson JE, Zeng L, He C, et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 19.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung SY, Malovannaya A, Wei J, O’Malley BW, Qin J. Proteomic analysis of steady-state nuclear hormone receptor coactivator complexes. Mol Endocrinol. 2005;19:2451–2465. doi: 10.1210/me.2004-0476. [DOI] [PubMed] [Google Scholar]
- 22.Daquinag A, Fadri M, Jung SY, Qin J, Kunz J. The yeast PH domain proteins Slm1 and Slm2 are targets of sphingolipid signaling during the response to heat stress. Mol Cell Biol. 2007;27:633–650. doi: 10.1128/MCB.00461-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Sadygov RG, Yates JR., III A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 24.Manes NP, Gustin JK, Rue J, Mottaz HM, et al. Targeted protein degradation by Salmonella under phagosome-mimicking culture conditions investigated using comparative peptidomics. Mol Cell Proteomics. 2007;6:717–727. doi: 10.1074/mcp.M600282-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Chen X, Fei Z, Smith LM, Bradbury EM, Majidi V. Stable-isotope-assisted MALDI-TOF mass spectrometry for accurate determination of nucleotide compositions of PCR products. Anal Chem. 1999;71:3118–3125. doi: 10.1021/ac9812680. [DOI] [PubMed] [Google Scholar]
- 26.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci USA. 1999;96:6591–6596. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 28.Taverna SD, Ilin S, Rogers RS, Tanny JC, et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Cox B, Kislinger T, Wigle DA, Kannan A, et al. Integrated proteomic and transcriptomic profiling of mouse lung development and Nmyc target genes. Mol Syst Biol. 2007;3:109. doi: 10.1038/msb4100151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng Q, Cao R, Xia L, Erdjument-Bromage H, et al. Identification and functional characterization of the p66/p68 components of the MeCP1 complex. Mol Cell Biol. 2002;22:536–546. doi: 10.1128/MCB.22.2.536-546.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng Q, Zhang Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 2001;15:827–832. doi: 10.1101/gad.876201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujita N, Jaye DL, Kajita M, Geigerman C, et al. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–219. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 34.Wade PA, Gegonne A, Jones PL, Ballestar E, et al. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23:62–66. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 35.Xue Y, Wong J, Moreno GT, Young MK, et al. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell. 1998;95:279–289. doi: 10.1016/s0092-8674(00)81758-4. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, et al. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quivy JP, Roche D, Kirschner D, Tagami H, et al. A CAF-1 dependent pool of HP1 during heterochromatin duplication. EMBO J. 2004;23:3516–3526. doi: 10.1038/sj.emboj.7600362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan RF, Schultz DC, Ayyanathan K, Singh PB, et al. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol. 1999;19:4366–4378. doi: 10.1128/mcb.19.6.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wysocka J, Swigut T, Milne TA, Dou Y, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 41.Dou Y, Milne TA, Ruthenburg AJ, Lee S, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 42.Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Khorasanizadeh S, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 43.Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, et al. The parafibromin tumor suppressor protein is part of a human Paf1 complex. Mol Cell Biol. 2005;25:612–620. doi: 10.1128/MCB.25.2.612-620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sims RJ, III, Millhouse S, Chen CF, Lewis BA, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and premRNA splicing. Mol Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi X, Hong T, Walter KL, Ewalt M, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wysocka J, Swigut T, Xiao H, Milne TA, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 47.Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, et al. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zegerman P, Canas B, Pappin D, Kouzarides T. Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J Biol Chem. 2002;277:11621–11624. doi: 10.1074/jbc.C200045200. [DOI] [PubMed] [Google Scholar]
- 49.Schneider R, Bannister AJ, Weise C, Kouzarides T. Direct binding of INHAT to H3 tails disrupted by modifications. J Biol Chem. 2004;279:23859–23862. doi: 10.1074/jbc.C400151200. [DOI] [PubMed] [Google Scholar]
- 50.Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, et al. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. 2000;20:7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lan F, Collins RE, De Cegli R, Alpatov R, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peng J, Wysocka J. It takes a PHD to SUMO. Trends Biochem Sci. 2008;33:191–194. doi: 10.1016/j.tibs.2008.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.