

Abstract
Treatment of neuropathic pain is a major clinical challenge that has been met with minimal success. After peripheral nerve injury, a decrease in the expression of the K–Cl cotransporter KCC2, a major neuronal Cl− extruder, leads to pathologic alterations in GABAA and glycine receptor function in the spinal cord. The down-regulation of KCC2 is expected to cause a reduction in Cl− extrusion capacity in dorsal horn neurons, which, together with the depolarizing efflux of anions via GABAA channels, would result in a decrease in the efficacy of GABAA-mediated inhibition. Carbonic anhydrases (CA) facilitate intracellular generation and hence, we hypothesized that inhibition of CAs would enhance the efficacy of GABAergic inhibition in the context of neuropathic pain. Despite the decrease in KCC2 expression, spinal administration of benzodiazepines has been shown to be anti-allodynic in neuropathic conditions. Thus, we also hypothesized that spinal inhibition of CAs might enhance the anti-allodynic effects of spinally administered benzodiazepines. Here, we show that inhibition of spinal CA activity with acetazolamide (ACT) reduces neuropathic allodynia. Moreover, we demonstrate that spinal co-administration of ACT and midazolam (MZL) act synergistically to reduce neuropathic allodynia after peripheral nerve injury. These findings indicate that the combined use of CA inhibitors and benzodiazepines may be effective in the clinical management of neuropathic pain in humans.
Keywords: Neuropathic pain, GABA, Benzodiazepine, Carbonic anhydrase, Spinal cord
1. Introduction
Neuropathic pain is a major clinical problem that, despite increasing basic science understanding of the disorder, is still poorly managed by available therapeutics. Recent preclinical work in neuropathic pain models suggests that peripheral nerve injury (PNI) results in a pathology of the spinal GABAergic system. This may include decreases in spinal dorsal horn GABAergic interneurons [24], decreases in GABA release [24] (but see [27,34]) or alterations in GABAA and glycine receptor reversal potentials [9,10,28] attributed to a decrease in K+–Cl− cotransporter type 2 (KCC2) expression in spinal dorsal horn neurons [10]. A possible mechanism to reverse these effects is through treatment with benzodiazepines [20,21,41] which augment spinal GABAergic neurotransmission postsynaptically. This notion is supported by the efficacy of spinally applied benzodiazepines for chronic pain conditions in humans (for review see [12]); and their efficacy in preclinical pain models [20,21].
While spinally administered benzodiazepines are clearly antiallodynic in neuropathic conditions [20] it is also true that reduced KCC2 expression after peripheral nerve injury causes a decrease in the efficacy of inhibition mediated by GABAA and glycine receptors in a subset of dorsal horn neurons [10,28]. This reduction of KCC2 expression would reduce the Cl− extrusion capacity of dorsal horn neurons leading to a reduction of Cl− mediated hyperpolarization through GABAA and glycine channels. Under physiological conditions, GABAA receptor channels mediate substantial currents carried not only by Cl− but also by [15,16] as do glycine receptors [4]. Hence, a reduced Cl− extrusion capacity will favor the depolarizing channel-mediated efflux of , especially in neurons equipped with cytoplasmic carbonic anhydrase (CA). Because this enzyme is able to quickly replenish intracellular during the channel-mediated net efflux, the -dependent depolarization can become large enough to result in GABAergic or glycinergic excitation during ongoing neuronal network activity, as previously demonstrated in the adult rat hippocampus [33,38]. Notably, synchronous network activity in the dorsal horn of the spinal cord is enhanced after PNI [35]. Therefore, inhibition of CA after PNI may be expected to reduce -mediated excitatory GABAergic and/or glycinergic currents in the context of neuropathic pain.
Using a model of PNI based on spinal nerve ligation (SNL) [19], we have addressed two questions: (1) does inhibition of spinal CA activity reduce neuropathic allodynia? and (2) can inhibition of spinal CA augment the anti-allodynic activity of spinally administered benzodiazepines? Our results show that acetazolamide (ACT), a widely used CA inhibitor [37], reduces neuropathic allodynia, and that spinally administered ACT and midazolam (MZL) act synergistically to reduce allodynia as a result of PNI. These findings suggest that these two clinically used drugs may be utilized in combination to achieve relief of neuropathic pain in patients.
2. Materials and methods
2.1. Animals
Male, Sprague–Dawley rats (Harlan, Indianapolis, IN), 250–275 g, approximately 2 months old, at the time of surgery, were maintained in a climate-controlled room on a 12-h light/dark cycle (lights on at 6 AM), and food and water were available ad libitum. All testing was performed in accordance with the policies and recommendations of the International Association for the Study of Pain and the National Institutes of Health guidelines for the handling and use of laboratory animals. Prior approval for the project was received from the Institutional Animal Care and Use Committee of the University of Arizona.
2.2. Solutions and drugs
Acetazolamide (ACT) and midazolam (MZL) were purchased from Sigma–Aldrich (St. Louis, MO). ACT was made fresh as described below and 100 mM stock solutions of MZL was made in ddH2O. For all experiments the ACT was diluted into physiological buffer (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 0.5 mM CaCl2, 0.4 mM Na2HPO4, 25 mM NaHCO3 and 5 mM glucose) at a concentration of 10 mM with a starting pH of 8.2 to allow for ACT solubility. The pH was then adjusted down to 7.4 prior to use. The physiological buffer was used at pH 7.4 for MZL experiments and as a vehicle to control for any possible effects due to the alkalinity of the vehicle. Doses of ACT and MZL were based on [1,30].
2.3. SNL surgery
Prior to surgery all animals were assessed for mechanical withdrawal thresholds. SNL was done by tight ligation of the L5 and L6 spinal nerves as described by Kim and Chung [19]. Anesthesia was induced using ketamine/xylazine at 80 mg/kg. The dorsal vertebral column from L4 to S2 was exposed and the L5 and L6 spinal nerves of the left hindpaw were identified and carefully isolated. The L5 and L6 spinal nerves were tightly ligated distal to the dorsal root ganglion with a 4-0 silk suture and the incision was closed. Sham control rats underwent the same surgery and handling as the experimental animals but without the SNL. An intrathecal catheter was placed (as described previously [40]) at the time of SNL surgery. All animals were allowed to recover for at least 14 days and all testing was performed between 14 and 28 days post-SNL or sham. Following SNL, only animals that developed paw withdrawal thresholds less than 4.7 g by day 14 post-surgery were used. Any animals showing motor impairment from the intrathecal catheter placement were immediately euthanized.
2.4. Behavioral testing
2.4.1. Mechanical thresholds
Animals were placed in acrylic boxes with wire mesh floors and allowed to habituate for 1 h. After baseline or predrug mechanical thresholds were taken animals received intrathecal injections in a volume of 10 μl followed by flushing of the catheter with 6 μl sterile saline solution. Calibrated von Frey filaments (Stoelting, Wood Dale, IL) were used for mechanical stimulation of the plantar surface of the left hindpaw and withdrawal thresholds were calculated using the up-down method [5]. Stimulation frequency was 0.1 Hz or lower.
2.4.2. Rotarod
Animals were tested for their ability to balance on a slowly rotating rod (Rotamex 4/8, 6.5 cm diameter for rats, constant rate of rotation of 8 revolutions per min, Columbus Instruments, Columbus, OH) after intrathecal administration of compound. Prior to drug administration, rats were trained in four consecutive sessions to stay on the rod and reach a cut off time of 180 s. Once trained, the animals were given drugs and tested at time points matching mechanical threshold measures for the appropriate route of administration. Only doses that achieved peak effects in reversal of SNL-induced allodynia were used.
2.4.3. Statistics
All data are presented as means ± SEM unless otherwise noted. Percentile data are presented as withdrawal frequency for a given time point minus predrug withdrawal threshold divided by presurgery baseline. Significant differences between groups for area under curve measures were assessed by one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison post hoc test. Measures of dose by time and rotarod were done by two-way ANOVA with Bonferroni post hoc test. Dose–response curves were analyzed by variable slope nonlinear regression using Graph-Pad Prism 5.0 for Mac OS X (Graph-Pad, San Diego, CA). For dose–response curves with MZL, where an inverted U curve was observed, Emax was set to the most efficacious dose to avoid producing a right-ward shift in the EC50 calculation; however, all doses are shown for a full representation of the data obtained. The same strategy was also used for A50 analysis with MZL. Isobologram and A50 analysis were done with JFlashCalc [26], which is freely available for use at http://www.u.arizona.edu/~michaelo.
3. Results
3.1. Effects of intrathecal ACT on neuropathic allodynia
We first tested whether intrathecally administered ACT was capable of reducing neuropathic allodynia in the SNL model in rats. ACT was given in doses of 0.225 (n = 4), 2.25 (n = 6), 6.67 (n = 6) and 22.5 μg (n = 8) and mechanical withdrawal thresholds were measured at 30, 60, 90, 150 and 240 min post-injection of ACT or vehicle (n = 6). A significant main effect of ACT was observed (Fig. 1A, F(4, 144) = 6.95, p < 0.0001). ACT dose-dependently inhibited neuropathic allodynia compared to vehicle controls (Fig. 1C and D) with a peak effect at the highest dose (Fig. 1C) at 60 min post-injection (Fig. 1A). ACT, at the highest dose tested (n = 6), had no effect on mechanical withdrawal thresholds in sham animals compared to vehicle (n = 6, Fig. 1B). This dose of ACT did not, either, impair motor performance in the rotarod test compared to vehicle treatment (n = 7) at any of the time points tested (n= 5, Fig. 2). The EC50 for ACT was calculated as 1.85 ± 3.3 μg (Fig. 1D). Hence, intrathecal injection of ACT inhibits allodynia resulting from PNI indicating that spinal CA activity contributes to neuropathic mechanical allodynia.
Fig. 1.

Intrathecal acetazolamide (ACT) inhibits neuropathic allodynia in the spinal nerve ligation (SNL) model. (A) Predrug mechanical withdrawal thresholds were measured and ACT was injected intrathecally and mechanical withdrawal thresholds were reassessed at the indicated time points. A significant main effect of ACT was observed (F(4, 144) = 6.95, p < 0.0001). (B) ACT, at the highest dose given, did not alter mechanical withdrawal thresholds in sham animals. (C) ACT dose-dependently inhibited neuropathic allodynia with an EC50 (D) of 1.85 ± 3.3 μg. ***p < 0.001 with one-way ANOVA and Dunnett’s post hoc test.
Fig. 2.
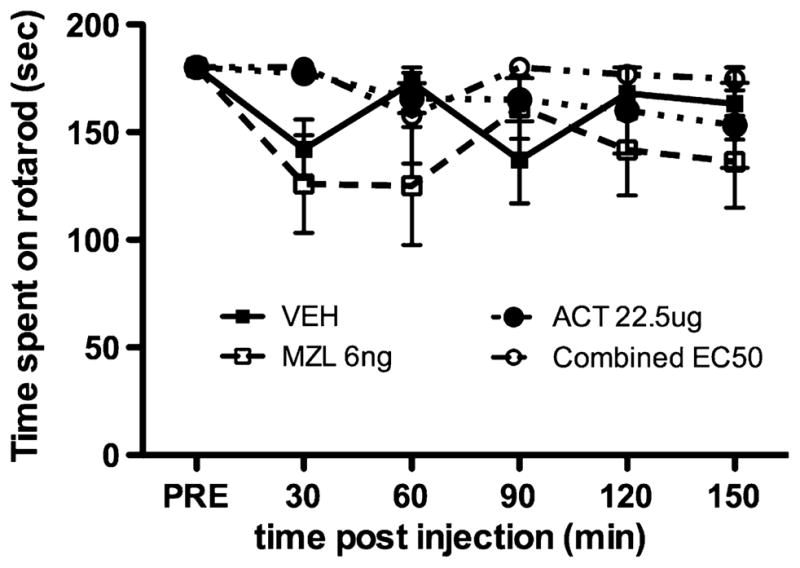
Acetazolamide (ACT), midazolam (MZL) and ACT and MZL in combination do not impair motor performance. The rotarod test was used to assess whether maximum effective doses of ACT or MZL impaired motor performance. Rats with intrathecal catheters were trained as described in the text and VEH, ACT, MZL or ACT + MZL (combined EC50) were administered and rotarod performance was assessed at the indicated time points. Neither ACT, MZL nor ACT (0.97 ng) + MZL (1.85 μg) impaired motor performance as compared to vehicle injection.
3.2. Effects of intrathecal MZL on neuropathic allodynia
We then tested whether intrathecally administered MZL was capable of reducing neuropathic allodynia. MZL was given in doses of 0.4 (n = 9), 2 (n = 7), 6 (n= 8), 20 (n = 9) and 60 ng (n = 6) and mechanical withdrawal thresholds were measured at 30, 60, 90, 150 and 240 min post-injection of MZL or vehicle (n= 8). A significant main effect of MZL was observed (Fig. 3A, F(5, 246) = 6.31, p < 0.0001). MZL, as expected, also dose-dependently inhibited neuropathic allodynia compared to vehicle controls (Fig. 3C and D) with a peak effect between the 6 and 20 ng doses (Fig. 3C) at 30–60 min post-injection (Fig. 3A). Area under the curve analysis indicated that the MZL dose–response curve shows an inverted U-shape suggesting the effectiveness of MZL may be dose limiting. MZL, at the highest effective dose observed with area under the curve analysis, 6 ng (n= 8), had no effect on mechanical withdrawal thresholds in sham animals compared to vehicle (n= 8, Fig. 3B). This dose of MZL did not, either, impair motor performance in the rotarod test at any of the time points tested (n = 7, Fig. 2). The EC50 for MZL was calculated as 0.97 ± 0.72 ng (Fig. 3D). Hence, intrathecal injection of MZL inhibits allodynia resulting from PNI but, apparently, in a dose-limiting fashion.
Fig. 3.

Intrathecal midazolam (MZL) inhibits neuropathic allodynia in the spinal nerve ligation (SNL) model. (A) Predrug mechanical withdrawal thresholds were measured and MZL was injected intrathecally and mechanical withdrawal thresholds were reassessed at the indicated time points. A significant main effect of MZL was observed (F(5, 246) = 6.31, p < 0.0001). (B) MZL, at the peak effect dose, did not alter mechanical withdrawal thresholds in sham animals. (C) MZL dose-dependently inhibited neuropathic allodynia with an EC50 (D) of 0.97 ± 0.72 ng. *p < 0.05, **p < 0.01 with one-way ANOVA and Dunnett’s post hoc test.
3.3. Effects of intrathecal ACT combined with MZL on neuropathic allodynia
We next tested the possibility that ACT and MZL might act synergistically to inhibit neuropathic allodynia. We used combined doses of ACT and MZL with a fixed dose ratio based on the EC50s of both compounds (in ng) given separately. Hence, animals received intrathecal injections of ACT 1850 ng + MZL 0.97 ng (n = 7), ACT 616.7 ng + MZL 0.32 ng (n = 6) or ACT 185 ng + MZL 0.097 ng (n = 5) representing EC50 values for each compound divided by 1, 3 and 10, respectively. At the highest combined dose motor performance was not affected (n = 5, Fig. 2). Mechanical withdrawal thresholds were measured at 30, 60, 90, 150 and 240 min post-injection of ACT + MZL or vehicle (n = 5). A significant main effect of ACT + MZL was observed (Fig. 4A, F(3, 126) = 6.29, p = 0.0005). Area under curve measures for ACT + MZL versus vehicle treatment are shown in Fig. 4B. The effect of the compounds administered together peaked at 60 min (Fig. 4A) and these values were used to construct a dose–response curve for ACT + MZL where the dose is shown as the combined amount of drug given (Fig. 4C). This was done in order to control for possible artefacts due to an expansion of the area under the curve measure from a prolonged action of the drugs given together, as was observed (Fig. 4A). The EC50 of ACT + MZL at this fixed dose ratio was 190 ± 141 ng. EC50s for ACT or MZL alone were also calculated under this paradigm using peak effects (60 min) for the compounds individually. The EC50 for ACT alone was 2.16 ± 1.75 μg and the EC50 for MZL was 1.13 ± 0.68 ng (Fig. 4C). These observed EC50s were consistent with EC50s calculated using the area under the curve measurements (Fig. 1D and Fig. 3D).
Fig. 4.

Intrathecal acetazolamide (ACT) combined with midazolam (MZL) acts synergistically to inhibit neuropathic allodynia in the spinal nerve ligation (SNL) model. (A) Predrug mechanical withdrawal thresholds were measured and ACT + MZL, at a fixed dose ratio of 1850–0.97 ng (see text for explanation) was injected intrathecally and mechanical withdrawal thresholds were reassessed at the indicated time points. A significant main effect of ACT + MZL was observed (F(3, 126) = 6.29, p = 0.0005). (B) ACT + MZL dose-dependently inhibited neuropathic allodynia with a combined dose EC50 (C) of 190 ± 141 ng, curves for ACT and MZL alone are shown for comparison. (D) Isobolographic analysis revealed that ACT and MZL act synergistically to inhibit neuropathic allodynia. The A50 for ACT given alone was 2.19 ± 0.75 μg. The A50 of MZL was 1.07 ± 0.88 ng. The theoretical additive A50 for a combined dose was 1058 ± 480 ng while the observed A50 for ACT and MZL administered together was 182 ± 65 ng. The theoretical additive mixture A50 differed from the observed A50 significantly (p < 0.001) indicating that ACT and MZL act synergistically to inhibit neuropathic allodynia. (E) MZL, ACT or MZL + ACT were given intrathecally and paw withdrawal thresholds were measured at 60 min post-drug administration to test whether ACT would restore efficacy of MZL at a dose where efficacy was lost. ACT alone and ACT + MZL significantly reversed neuropathic allodynia while MZL alone did not. *p < 0.05, **p < 0.01 with one-way ANOVA and Dunnett’s post hoc test.
Isobolographic analysis was used to determine whether ACT and MZL act synergistically to inhibit neuropathic allodynia. For this analysis we calculated A50s for ACT or MZL alone; a theoretical combined A50 if the drugs acted in an additive fashion; and an actual A50 from the observed effects of the drugs given together in the fixed dose ratio of 1850–0.97. As in the EC50 measurements shown in Fig. 4C, we used peak effects at 60 min for the drugs alone and in combination for these calculations. In this case the A50 for ACT given alone was found to be 2.19 ± 0.75 μg (Fig. 4D), again consistent with EC50s calculated by area under the curve or peak effect methods. The A50 of MZL was 1.07 ± 0.88 ng (Fig. 4D), also consistent with EC50s obtained by the methods described above. The theoretical additive A50 was found to be a combined dose of 1058 ± 480 ng whereas the actual A50 observed with ACT and MZL administered together was 182 ± 65 ng (Fig. 4D, closely matching the EC50 calculated from regression analysis of 190 ± 141 ng). The theoretical additive A50 differed from the observed A50 significantly (p < 0.001) indicating that ACT and MZL act synergistically to inhibit neuropathic allodynia (Fig. 4D).
Finally, our findings indicated that the efficacy of MZL may be limited at high doses. Hence, we tested if co-administration of ACT with MZL could reduce this effect. We administered MZL at 60 ng (n = 6), ACT at 22.5 μg (n = 7) and MZL at 60 ng + ACT at 22.5 μg (n = 8) or vehicle (n = 12) intrathecally to SNL rats and measured reversal of allodynia at 60 min post-drug injection, consistent with the peak time point for efficacy of these drugs given alone or in combination. We did not use the fixed dose ratio dosage schedule used for synergism studies in this experiment because that dose of ACT would have exceeded the maximum dose of ACT given in any other experiments. ACT or ACT + MZL significantly reversed neuropathic allodynia while MZL given alone did not show a significant reversal, consistent with a loss of efficacy for MZL at this dose (Fig. 4E). Hence, this finding suggests that a dose-limiting loss of efficacy for MZL at high doses in PNI animals can be avoided with co-administration of ACT.
4. Discussion
Our work shows that ACT reduces mechanic neuropathic allodynia which, together with previous observations on the alleviating actions of CA inhibition on thermal hyperalgesia [30], suggests that spinal CA is a promising target for novel drugs in the treatment of several types of chronic pain. Even more importantly, the present data show that ACT and MZL act synergistically to reduce allodynia as a result of PNI. Therefore, the combined use of benzodiazepines with CA inhibitors may represent a new therapeutic approach for treatment of neuropathic pain.
Previous experiments have shown that intrathecal ACT (at a 22.5 μg dose) inhibits thermal hyperalgesia after muscle inflammation [30]. Our present results indicate that ACT dose-dependently inhibits PNI-induced neuropathic allodynia and that ACT and MZL, when given in combination, act synergistically to alleviate the allodynia. Moreover, our findings suggest that the combined use of ACT and MZL may avoid possible dose-limiting effects of MZL which are illustrated by the inverted U shape of the MZL dose–response curve. These results lend support to a mechanistic scheme, described in detail below, wherein behavioral manifestations (allodynia) of the positive shift in reversal potential (Er) of GABAA that results from KCC2 down-regulation after PNI [10,28] can be blocked by reducing availability by CA inhibition. A potential anti-neuropathic pain role for CA inhibitors has been postulated previously [6] based largely on the CA inhibitor activity of topiramate, which alleviates PNI-induced allodynia in rats [2], shows synergism with tramadol after PNI [7] and reduces daily pain scores in humans with lumbar radicular pain [18]. Although topiramate has several molecular targets and inhibits some CAs, topiramate is a potent inhibitor of CAVII [39], an isozyme important for -mediated excitatory GABA actions in the hippocampus [31,33]. Our findings with ACT support the notion that CA inhibition is effective for the treatment of neuropathic allodynia. Further work is warranted to establish whether CA inhibition underlies the anti-neuropathic pain effects of topiramate.
Our work also shows that MZL alone dose-dependently inhibits neuropathic allodynia in the SNL model. Benzodiazepines require the presence of a γ2 subunit and different α subunit compositions can lead to different behavioral effects. Recent studies have elucidated that spinal GABAA receptors are composed of α2, α3 and α5 subunits [20] and that α2- and α3-containing subunits are critsome critical for benzodiazepine anti-allodynic effects in inflammatory, PNI [20] and anti-nociception in chemical pain models [21]. Critically, benzodiazepines that “spare” α1-containing subunits do not induce sedation yet achieve analgesia in all of these models [20,21,25]. Our results with intrathecal MZL are consistent with previous electrophysiological experiments in the SNL model [22]; with behavioral and biochemical experiments in the chronic constriction injury model of PNI [23]; with experiments in rats with chronic inflammation of the hindpaw from complete Freund’s adjuvant (CFA) injection [1]; and with reports of alleviation of pain in humans with spinal benzodiazepines [12]. Hence, these results provide further support for a spinal site of action for anti-allodynic effects of benzodiazepines after PNI.
A major question raised by these findings is: what are the mechanisms via which CA inhibitors and benzodiazepines act synergistically to inhibit neuropathic allodynia after PNI? PNI produces pathological changes in the spinal GABAergic and glycinergic network such that a decrease in the efficacy of inhibition is observed and this process may underlie the allodynic state that follows the injury [9,10,24,28]. PNI induces a positive shift in the reversal potential (Er) of GABAA receptors or glycine receptor-mediated currents in a subset of dorsal horn neurons [9,10,17,28]. The decrease in KCC2 expression after PNI would be expected to result in a decreased Cl− extrusion capacity in dorsal horn neurons; hence, the positive shift in Er for GABAA and glycine receptors may reflect a reduced or collapsed Cl− gradient with Cl− reversal potential (ECl) close to the resting membrane potential (Vrest) [10,28]. Under these conditions, -mediated currents would be expected to lead to a further positive shift in Er and, consequently, to strongly depolarizing and even excitatory GABAA or glycine channel responses that would be sensitive to CA inhibition.
While it is clear that PNI produces changes in the spinal GABAergic and glycinergic system that reduce the efficacy of GABAA-and glycine receptor-mediated hyperpolarizing inhibition, it is also true that spinally applied benzodiazepines are effective for reducing neuropathic allodynia and/or hyperalgesia [20,22,23,36]. According to the findings discussed above this would appear to be paradoxical; however, there are several scenarios that can resolve this potential paradox. The degree of positive shift in Er after PNI is paramount to whether GABAA activity would be expected to produce gross excitation or shunting inhibition [3,13,28,29], and with smaller positive shifts in Er, benzodiazepine-mediated augmentation of GABAA currents would be expected to produce a larger shunting inhibition. However, this would clearly be dependent on the intensity and duration of GABAergic network activity (see [35]), as prolonged activity would move Er progressively more positive as a result of Cl− accumulation driven by the -mediated depolarization [14]. Under this scenario, CA inhibition is expected to augment the anti-allodynic activity of benzodiazepines because it reduces the -dependent progressive positive shift in Er. Moreover, modeling studies have shown that an increase in shunting conductance can lead to an increase in excitability [28]. Such findings may explain the inverted U shape of the MZL dose–response curve found here and, according to the model postulated above, and our findings, this would be reversed by inhibition of CA activity.
A further point of importance in the present context is that phasic GABAA receptors are found preferentially in the postsynaptic membrane whereas tonic GABAA subunits, which possess high affinity for GABA, are found extrasynaptically in CNS neurons [11]. Phasic, synaptic GABAA receptors preferentially contain α1, α2 or α3 and γ2 subunits and are benzodiazepine sensitive while tonic, extrasynaptic channels contain a ∂ subunit and, with the exception of those that also possess α5 and γ2 subunits, are benzodiazepine- insensitive [11]. Tonic receptors generate persistent currents as observed in dorsal horn neurons with prolonged GABA application [8]. On the other hand, after PNI, benzodiazepines generate anti-allodynia on the spinal level largely via α2- and α3-containing subunits suggesting that synaptic, or phasic, receptors are responsible for this effect [20]. Hence, during network activity, which can be generated in the spinal cord either by the convulsant 4-aminopyridine [32] or PNI [35], the large increase in conductance caused by the benzodiazepine sensitive, phasic postsynaptic events might well lead to a shunting inhibition despite a substantial depolarization of the target cell, while the low-conductance, tonic depolarization mediated by the extrasynaptic GABAA receptors is expected to provide a tonic excitatory drive that is sensitive to CA blockade [3,31]. Consistent with this, we observed striking synergism between benzodiazepines and CA inhibitors after PNI suggesting that CA inhibition augments the inhibitory effects of benzodiazepine action by suppressing -dependent excitatory events.
In conclusion, the major finding of the present work is that ACT and MZL act synergistically to inhibit neuropathic allodynia. In light of the available in vitro data reviewed above, a parsimonious way to explain this synergism is that CA inhibition blocks an -dependent positive shift in the Er of GABA and/or glycine-mediated currents and the consequent tonic excitatory drive mediated by extrasynaptic GABAA receptors, while preserving shunting inhibition that is augmented by benzodiazepine actions at postsynaptic GABAA receptors. Obviously, further work is needed at the in vitro level in order to directly examine the cellular and synaptic basis of the ACT-MZL synergism and clinical studies are required to determine the safety of intrathecally applied CA inhibitors in humans. Since MZL and ACT, as well as several other inhibitors of CA [37], are clinically approved, we propose that their use in combination opens up a novel approach for the treatment of chronic neuropathic pain.
Acknowledgments
The authors thank Ning Qu and Janice Oyarzo for expert technical assistance. This work was supported by funds from The University of Arizona (T.J.P.), The University of Arizona School of Medicine (T.J.P.), The Rita Allen Foundation(T.J.P.), The American Pain Society (T.J.P.), The Academy of Finland (K.K.) and from The Sigrid Jusélius Foundation (K.K.).
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- 1.Anseloni VC, Gold MS. Inflammation-induced shift in the valence of spinal GABA-A receptor-mediated modulation of nociception in the adult rat. J Pain. 2008;9:732–8. doi: 10.1016/j.jpain.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benoliel R, Tal M, Eliav E. Effects of topiramate on the chronic constriction injury model in the rat. J Pain. 2006;7:878–83. doi: 10.1016/j.jpain.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–38. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol. 1987;385:243–86. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 6.Chong MS, Libretto SE. The rationale and use of topiramate for treating neuropathic pain. Clin J Pain. 2003;19:59–68. doi: 10.1097/00002508-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Codd EE, Martinez RP, Molino L, Rogers KE, Stone DJ, Tallarida RJ. Tramadol and several anticonvulsants synergize in attenuating nerve injury-induced allodynia. Pain. 2008;134:254–62. doi: 10.1016/j.pain.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 8.Cordero-Erausquin M, Coull JA, Boudreau D, Rolland M, De Koninck Y. Differential maturation of GABA action and anion reversal potential in spinal lamina I neurons: impact of chloride extrusion capacity. J Neurosci. 2005;25:9613–23. doi: 10.1523/JNEUROSCI.1488-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 10.Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–42. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- 11.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–29. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- 12.Jasmin L, Wu MV, Ohara PT. GABA puts a stop to pain. Curr Drug Targets CNS Neurol Disord. 2004;3:487–505. doi: 10.2174/1568007043336716. [DOI] [PubMed] [Google Scholar]
- 13.Jean-Xavier C, Mentis GZ, O’Donovan MJ, Cattaert D, Vinay L. Dual personality of GABA/glycine-mediated depolarizations in immature spinal cord. Proc Natl Acad Sci USA. 2007;104:11477–82. doi: 10.1073/pnas.0704832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–72. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaila K, Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–5. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- 16.Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J Physiol. 1993;464:273–89. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller AF, Beggs S, Salter MW, De Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol Pain. 2007;3:27. doi: 10.1186/1744-8069-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khoromi S, Patsalides A, Parada S, Salehi V, Meegan JM, Max MB. Topiramate in chronic lumbar radicular pain. J Pain. 2005;6:829–36. doi: 10.1016/j.jpain.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 20.Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Mohler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451:330–4. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- 21.Knabl J, Zeilhofer UB, Crestani F, Rudolph U, Zeilhofer HU. Genuine antihyperalgesia by systemic diazepam revealed by experiments in GABAA receptor point-mutated mice. Pain. 2009;141:233–8. doi: 10.1016/j.pain.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Kontinen VK, Dickenson AH. Effects of midazolam in the spinal nerve ligation model of neuropathic pain in rats. Pain. 2000;85:425–31. doi: 10.1016/S0304-3959(99)00298-5. [DOI] [PubMed] [Google Scholar]
- 23.Lim J, Lim G, Sung B, Wang S, Mao J. Intrathecal midazolam regulates spinal AMPA receptor expression and function after nerve injury in rats. Brain Res. 2006;1123:80–8. doi: 10.1016/j.brainres.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–31. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munro G, Lopez-Garcia JA, Rivera-Arconada I, Erichsen HK, Nielsen EO, Larsen JS, Ahring PK, Mirza NR. Comparison of the novel subtype-selective GABAA receptor-positive allosteric modulator NS11394 [3′-[5-(1-hydroxy-1-methylethyl)- benzoimidazol-1-yl]-biphenyl-2-carbonitrile] with diazepam, zolpidem, bretazenil, and gaboxadol in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2008;327:969–81. doi: 10.1124/jpet.108.144568. [DOI] [PubMed] [Google Scholar]
- 26.Ossipov MH, Lozito R, Messineo E, Green J, Harris S, Lloyd P. Spinal antinociceptive synergy between clonidine and morphine, U69593, and DPDPE: isobolographic analysis. Life Sci. 1990;47:PL71–6. doi: 10.1016/0024-3205(90)90530-5. [DOI] [PubMed] [Google Scholar]
- 27.Polgar E, Hughes DI, Riddell JS, Maxwell DJ, Puskar Z, Todd AJ. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain. 2003;104:229–39. doi: 10.1016/s0304-3959(03)00011-3. [DOI] [PubMed] [Google Scholar]
- 28.Prescott SA, Sejnowski TJ, De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Mol Pain. 2006;2:32. doi: 10.1186/1744-8069-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev. 2009;60:149–70. doi: 10.1016/j.brainresrev.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radhakrishnan R, Sluka KA. Acetazolamide, a carbonic anhydrase inhibitor, reverses inflammation-induced thermal hyperalgesia in rats. J Pharmacol Exp Ther. 2005;313:921–7. doi: 10.1124/jpet.104.082776. [DOI] [PubMed] [Google Scholar]
- 31.Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+–Cl-cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol. 2005;562:27–36. doi: 10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruscheweyh R, Sandkuhler J. Epileptiform activity in rat spinal dorsal horn in vitro has common features with neuropathic pain. Pain. 2003;105:327–38. doi: 10.1016/s0304-3959(03)00248-3. [DOI] [PubMed] [Google Scholar]
- 33.Ruusuvuori E, Li H, Huttu K, Palva JM, Smirnov S, Rivera C, Kaila K, Voipio J. Carbonic anhydrase isoform VII acts as a molecular switch in the development of synchronous gamma-frequency firing of hippocampal CA1 pyramidal cells. J Neurosci. 2004;24:2699–707. doi: 10.1523/JNEUROSCI.5176-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schoffnegger D, Heinke B, Sommer C, Sandkuhler J. Physiological properties of spinal lamina II GABAergic neurons in mice following peripheral nerve injury. J Physiol. 2006;577:869–78. doi: 10.1113/jphysiol.2006.118034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schoffnegger D, Ruscheweyh R, Sandkuhler J. Spread of excitation across modality borders in spinal dorsal horn of neuropathic rats. Pain. 2008;135:300–10. doi: 10.1016/j.pain.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 36.Shih A, Miletic V, Miletic G, Smith LJ. Midazolam administration reverses thermal hyperalgesia and prevents gamma-aminobutyric acid transporter loss in a rodent model of neuropathic pain. Anesth Analg. 2008;106:1296–302. doi: 10.1213/ane.0b013e318164f1e9. [table of contents] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–81. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 38.Uusisaari M, Smirnov S, Voipio J, Kaila K. Spontaneous epileptiform activity mediated by GABA(A) receptors and gap junctions in the rat hippocampal slice following long-term exposure to GABA(B) antagonists. Neuropharmacology. 2002;43:563–72. doi: 10.1016/s0028-3908(02)00156-9. [DOI] [PubMed] [Google Scholar]
- 39.Vullo D, Voipio J, Innocenti A, Rivera C, Ranki H, Scozzafava A, Kaila K, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett. 2005;15:971–6. doi: 10.1016/j.bmcl.2004.12.052. [DOI] [PubMed] [Google Scholar]
- 40.Yaksh TL, Stevens CW. Simple catheter preparation for permitting bolus intrathecal administration during chronic intrathecal infusion. Pharmacol Biochem Behav. 1986;25:483–5. doi: 10.1016/0091-3057(86)90028-6. [DOI] [PubMed] [Google Scholar]
- 41.Zeilhofer HU, Witschi R, Hosl K. Subtype-selective GABA(A) receptor mimetics-novel antihyperalgesic agents? J Mol Med. 2009 doi: 10.1007/s00109-009-0454-3. [DOI] [PubMed] [Google Scholar]