

SUMMARY
The activation of the tumor suppressor p53 facilitates the cellular response to genotoxic stress; however, the p53 response can only be executed if its interaction with its inhibitor Mdm2 is abolished. There have been conflicting reports on the question of whether p53 posttranslational modifications, such as phosphorylation or acetylation, are essential or only play a subtle, fine-tuning role in the p53 response. Thus, it remains unclear whether p53 modification is absolutely required for its activation. We have now identified all major acetylation sites of p53. Although unacetylated p53 retains its ability to induce the p53-Mdm2 feedback loop, loss of acetylation completely abolishes p53-dependent growth arrest and apoptosis. Notably, acetylation of p53 abrogates Mdm2-mediated repression by blocking the recruitment of Mdm2 to p53-responsive promoters, which leads to p53 activation independent of its phosphorylation status. Our study identifies p53 acetylation as an indispensable event that destabilizes the p53-Mdm2 interaction and enables the p53-mediated stress response.
INTRODUCTION
The p53 tumor suppressor is a key component of a regulatory circuit that monitors signaling pathways from diverse sources, including DNA damage responses, abnormal oncogenic events, and everyday normal cellular processes (Vogelstein et al., 2000; Prives and Hall, 1999). p53 is tightly regulated, such that its protein product usually exists in a latent form, and at low levels, in unstressed cells. However, the steady-state levels and transcriptional activity of p53 increase dramatically in cells that sustain various types of stress. While the precise mechanisms of p53 activation are not fully understood, they are generally thought to entail posttranslational modifications, such as ubiquitination, phosphorylation, methylation, and acetylation, of the p53 polypeptide (Brooks and Gu, 2003; Vousden and Lane, 2007).
The functions of p53 are downregulated by the Mdm2 onco-protein and a related protein Mdmx (also called Mdm4), at least in part by ubiquitin-mediated proteolysis (Brooks and Gu, 2006; Michael and Oren, 2003; Marine and Jochemsen, 2005). The central role of Mdm2 in this process is best illustrated by studies carried out in mice where inactivation of p53 was shown to completely rescue the embryonic lethality caused by loss of Mdm2 function (Jones et al., 1995; Montes de Oca Luna et al., 1995). Nonetheless, the molecular mechanisms by which p53 activity is controlled are complex. Although Mdm2, a really interesting new gene (RING) oncoprotein, was once thought to be the sole E3 ubiquitin ligase for p53, recent studies have shown that p53 is degraded in the tissues of Mdm2 null mice (Ringshausen et al., 2006) and that other E3 ligases can also induce p53 ubiq-uitination, such as ARF-BP1, COP1, and Pirh2 (Leng et al., 2003; Dornan et al., 2004; Chen et al., 2005). In contrast, Mdmx does not have intrinsic E3 ligase activity but Mdmx knockout mice die despite having functional Mdm2, and this lethality is also rescued by inactivation of p53 (Marine and Jochemsen, 2005). Thus, the role of Mdmx in repressing p53 function is as critical as that of Mdm2. Moreover, accumulating evidence indicates that degradation-independent mechanisms are crucial for both Mdm2 and Mdmx in controlling p53 activities. Recent studies suggest that Mdm2 mediates transcriptional repression by forming a protein complex with p53 on the promoters of specific p53-responsive genes (Minsky and Oren, 2004; Arva et al., 2005; Ohkubo et al., 2006). Nevertheless, it remains unclear whether similar mechanisms are also used for Mdmx-mediated transcription repression.
Histone acetyltransferases (HATs) represent an important layer of p53 regulation, particularly in transcription (Brooks and Gu, 2003). The covalent linkage of an acetyl group to lysine, the enzymatic process of acetylation, was first discovered on histones, and the significance of histone acetylation in transcriptional regulation is well accepted (Jenuwein and Allis, 2001; Berger, 2007). However, histones are not the only proteins that can be acetylated. p53 was the first nonhistone protein known to be regulated by acetylation and deacetylation (Gu and Roeder, 1997; Luo et al., 2000). The acetylation levels of p53 are significantly enhanced in response to stress and correlate well with p53 activation and stabilization (Luo et al., 2000, 2001; Vaziri et al., 2001; Ito et al., 2001; Barlev et al., 2001; Knights et al., 2006; Li et al., 2007; Zhao et al., 2008; Kim et al., 2008). Recently, an acetylation-deficient missense mutant (p53-6KR) was successfully introduced into the endogenous p53 gene by a knockin approach. Although p53-mediated transcriptional activation upon DNA damage is partially impaired in the ESCs and thymocytes of these mice, loss of p53 acetylation at its C terminus by CBP/p300 is apparently not as essential as originally anticipated (Feng et al., 2005; Krummel et al., 2005). Thus, it is possible that other coactivators or additional acetylation sites of p53 may compensate for the loss of p53 acetylation at its C terminus. Indeed, we and others have shown that the Tip60/hMOF protein induces p53 acetylation at lysine 120 (K120) within the DNA-binding domain, and that Tip60/hMOF, which shares no homology with the CBP/p300 or PCAF acetyltransferases, is required for p53-mediated transcriptional activation (Tang et al., 2006; Sykes et al., 2006; Berns et al., 2004). Interestingly, K120 is a recurrent site for p53 mutation in human cancer and Tip60/MOF-dependent acetylation at K120 is important for p53-mediated apoptosis. Nevertheless, loss of K120 acetylation has no effect on p53-mediated activation of p21, and the tumor-derived mutant p53-K120R retains its ability to induce cell growth arrest (Tang et al., 2006; Sykes et al., 2006).
Although numerous studies validate the crucial role of p21 in p53-mediated tumor suppression (el-Deiry et al., 1993; Vogel-stein et al., 2000), the molecular mechanism by which p53 induces p21 expression is not well understood. Here we have identified K164 as a new site for in vivo acetylation of p53 by CBP/p300 and evaluated its function in these processes. Although acetylation defects at each individual site (K164, K120, and C terminus) can be compensated by the modification of other sites, loss of acetylation at all these major sites completely abolishes its ability to activate p21 and suppress cell growth. Moreover, acetylation blocks the interaction of p53 with its cognate repressors (Mdm2 and Mdmx) on DNA, and this event directly results in p53 activation regardless of its phosphorylation status. Notably, the transcriptional functions of unacetylated p53 can be restored by inactivation of Mdm2 and Mdmx. These data have significant implications regarding an essential role of acetylation in p53 regulation.
RESULTS
K164 Is a New Acetylation Site Catalyzed by CBP/p300
To further elucidate acetylation-mediated effects on p53, we used mass spectrometric (MS/MS) analysis to identify all modification sites induced by CBP/p300. To increase the yield of acetylated forms of p53, we treated the cells with deacetylase inhibitors such as trichostatin A (TSA) and nicotinamide for 6 hr before isolating acetylated p53 from these cells. Consistent with previous studies, mass spectrometric analysis revealed modification of the known acetylation sites, including the six C-terminal lysine residues: K370, K372, K373, K381, K382, and K386. Interestingly, however, a p53 peptide with a molecular weight consistent with acetylation of lysine 164 (K164) was also identified (Figures 1A and 1B), suggesting the existence of a novel modification site within the DNA-binding core domain of p53. K164, which is located in the L2 loop of the DNA-binding core domain of p53, is conserved in all the species known to encode p53— including human, mouse, Xenopus, and zebrafish—as well as in p53-related proteins p63 and p73 (Figure 1C). Moreover, tumor-associated mutations of K164 have been observed in several different types of human tumors (http://p53.free.fr). To confirm acetylation of p53 proteins at K164, we developed an antiserum that specifically recognizes K164-acetylated p53 (AcK164-p53) (see Experimental Procedures and Figure S1). To avoid crossreactivity with other acetylation sites by this antibody, we compared its reactivity to two substrate proteins: the p53-7KR mutant, in which the seven known acetylated sites (K120 and the six C-terminal lysine residues) were all substituted with arginine; and the p53-8KR mutant, in which the seven known sites and K164 are all substituted with arginine. Upon in vitro acetylation with recombinant CBP and p300 proteins, the p53-7KR but not the p53-8KR polypeptide was recognized by the AcK164-p53 antibody (Figure 1D). To examine acetylation of endogenous p53, we used the acetylation-specific antibody to measure the levels of K164-acetylated p53 in untreated and DNA-damaged cells. As shown in Figure 1E, upon stabilization of p53 proteins, the steady-state levels of K164 acetylation were also enhanced. These data indicate that K164 is acetylated both in vitro and in vivo.
Figure 1. Identification of K164 within the Human p53 DNA-Binding Domain as a Novel Acetylation Site by p300/CBP.

(A) Schematic representation of the human p53 protein with known acetylation sites indicated, including K120, K370, K372, K373, K381, K382, and K386. In this study, K164 was identified to be acetylated by p300/CBP.
(B) Mass spectrometry analysis of the p53-derived peptides containing acetylated K164 (AcK164). The protein was prepared as described in the Experimental Procedures.
(C) Alignment of the K164 flanking region of the human p53 protein with those of p53 from other species and of human p63 and p73. The conserved lysine residue is marked in bold; h: human; m: mouse; c: chicken; x: Xenopus; and z: zebrafish.
(D) In vitro acetylation of p53 by p300/CBP. The Flag-p53-K120R/6KR (lanes 2, 4, and 6) or Flag-p53-K120R/K164/6KR (lanes 1, 3, and 5) recombinant protein was incubated alone (lanes 1 and 2), with p300 (lanes 3 and 4), or with CBP (lanes 5 and 6). The reaction products were resolved by SDS-PAGE and analyzed by western blot using the site-specific polyclonal antibody against acetylated K164 (anti-AcK164-p53) (top panel). The levels of the p53 recombinant protein substrates are shown in the bottom panel by Ponceau red staining.
(E) Acetylation of the endogenous p53 protein at K164. HCT116 cells were treated with 20 μM etoposide (0, 4, and 6 hr). The endogenous p53 proteins were immunoprecipitated by the anti-p53 (1801) antibody and assayed for acetylation by western blot using the anti-AcK164-p53 antibody (top panel). The levels of p53 in the cell extracts are shown in the middle panel.
Simultaneous Mutation at All the Major Acetylation Sites Completely Abolishes p53-Mediated Activation of p21
We and others recently showed that K120 acetylation is crucial for p53-mediated apoptosis but has no effect on the induction of p21 and cell growth arrest (Tang et al., 2006; Sykes et al., 2006). To elucidate the effect of K164 acetylation on p53-dependent transcriptional activation, we examined whether loss of K164 acetylation influences p53-mediated p21 activation. As shown in Figure 2B, expression of wild-type p53 led to strong induction of p21, and a slight reduction but comparable degree of p21 induction was also obtained upon expression of K164R mutant p53. Moreover, consistent with previous studies (Tang et al., 2006; Sykes et al., 2006), p53 mutants that lack C-terminal lysines or K120, but retain K164, readily activate p21 under these conditions (Figures 2B and S2). Strikingly, loss of acetylation only at all eight sites (p53-8KR) completely abolished p21 induction (Figures 2A, 2B, and 2C). Thus, although individual mutations at each acetylation site have no significant effect on its ability to induce p21, loss of acetylation at all sites (p53-8KR) greatly impairs p53-dependent transcription.
Figure 2. Functional Characterization of Acetylation-Defective p53 Mutants.

(A) Scheme of the human p53 lysine-to-arginine mutants used in this study.
(B) Induction of p21 and Mdm2 by p53 lysine-to-arginine mutants. H1299 cells were transfected with increasing amount of plasmid DNA expressing different p53 mutants. The total cell extracts were subjected to western blot for p53, p21, and Mdm2, and GFP was used as a transfection control.
(C) As in (B), two additional p53 lysine-to-arginine mutants were examined.
(D) In vitro DNA-binding activities of p53 and p53-8KR. Gel shift assay was performed using a fragment containing a p53-binding site from the p21 promoter, as described in the Experimental Procedures.
(E) Degradation of the p53 and p53-8KR proteins by Mdm2. H1299 cells were cotransfected with CMV-p53 expressing either p53 or p53-8KR and an increased amount of CMV-Mdm2. The cellular levels of p53 were determined by western blot using anti-p53 antibody (DO-1).
The Acetylation-Defective p53 Mutant Retains Its Ability to Act as a DNA-Binding Transcription Factor that Induces the p53-Mdm2 Feedback Loop
Lysine-to-arginine mutants are commonly used to mimic unacetylated forms of histone in transcriptional regulation (Jenuwein and Allis, 2001). Nevertheless, two of the p53 acetylation sites (K120 and K164) reside within the DNA-binding domain, and it is known that many tumor-associated mutations in this domain can inactivate p53 function by ablating its DNA-binding activity (Vogelstein et al., 2000). Thus, we used an electrophoretic mobility shift assay (EMSA) to ascertain whether loss of these acetylation sites affects sequence-specific DNA binding by p53. As shown in Figure 2D, p53/DNA complexes were obtained by incubation of highly purified, recombinant full-length human wild-type p53 with a radiolabeled probe containing the p53-binding site from the p21 promoter. Significantly, the DNA-binding activity of the wild-type and 8KR mutant forms of p53 were indistinguishable, indicating that the eight lysine-to-arginine substitutions, including K120R and K164R, do not abolish the DNA binding. Moreover, both forms of p53 also bound equally to the transcriptional coactivators/acetylases CBP and Tip60 (Figures S3A and S3B), as well as with Mdm2 and Mdmx (Figure S4).
Consistent with its ability to bind DNA and to associate with its transcriptional coactivators, p53-8KR retained its full capacity to transactivate Mdm2 expression (Figure 2B). We also tested whether Mdm2 can induce degradation of p53-8KR in human cells. As shown in Figure 2E, although some effects were expected given that the p53-8KR protein lacks the major C-terminal ubiquitination sites, p53-8KR was degraded by Mdm2 in human cells. Taken together, these results indicate that the acetylation-defective mutant p53-8KR retains its abilities to act as a DNA-binding transcriptional factor that induces the p53-Mdm2 feedback loop.
Acetylation Is Essential for p53-Mediated Cell Growth Arrest and Apoptosis
To evaluate the role of p53 acetylation under physiological conditions, we established inducible cell lines expressing tetracy-cline-regulated wild-type p53 or acetylation-defective 8KR mutant p53. These cells express relatively low levels of p53 upon withdrawal of tetracycline (Zupnick and Prives, 2006). As shown in Figure 3A, the levels of p53 induced were very close to those of endogenous p53 in HCT116 cells upon DNA damage treatment. Moreover, after removing tetracycline from the medium, the levels of both wild-type p53 and p53-8KR were increased to a similar degree. As expected, wild-type p53, but not p53-8KR, was acetylated at all sites examined by acetylation-specific antibodies (Figure 3B). In addition, both wild-type p53 and p53-8KR were phosphorylated at similar levels (lane 4 versus 2, Figure 3B), indicating that the acetylation mutations do not affect on the phosphorylation status of p53.
Figure 3. Lack of Acetylation at Its DNA-Binding Domain and C Terminus Abolishes p53 Ability to Induce Cell Growth Arrest.

(A) Expression of the human p53 protein at physiological levels in Tet-off-p53 H1299 cells. The total cell extracts from Tet-off-p53 cells (before and after induction) and HCT116 cells (treated without and with etoposide) were assayed by western blot using the antibodies against p53 (DO-1) and actin.
(B) Acetylation of p53 in Tet-off-p53 cells. The Flag-p53 proteins enriched from Tet-off-p53 and Tet-off-p53-8KR cells by M2 immunoprecipitation were subjected to western blot analysis using acetylation- and phosphorylation-specific antibodies, as indicated, and anti-p53 (1801) to determine the levels of total p53.
(C) Tet-off-p53 and Tet-off-p53-8KR cells were induced for 0, 1, 2, 3, and 4 days. The total cell extracts were analyzed by western blot using antibodies against p53 (DO-1), Mdm2, Bax, Puma, Pig3, p21, and actin.
(D) Four days after induction, Tet-off-p53 and Tet-off-p53-8KR cells were treated with 10 μM Brdu for 1 hr and immunostained with the anti-BrdU antibody. The nuclei are in blue (DAPI), and Brdu-positive nuclei are shown in red.
(E) Tet-off-p53 and Tet-off-p53-8KR cells were induced as in (C), and incorporation of BrdU was assayed as in (D).
To examine the transcriptional activity of p53 at low levels of expression, we monitored p21 expression during the course of a 4-day Tet-off induction. Although Mdm2 and p21 were both gradually activated in wild-type p53 cells, Mdm2 (but not p21) was induced in p53-8KR cell lines (Figure 3C). We also compared the effects of wild-type p53 and p53-8KR on cell growth by monitoring BrdU incorporation. Less than 5% BrdU-positive cells were detected in the Tet-off-p53 cells at 4 days after p53 induction. In contrast, more than 50% BrdU-positive cells were observed in the p53-8KR Tet-off cells (Figures 3D and 3E). Moreover, consistent with previous studies (Tang et al., 2006; Sykes et al., 2006), loss of acetylation abrogates p53-mediated activation of proapoptotic targets such as BAX, PUMA, and Pig3 (Figure 3C), and no apoptosis was induced by the acetylation mutant p53-8KR (Figure S5). Together, these data suggest that modification of these sites is required for both growth arrest and apoptosis induced by p53.
Acetylation of p53 Blocks the Recruitment of Mdm2 to the p21 Promoter
To elucidate the mechanism of p53 acetylation in p21 activation in cells, we performed chromatin immunoprecipitation (ChIP) assays with cells expressing either wild-type or acetylation-defective p53 proteins. Thus, the cells were transfected with expression vectors encoding either wild-type p53 or p53-8KR, together with expression vectors for Mdm2 and the known p53 acetylases (CBP and Tip60). At 36 hr after transfection, the cells were fixed with 1% formaldehyde for ChIP analysis. Immunoprecipitation with the p53-specific antibody revealed that both wild-type p53 and p53-8KR can be recruited to the p21 promoter (Figure 4A). Moreover, in the presence of Mdm2 expression, the levels of recruitment were decreased for both wild-type p53 and p53-8KR (Figure 4A), which reflects degradation of p53 induced by Mdm2 (Figure 4B). Since several recent studies showed that the transcriptional repression activity of Mdm2 requires p53-mediated recruitment of Mdm2 to the p21 promoter (Minsky and Oren, 2004; Ohkubo et al., 2006; Arva et al., 2005), we examined whether Mdm2 recruitment is modulated by p53 acetylation. As expected, immunoprecipitation with the Mdm2-specific antibody revealed that both wild-type p53 and p53-8KR can form protein complexes with Mdm2 on the p21 promoter in human cells (lanes 3 and 6, Figure 4A). Interestingly, coexpression of the CBP and Tip60 acetylases with wild-type p53, which led to increasing levels of p53 acetylation (lane 4, Figure 4B), severely ablated Mdm2 recruitment to the p21 promoter (lane 4, Figure 4A); conversely, however, Mdm2 recruitment was unaffected by coexpression of the acetylases with acetylation-defective p53 (lane 7, Figure 5A). Thus, these data suggest that p53 acetylation represses recruitment of Mdm2 to the p21 promoter.
Figure 4. Acetylation of p53 Abrogates the Promoter-Specific Recruitment of Mdm2 and Mdmx.

(A) H1299 cells transfected by various plasmids, as indicated, were treated with 1% formaldehyde for 10 min and processed for ChIP analysis. The occupancy of p53, Mdm2, CBP, and Tip60 of the p21 promoter was detected by PCR.
(B) Acetylation of p53 in the presence of CBP and Tip60. H1299 cells were transfected as in (A), and Flag-p53 immunoprecipitated with M2 beads was assayed for acetylation at specific sites by western blot. Expression of Mdm2, CBP, and Tip60 was detected in the total cell extracts.
(C) H1299 cells transfected by the indicated plasmids were treated with 1% formaldehyde for 10 min and processed for ChIP analysis as in (A). The occupancy of p53, Mdm2, Mdmx, CBP, and Tip60 of the p21, Pig3, and Mdm2 promoters was detected by PCR-agarose gel electrophoresis. The relative levels of recruitment of Mdm2 and Mdmx were further quantitated by real-time PCR as shown in Figures S7, S8, and S9.
Figure 5. Acetylation Disrupts the p53-Mediated Mdm2-p53-DNA Complex Formed In Vitro.
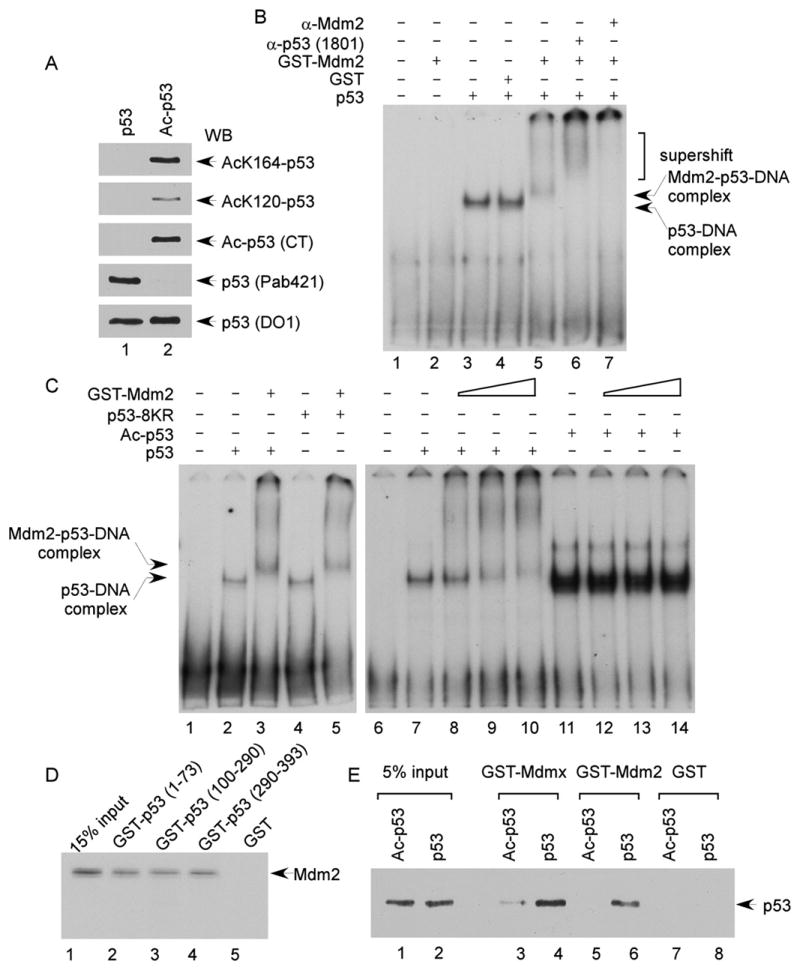
(A) The fully acetylated Flag-p53 proteins were purified from H1299 cells cotransfected with plasmid DNA expressing CMV-Flag-p53, CMV-Tip60, and CBP-HA. Acetylation levels were examined by western blot using site-specific acetylation antibodies, as indicated.
(B) Gel shift assay was performed, according to the Experimental Procedures, to analyze Mdm2-p53-DNA complex. A 32P-labeled 159 bp DNA fragment containing the p53-binding site from the human p21 promoter was incubated with proteins, as indicated. The reaction was resolved by 4% PAGE and protein-DNA complexes were visualized by autoradiography.
(C) Gel shift assays were carried out as in (B). The DNA probe was incubated with the Flag-p53, Flag-p53-8KR, or acetylated Flag-p53 proteins (20 ng) in the absence or presence of an increasing amounts of GST-Mdm2.
(D) In vitro binding of Mdm2 to the various domains of p53. The in vitro translated 35S-methionine-labeled Mdm2 protein was used in the pull-down assay with GST (lanes 5), GST-p53 (1–73) (lane 2), GST-p53 (100–290) (lane 3), or GST-p53 (290–393) (lane 4). The glutathione elutes and 15% input materials (lane 1) were resolved in SDS-PAGE gel and visualized by autoradiography. Levels of the recombinant proteins were shown in the Figure S6B.
(E) In the GST-pull-down assays, the purified p53 or acetylated p53 proteins were incubated with GST (lanes 7 and 8), GST-Mdm2 (lanes 5 and 6), or GST-Mdmx (lanes 3 and 4). The glutathione eluted material and 5% input materials (lanes 1 and 2) were analyzed by western blot using anti-p53 antibody (DO-1). GST fusion proteins were shown in Figures S6C and S6D.
Acetylation of p53 Inhibits Recruitment of Mdm2 and Mdmx to the p21 Promoter in a Ubiquitination-Independent Manner
Numerous studies demonstrate that Mdm2 employs both ubiquitination-dependent and ubiquitination-independent mechanisms to control p53 activity in vivo. In contrast, since Mdmx cannot directly catalyze p53 ubiquitination, it is thought to repress p53 function in a degradation-independent manner. To examine ubiquitination-independent p53 regulation by Mdm2 and Mdmx, we tested the effects of p53 acetylation on promoter recruitment of both Mdmx and Mdm2-C464A, an ubiquitin ligase-defective mutant of Mdm2. Thus, the cells were transfected with expression vectors encoding wild-type p53 or p53-8KR, together with Mdmx and Mdm2-C464A in the presence or absence of acetylases (CBP, Tip60). As shown in Figure 4C, both Mdmx and Mdm2-C464A can be recruited to the p21 promoter. Again, these recruitments were abrogated in cells expressing wild-type p53, but not p53-8KR, upon coexpression of CBP and Tip60 (lanes 4 and 7, Figure 4C). These data indicate that acetylation of p53 inhibits recruitment of Mdm2 and Mdmx to the p21 promoter in an ubiquitination-independent manner.
We then examined whether similar acetylation-mediated effects influence transactivation of other p53 target promoters. Previous studies indicated that Mdm2 can also be recruited to the Pig3 promoter (Ohkubo et al., 2006). Similar to p21 expression, we observed Pig3 induction by wild-type p53 but not by the acetylation-defective p53-8KR (Figure 3C). ChIP analysis revealed that Mdm2 and Mdmx were recruited to the Pig3 promoter to a comparable degree by both wild-type p53 and p53-8KR. In contrast, the recruitment of Mdm2 and Mdmx by wild-type p53, but not p53-8KR, was severely inhibited by coex-pression of CBP and Tip60 (Pig3 promoter, Figure 4C).
Finally, since acetylation-defective p53-8KR retains the ability to induce Mdm2 (Figure 2B), we tested the effects of p53 acetylation on recruitment of Mdm2 and Mdmx to the Mdm2 promoter. As expected, ChIP analysis demonstrates that both wild-type p53 and p53-8KR can be recruited to the Mdm2 promoter (Mdm2 promoter, Figure 4C). Consistent with our previous studies (Tang et al., 2006), Tip60 was recruited to the p21 promoter—but not Mdm2 promoter—in the presence of wild-type p53 or p53-8KR, whereas CBP was recruited to both promoters in the presence of wild-type p53 or p53-8KR. Interestingly, however, the recruitment of both Mdm2 and Mdmx to the Mdm2 promoter was extremely weak, and this modest level of recruitment was unaffected by coexpression of Tip60 and CBP (Mdm2 promoter in Figures 4C and S9). These results suggest that the recruitment of Mdm2 or Mdmx by p53 to the Mdm2 promoter is not significantly affected by the acetylation status of p53. To extend these findings, we identified Pirh2 as another p53 target (Leng et al., 2003), whose induction by p53 is independent of the acetylation status (Figure S10). Taken together, these data demonstrate that loss of acetylation does not affect p53’s ability to act as a transcription factor but significantly abrogates acetylation-induced inhibition of Mdm2 and Mdmx recruitment in a promoter-specific manner.
Acetylation of p53 Destabilizes the p53-Mdm2 Complex Formation
To provide direct evidence for the above hypothesis, we used an in vitro system to test whether acetylation blocks the interactions of DNA-bound p53 with Mdm2 and Mdmx. To this end, p53, p53-8KR, Mdm2, and Mdmx were each purified to near homogeneity (Figure S6A). Acetylated p53 was prepared in the presence of Tip60 and CBP essentially as previously described (Luo et al., 2004), and the acetylation status of p53 was verified with acetylation-specific antibodies (Figure 5A). An EMSA was then used to examine whether Mdm2 can form a protein complex with DNA-bound p53. As shown in Figure 5B, recombinant p53 readily forms a p53-DNA complex with a 159 base pair DNA fragment derived from the p21 promoter, as previously reported (Luo et al., 2004). In the presence of Mdm2, a more slowly migrating complex was formed (lane 5, Figure 5B), and this complex was either supershifted by the p53-specific antibody or blocked by the Mdm2-specific antibody (lanes 6 and 7, Figure 5B), suggesting the formation of an Mdm2-p53-DNA ternary complex. To examine the effect of p53 acetylation on Mdm2 recruitment, we compared complex formation by unacetylated and acetylated p53 polypeptides. As expected, when increasing amounts of Mdm2 were introduced in the reactions, unacetylated p53 formed the slowly migrating Mdm2-p53-DNA ternary complex (lanes 8–10, Figure 5C). The same complex was formed with the p53-8KR protein (lane 5, Figure 5C), implying that p53-8KR behaves like unacetylated p53 with respect to Mdm2 recruitment. In contrast, although acetylated p53-bound DNA more effectively than unacetylated p53 (lane 11, Figure 5C), as previously reported (Luo et al., 2004), Mdm2 was unable to form the Mdm2-p53-DNA ternary complex with acetylated p53 (lanes 12–14, Figure 5C). Similar results were also obtained with the inhibitory effect of p53 acetylation on formation of an Mdmx-p53-DNA ternary complex (Figure S11).
Finally, to extend these observations, we tested whether acetylation affects the p53-Mdm2 interaction in the absence of DNA. Although it is well accepted that the N terminus of p53 is crucial for interactions with Mdm2, our data indicate that p53 acetylation outside the N terminus modulates the p53/Mdm2 interaction. Thus, we first examined whether other regions of p53 are also involved in this interaction. To this end, the recombinant p53 fusion proteins containing different regions (N terminus, aa 1–73; core domain, aa 100–290; C terminus, aa 290–393) were expressed and purified to near homogeneity for in vitro binding assays (Figure S6B). As expected, 35S-labeled in vitro-translated Mdm2 bound the recombinant protein GST-p53 (aa 1–73) (lane 2, Figure 5D) but not GST (lane 5, Figure 5D). Surprisingly, both the core domain and the C terminus of p53 showed similar binding affinities for Mdm2 (lanes 3 and 4, Figure 5D), indicating that these two regions are also involved in the stable interaction between p53 and Mdm2. More importantly, to examine the effect of acetylation in this interaction, we performed similar GST-pull-down assays with either purified unacetylated p53 or acetylated p53 proteins. As shown in Figure 5E, western blot analysis revealed that unacetylated forms bound strongly to both Mdm2 and Mdmx; however, acetylation of p53 strongly abrogated these interactions. Taken together, these data indicate that there are multiple regions of p53 involved in its interaction with Mdm2 or Mdmx and that acetylation of p53 significantly destabilizes the complex formation with Mdm2 or Mdmx.
p53 Acetylation Is Essential for Inhibiting the p53-Mdm2 Complex Formation During the DNA Damage Response
Both phosphorylation and acetylation of p53 are commonly induced upon many types of stress (Brooks and Gu, 2003). In order to demonstrate the essential role of p53 acetylation under physiological settings, we had to test whether acetylation of p53 is crucial for abrogating Mdm2-mediated repression during the stress response independent of p53 phosphorylation status. Interestingly, it was reported that p53 can be activated normally without inducing its phosphorylation at the N terminus upon certain types of DNA damage such as actinomycin D treatment (Ashcroft et al., 1999, 2000; Ito et al., 2001). To dissect the role of p53 acetylation alone during the DNA damage response, we examined the effect of p53 acetylation in human osteosarcoma U20S cells in the presence of actinomycin D treatment. As shown in Figure 6A, upon the treatment, p53 was stabilized and p53-mediated activation of p21 was significantly induced. Indeed, despite a robust elevation of p53 acetylation levels, no significant increase of its phosphorylation levels could be detected at any time point (Figure 6A). To evaluate the role of p53 acetylation in modulating the p53-Mdm2 complex on chromatin, we performed chromatin immunoprecipitation (ChIP) assays with these cells. As expected, immunoprecipitation with the p53 antibody showed that an increasing amount of p53 associated with the p21 promoter (Figure 6B); conversely, although the levels of Mdm2 were elevated (Figure 6A), the recruitment of Mdm2 to the p21 promoter by p53 was severely inhibited when p53 was acetylated (Figure 6B).
Figure 6. Recruitment of Mdm2 to the p21 Promoter in Response to DNA Damage.

(A) Cell extracts from U2OS cells treated with actinomycin D (10 nM) for 0, 4, 8, 16, and 24 hr were fractionated in SDS-PAGE gel and analyzed by western blot using antibodies against p53 (FL), Mdm2, ser-15-phospho-p53, and actin. Acetylation levels of p53 were determined by immunoprecipitation with site-specific acetylation antibodies followed by western blot using a monoclonal p53 antibody DO-1.
(B) U2OS cells were treated as in (A) and processed for ChIP assay. The binding of p53 and Mdm2 to the p21 promoter was detected by PCR.
(C) The Flag-p53 proteins were immunoprecipitated with M2 beads from the total cell extracts of H1299 cells infected with the indicated adenoviruses for 24 hr and treated with or without actinomycin D (10 nM) for 16 hr. The levels of acetylation or phosphorylation were detected by western blot using site-specific antibodies.
(D) H1299 cells were infected and treated as in (C) and processed for ChIP assay. The binding of p53 and Mdm2 to the p21 promoter was detected by PCR.
Moreover, to further elucidate the requirement of acetylation during this event, we performed the same assay by using the acetylation-defective mutant (p53-8KR). Thus, p53 null H1299 cells were first infected with either Ad-p53 or Ad-p53-8KR and were then treated with actinomycin D for further analysis. As shown in Figure 6C, acetylation of wild-type p53 at all different sites was significantly induced whereas no obvious enhancement of p53 phosphorylation was detected after the treatment with actinomycin D. Using ChIP, we found that Mdm2 recruitment at the p21 promoter was strongly repressed upon actinomycin D treatment in a manner that correlated with the acetylation levels of wild-type p53 (lane 2 versus lane 1, Figure 6D). In contrast, no obvious difference in Mdm2 recruitment at this promoter was detected by the acetylation defective mutant p53-8KR (lane 4 versus lane 3, Figure 6D). Taken together, these data demonstrate that p53 acetylation is essential for inhibiting the p53-Mdm2 complex formation on the p21 promoter during DNA damage responses.
The Functional Defects of Unacetylated p53 Can Be Rescued by Removing Mdm2 and Mdmx from Cells
The above results suggest that unacetylated p53 is more vulnerable to the repressive effects of Mdm2 and Mdmx and that acetylation can activate p53 function by blocking Mdm2/Mdmx-mediated downregulation. To elucidate the physiological significance of this mechanism, we tested whether the transactivation potential of acetylation-defective p53 can be restored by RNAi-mediated knockdown of Mdm2 and Mdmx. To this end, the Tet-off p53-8KR cells were transfected with control siRNA or with siRNAs specific for Mdm2 or Mdmx. Two days after transfection, p53 expression was induced by removing tetracycline from the medium. As expected, p21 induction was not observed in p53-8KR cells transfected with control RNAi; however, significant levels of p21 and Pig3 were induced in cells subject to siRNA-mediated depletion of Mdm2, Mdmx, or the combination of both Mdm2 and Mdmx (lanes 8–10, Figure 7A). To avoid off-target effects by RNAi-mediated knockdown, we also obtained the same results with different RNAi oligos against the mRNA of either Mdm2 or Mdmx (data not shown). As a negative control, we performed the similar experiments with two additional p53 mutants, which are unable to interact with Mdm2 (Figure S12), to support the same conclusion.
Figure 7. Cell Growth Repression Function of p53 Is Abrogated by Loss of Acetylation but Rescued by siRNA-Mediated Ablation of Mdm2 and Mdmx.

(A) Tet-off-p53 cells and Tet-off-p53-8KR cells treated with control siRNA, Mdm2 siRNA, Mdmx siRNA, or a combination of Mdm2 siRNA and Mdmx siRNA, were induced by removal of tetracycline from the media. The total cell extracts were analyzed by western blot using antibodies against p53 (DO-1), Mdmx, Mdm2, Pig3, p21, and actin.
(B) In the upper panel, p53-8KR is unable to induce cell death and growth arrest. Tet-off-p53 and Tet-off-p53-8KR cells were plated at 10% confluence and incubated in the presence (uninduced) or absence (induced) of tetracycline for 6 days. The plates were washed with PBS and stained with crystal violet. In the lower panel, Tet-off-p53-8KR treated with either control siRNA or Mdm2/Mdmx siRNA were split and incubated in the presence (uninduced) or absence (induced) of tetracycline for 6 days. The plates were stained with crystal violet.
Next, we examined the effects of Mdm2 and Mdmx depletion on the growth suppression activity of p53 and p53-8KR. Both the wild-type p53 and p53-8KR cell lines were cultured in Tet-free medium to induce p53 expression, and cells were stained with crystal violet 5 days after tetracycline withdrawal. As expected, induction of wild-type p53—but not p53-8KR—strongly suppressed tumor cell growth (upper panel in Figure 7B). Significantly, the ability of acetylation-defective p53-8KR to suppress cell growth was partially rescued by siRNA-depletion of Mdm2 and Mdmx (lower panel in Figure 7B). Taken together, these data indicate that the acetylation defective mutant of p53 does not lose its activity permanently and that its mediated activation of p21 and cell growth repression can be at least in part restored by inactivation of Mdm2 and Mdmx in human cells.
DISCUSSION
p53 is regulated by a variety of posttranslational modifications, including phosphorylation, acetylation, methylation, ubiquitination, sumoylation , and neddylation (Vousden and Lane, 2007; Brooks and Gu, 2006; Berger, 2007). While ubiquitin-mediated proteasomal degradation is well accepted as a key mechanism for controlling p53 levels, it remains controversial how p53 is activated. Indeed, numerous studies indicate potential roles for phosphorylation and acetylation in p53 regulation; nevertheless, it was under debate whether protein modification is absolutely required for p53 activation. Following our early findings of C terminus p53 acetylation, we and others recently showed that p53 is also acetylated by Tip60/hMOF at residue K120 within the DNA-binding domain (Tang et al., 2006; Sykes et al., 2006). K120 acetylation is crucial for p53-mediated apoptosis but has no obvious effect on p21 expression, an essential target of p53-mediated growth arrest (el-Deiry et al., 1993). Although the molecular mechanism of p21 transactivation is not well understood, the p21 gene is induced by p53 in almost all cell types by a variety of stresses, suggesting that multiple pathways can promote its activation. Consistent with this notion, while loss of acetylation at each individual site can be compensated by the modification of other sites, loss of acetylation at all sites completely abolishes p53-mediated transactivation of p21. Thus, although expression of p21 likely is redundantly regulated by p53 through modifications of multiple modification sites, our data demonstrate that acetylation is an indispensable event for p53-mediated activation of p21 and subsequent growth arrest.
Earlier studies from us and others showed that acetylation modulates p53 function through multiple mechanisms, including DNA binding, stability control, and coactivator recruitment (Brooks and Gu, 2003). Although we indeed observed these acetylation-mediated effects contribute, in part, to overall p53 activation (Figures 4A, 4B, and 5C), the dramatic loss of p53 functions in both growth arrest and apoptosis (but not in Mdm2 induction) with acetylation-defective p53 suggests the involvement of additional mechanisms. Recent studies have established crucial roles for both Mdm2 and Mdmx in controlling p53 activity in vivo (Michael and Oren, 2003; Marine and Jochemsen, 2005). Since Mdm2 can induce p53 degradation by ubiquitin-mediated proteolysis and downregulate p53 function by repressing p53-mediated transactivation, both ubiquiti-nation-dependent and ubiquitination-independent mechanisms have been proposed for Mdm2-mediated regulation of p53. In contrast, the molecular mechanism by which Mdmx, which is enzymatically inactive as an E3 ubiquitin ligase, downregulates p53 is unclear. Here we show that Mdmx, in a manner similar to Mdm2, can be recruited to p53 target promoters to serve as a transcriptional repressor. Importantly, acetylation prevents downregulation of p53 function by blocking the recruitment of both Mdm2 and Mdmx to p53 responsive promoters.
Inhibition of the p53-Mdm2 interaction is well accepted as a central event for p53 activation during the stress response. Several earlier reports indicated that phosphorylation of the N terminus is essential for p53 activation as the phosphorylation sites lie in or close to the MDM2 binding site on p53; however, a number of other studies have recently shown that p53 can be activated regardless of its phosphorylation status (Ashcroft et al., 1999, 2000; Dumaz and Meek, 1999; Blattner et al., 1999; Wu et al., 2002; Thompson et al., 2004; Prives and Hall., 1999). Thus, it remains a major issue regarding how p53 is activated without inducing its phosphorylation. Our study demonstrates that acetylation of p53 is sufficient for abrogating Mdm2-mediated repression during the stress response in the absence of phosphorylation. The antirepression mechanism described here explains why the function of unacetylated p53 can be restored in human cells by the loss of the p53 repressors (Mdm2 and Mdmx). Other lines of evidence also support this mechanism. For example, the unexpected activity of unacety-lated p53 in reconstituted in vitro transcription reactions with purified components can now be explained by the absence of Mdm2 and Mdmx in this system (An et al., 2004; Espinosa and Emerson, 2001). Moreover, the mechanism presented here is also consistent with recent studies showing that p53 is spontaneously activated in Mdm2 null mice (Ringshausen et al., 2006).
Notably, in addition to the N-terminal domain, the DNA-binding domain of p53 was recently identified as a second binding site for Mdm2 interaction (Yu et al., 2006; Kulikov et al., 2006). Surprisingly, we found that all three major domains of p53 (N terminus, core region, and C terminus) show similar binding affini-ties for Mdm2, which raised the possibility that multiple contacting sites are involved in the stable interaction between p53 and Mdm2. The relationship among phosphorylation, acetylation, and other modifications in p53 regulation warrants further investigations. It is very likely that phosphorylation (e.g., the N terminus) and acetylation of p53 have synergistic effects on inhibiting the p53-Mdm2 interaction, the most crucial step for p53 activation. Indeed, agents that block this interaction, including Nutlin-3, a small molecule antagonist of Mdm2, attract huge interests for their potentials in cancer therapy (Vassilev, 2007). However, blocking the p53-Mdm2 interaction is apparently not sufficient to turn on all p53-responsive promoters. Recent studies showed that the treatment of Nutlin-3 strongly induces p53-dependent growth arrest in cancer cells but has a much-reduced ability to activate proapoptotic targets of p53 (Tovar et al., 2006).
Thus, p53 acetylation may have additional functions besides destabilizing the p53-Mdm2 interaction. Since two key acetylation sites (K120 and K164), in addition to a cluster of acetylation sites at the C terminus reside in the DNA-binding core domain of p53, it will be interesting to know whether acetylation of these sites changes the conformation of p53 in a manner that attracts other promoter-specific regulators. For example, there is accumulating evidence showing that the core domain and the C terminus may be the docking sites for several important p53 coactivators such as ASPPs, 53BP1, Tip60/hMOF, hCAS/CSE1L, and HZF, which are all critically involved in the induction of different targets by p53 (Sullivan and Lu, 2007; Das et al., 2007; Tanaka et al., 2007; Li et al., 2007; Tang et al., 2006; Sykes et al., 2006; Joo et al., 2002). The interactions between p53 and these factors may be modulated by acetylation. Moreover, the differential effect of p53 acetylation on Mdm2/Mdmx recruitment to p53 target promoters needs further elucidation. We failed to detect a significant effect on the Mdm2 promoter by p53 acetylation, which is consistent with normal Mdm2 induction by the acetylation-defective p53 mutant. Thus, the consequences of p53 acetylation are promoter specific and different architectures of the p53-DNA complexes at various p53 target sequences may also contribute to such specificities.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
H1299, U2OS, and 293cells were maintained in DMEM medium and HCT116 cells in McCoy’s 5A medium. All media were supplemented with 10% fetal bovine serum. The inducible cell lines (Tet-off-p53 and Tet-off-p53-8KR) were established by transfection of H1299 cells with the plasmid DNA (pTRE2-hyg-Flag-p53 or pTRE2hyg-Flag-p53-8KR) and selection in DMEM medium supplemented with 5 μg/ml doxycycline (Sigma), 0.2 mg/ml G418 (EMD Biosciences), and 0.4 mg/ml hygromycin B (Roche) for 3 weeks. Transfections with plasmid DNA were performed using the calcium phosphate method and siRNA transfections by Lipofectamine2000 (Invitrogen) according to the manufacturer’s protocol.
DNA Binding
The highly purified proteins used in EMSA include Flag-p53, Flag-p53-8KR, acetylated Flag-p53, GST-Mdm2, and GST-Mdmx. Flag-p53 and Flag-p53-8KR were purified from the transfected 293 cells, GST-Mdm2, and GST-Mdmx from bacteria, and the acetylated Flag-p53 from H1299 cells cotrans-fected with CMV-Flag-p53, CMV-Tip60, and CMV-CBP-HA (Luo et al., 2004). The protein-DNA binding reactions (20 μl) contained 20 mM HEPES (pH 7.6), 80 mM NaCl, 0.1 mM EDTA, 12.5% glycerol, 2 mM MgCl2, 2 mM spermidine, 0.7 mM DTT, 200 ng/μl BSA, 20 ng/μl sheared sperm DNA, 10–20 fmol DNA probe, 20 ng Flag-p53, Flag-p53-8KR, or acetylated Flag-p53. In supershift assays, antibodies (α-p53 [1801], 200 ng; α-Mdm2, 1 μg; α-Mdmx, 1 μg) and an increasing amount of GST-Mdm2 or GST-Mdmx (?30, 100, or 250 ng) were added to the reactions, accordingly. The 159 bp DNA fragment used as probe was obtained by PCR amplification from the human p21 promoter, labeled by T4 kinase (NEB, M0201S) and purified using the Bio-Spin 30 columns (Bio-Rad).
Chromatin Immunoprecipitation (ChIP)
ChIP assay was performed essentially according to the protocol (Aparicio et al., 2004) with some modifications. Briefly, adherent cells were incubated in culture media containing 1% formaldehyde with gentle shaking for 10 min at room temperature, and crosslinking was stopped by addition of 2.5 M glycine to a final concentration of 0.125 M glycine. After two washes with cold PBS, cells were harvested in ice cold lysis buffer (10 mM Tris-Cl [pH 8.0], 85 mM KCl, 0.5% NP-40, 5 mM EDTA, and fresh proteinase inhibitor cocktail) and incubated on ice for 10 min. Nuclei were collected, suspended in cold RIPA buffer (10 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.1% DOC, 1% Triton X-100, 5 mM EDTA, and fresh proteinase inhibitor cocktail), and sonicated to shear the genomic DNA to an average of 300 bp. Cleared extracts were blocked with protein A/G beads (Upstate Biotechnology), and aliquots of the supernatants were used for immunoprecipitation by various antibodies. After seven washes by RIPA buffer with gentle rotation for 5 min each time, the proteins were eluted from the beads by 0.5 ml elution buffer (0.1 M NaHCO3 and 1% SDS). The DNA samples were recovered by phenol extraction and ethanol precipitation after reversal of crosslinking. The purified DNA was then analyzed either by PCR within linear amplification range followed by agarose gel electrophoresis or by quantitative real-time PCR using Applied Biosystems 7300.
Supplementary Material
Acknowledgments
We especially thank C. Prives for valuable suggestions and reagents and R. Baer for critical comments on this manuscript. We also thank E. McIntush from Bethyl, Inc. for developing the Ac-K164 p53 antibody. This work was supported in part by grants from NIH/NCI to Y.Z. and W.G. W.G. is an Ellison Medical Foundation Senior Scholar in Aging.
Footnotes
Supplemental Data include Supplemental Experimental Procedures and 12 figures and can be found online at http://www.cell.com/cgi/content/full/133/4/612/DC1/.
References
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Aparicio O, Geisberg JV, Struhl K. Chromatin Immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Curr Protoc Cell Biol. 2004;17:7.1–7.23. doi: 10.1002/0471143030.cb1707s23. [DOI] [PubMed] [Google Scholar]
- Arva NC, Gopen TR, Talbott KE, Campbell LE, Chicas A, White DE, Bond GL, Levine AJ, Bargonetti J. A chromatin-associated and transcriptionally inactive p53-Mdm2 complex occurs in Mdm2 SNP309 homozygous cells. J Biol Chem. 2005;280:26776–26787. doi: 10.1074/jbc.M505203200. [DOI] [PubMed] [Google Scholar]
- Ashcroft M, Kubbutat MH, Vousden KH. Regulation of p53 function and stability by phosphorylation. Mol Cell Biol. 1999;19:1751–1758. doi: 10.1128/mcb.19.3.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol. 2000;20:3224–3233. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcript. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Hei-merikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P. DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene. 1999;18:1723–1732. doi: 10.1038/sj.onc.1202480. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Das S, Raj L, Zhao B, Bernstein A, Aaronson SA, Lee SW. Hzf determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell. 2007;130:624–637. doi: 10.1016/j.cell.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O’Rourke K, Koeppen H, Dixit VM. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Meek DW. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999;18:7002–7010. doi: 10.1093/emboj/18.24.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–5395. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20:1331–1340. doi: 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes Dev. 2002;16:583–593. doi: 10.1101/gad.959202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, Quong AA, Zhang X, Beerman T, Pestell RG, Avantaggiati ML. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci USA. 2005;102:10188–10193. doi: 10.1073/pnas.0503068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov R, Winter M, Blattner C. Binding of p53 to the central domain of Mdm2 is regulated by phosphorylation. J Biol Chem. 2006;281:28575–28583. doi: 10.1074/jbc.M513311200. [DOI] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- Li AG, Piluso LG, Cai X, Gadd BJ, Ladurner AG, Liu X. An acetylation switch in p53 mediates holo-TFIID recruitment. Mol Cell. 2007;28:408–421. doi: 10.1016/j.molcel.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci USA. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, Jochemsen AG. Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun. 2005;331:750–760. doi: 10.1016/j.bbrc.2005.03.151. [DOI] [PubMed] [Google Scholar]
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell. 2004;16:631–639. doi: 10.1016/j.molcel.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in Mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- Ohkubo S, Tanaka T, Taya Y, Kitazato K, Prives C. Excess HDM2 impacts cell cycle and apoptosis and has a selective effect on p53-dependent transcription. J Biol Chem. 2006;281:16943–16950. doi: 10.1074/jbc.M601388200. [DOI] [PubMed] [Google Scholar]
- Prives C, Hall PA. The p53 pathway. J Pathol. 1999;187:112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 2006;10:501–514. doi: 10.1016/j.ccr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Sullivan A, Lu X. ASPP: a new family of oncogenes and tumour suppressor genes. Br J Cancer. 2007;96:196–200. doi: 10.1038/sj.bjc.6603525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–650. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, Vassilev LT. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem. 2004;279:53015–53022. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Wu Z, Earle J, Saito S, Anderson CW, Appella E, Xu Y. Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol. 2002;22:2441–2449. doi: 10.1128/MCB.22.8.2441-2449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GW, Rudiger S, Veprintsev D, Freund S, Fernandez-Fernandez MR, Fersht AR. The central region of HDM2 provides a second binding site for p53. Proc Natl Acad Sci USA. 2006;103:1227–1232. doi: 10.1073/pnas.0510343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupnick A, Prives C. Mutational analysis of the p53 core domain L1 loop. J Biol Chem. 2006;281:20464–20473. doi: 10.1074/jbc.M603387200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.